0023 - 6837 / I 4 / 7 001 -0039$03.00/0 L.n sonAronY INvESTIGATIoN
Copyright @ 1994 by The United States and Canadian Academy of Pathology, Inc.
Vol. 70, No. 1, p. 39, 1994 Printed in U.S.A.
The
Blood-Retinal
Barrier
in
Experimental
Autoimmune
lJveoretinitis
Leukocyte Interactions
and
Functional
Damage
J.
GnnpNwooD,
R. Howns,
AND S.
Ltcsrue,N
Department
of
Clinical
Science,Institute
of Ophthalmology,
Bath
Street, London
ECLV gEL,
UK
BACKGROUND:
In
posterior uveitis the blood-retinalbarrier
(BRB) plays an important role inthe pathogenesis of the disease. However, the morphologic correlate of BRB breakdown and the
route of leukocyte migration remain poorly defined.
EXPERIMENTAL DESIGN: Using an experimental model of autoimmune uveoretinitis in the rat,
we have examined the ultrastructural alterations and leukocyte interactions occurring at the BRB.
By
employing electron-dense tracers, the developmentof
BRB breakdown, and the route of extravasation were investigated.RESULTS: No increase in BRB permeability was found before lymphocytic infiltration. At day 1O
postimmunization with retinal-soluble antigen and beyond, inflammatory cells could be seen within
the retina that was quickly followed by an extensive increase in the permeability of the retinal
vasculature to lanthanum and horseradish peroxidase. Occasionally, horseradish peroxidase re-action product could be seen extending throughout the 'tight junctions' of the retinal endothelia, but not those of the retinal pigment epithelia. Inflammatory cells, particularly mononuclear cells,
were seen forming perivascular cuffs and extending posteriorly towards the outer retina. Retinal
damage was
initially
restricted to the outer nuclear and photoreceptor layers that werein
closeproximity to these vessels. Leukocytes could be seen adhering to the retinal vessels and penetrating
the endothelial cell cytoplasm close to tight junctions, but were never seen probing the junctions
directly.
At
the retinal pigment epithelium, however, there waslittle
evidence of migration into the retina during the early stages of the disease, even though the choroid often became packedwith inflammatory cells. At later stages, oceasional inflammatory cells could be seen between, and
apparently
within,
retinal pigment epithelium cellsin
areas overlying sites of severe choroidalinfiltration.
CONCLUSIONS: The prime site of leukocyte
infiltration
and damage to the BRB in autoimmuneuveoretinitis occurred
at
the levelof
the vascular endothelia andthat
diapedesis takes placeprimarily via an intraendothelial process.
Additional key words: Endothelium, Lymphocyte, Migration.
The blood-retinal barrier (BRB) forms a selective
cell-ular interface between the blood and the retinal
paren-chyma and
is
functionally identicalwith that of
the blood-brain barrier (BBB). The retinal vascularendo-thelium and retinal pigment epithelium (RPE) that form
the BRB restrict the movement of polar solutes, and also
play a significant part
in
controlling leukocyteextrava-sation. Under normal conditions, the level of leukocyte
traffic through the retina is low, but becomes markedly increased during inflammatory diseases
of
the
retina.This increase
in
cellularinfiltration is
also associated with breakdown of theBRB
(1) and edema formation(2). These two separate but related processes play a major part in the pathogenesis of both human posterior uveitis,
and the animal model of this disease, experimental
au-toimmune uveoretinitis (EAU) (3).
Adoptive transfer studies have shown that EAU is a
CD4* T cell-mediated disease and thus is similar in many
respects to the animal model of multiple sclerosis,
exper-imental autoimmune encephalomyelitis (EAE) (4). The inflammatory cells that mediate these diseases must first be recruited from the circulation via interactive
mecha-nisms
with
the vascular endothelium.This
process isthought
to
be regulatedby the
levelof
expression ofadhesion molecules on both the leukocyte and
endothe-lium and the degree of avidity between the receptor pairs
(5-9). Evidence also exists for endothelial cell (EC)
GREENWOOD, HOWES, AND LIGHTMAN LesoRlrony ltvpstrcltror.r
vein (HEV)-like endothelia (1, 11-19) in both EAE and
EAU, although
it
is not clear whether this phenomenonplqys a significant role in leukocyte recruitment.
The retinal vascular endothelii is in close contact with circulating inflammatory cells unlike the RpE which is
separated from the blood
by
the choroidal vessel walland Bruch's membrane. As a consequence of this ana_
tomical arrangement,
it
is likely that the retinal endo_ thelia is a prime site_of leukocyte recruitment and entry, particularly during the early stages of inflammatorydis-ease (1, 3). For circulating leukoiytes to enter the retina across the retinal vasculature, they must first adhere to
the endothelium followed by diapedesis and migration through
the
extracellular spaceof
tne parenchlima.It
still
remains unclear, however, whether leukocvtes mi_grate through
the tight
junctions between endothelialcells or via an intracellular route.
It
is possible that thepath of migration is dependent upon the cell phenotype
and state of cell activation, and that both iniercelluLr
and intracellular routes of diapedesis occur. For leuko_ c.ft9s t_o cross the posterior aspect of the BRB, the RpE,
their flow must
first
be halted by interactions with the choroidal vascular endothelium, followedby
adhesion and migration out into the extracellula..pu.".It
is onlywhen they have crossed the choroidal vasculature thai
they are able
to
interactwith
the RpE. Recruitment,therefore, at this site is governed not by the RpE, but by
the choroidal endoth_elia. The migration of inflammatory
cells across the RPE, and the route they take, has noi
been well documented, although there is sbme ultrastruc_ tural evidence to suggest that migration into the retina from the choroid does occur (10, 14). The mechanisms
involved in inflammatory cell extravasation in EAU still
requires further investigation and the differential role
played by the two barrier sites in the pathogenesis of this
disease need clarifying.
The alteration
in
functional integrity of the BRB, asa result of both cellular
infiltration
and the release ofvasoactive substances, is a well established phenomenon
that c-linically leads to edema and loss of vision (1b). The
path by which extravasation of plasma constituents oc_
curs, needs to be defined. Unlike that of the BBB, there
are two major areas of the BRB across which leakage
may occur. Furthermore, the surface area of these t#o
barrier sites is collectively much greater than an equiv_
alent area of cerebral cortex (16),
ind
thus has ugr""t.,
potential for leakage of plasma constituents andidema
formation. A further question regarding BRB disruption,
as
with that of
the BBB,
is
the
meihanism throughw^high leakage occurs (17). Whether this is via disrupti6n
of
thetight
junctions, pole formation,or
transcytosisremains a contentious issue.
EXPERIMENTAL
DESIGN, This study was undertaken to investigate the related
phenomena of leukocyte extravasatior,
""rrd BRB break_
down
in
EAUin
rats and to assess the differential roleql-ayed by the two sites of the BRB
in
these processes.The _temporal sequence of events
in
rats induced withEAU by systemic immunization with bovine retinal S_
antigen was determined ultrastructurally with transmis_
sion and scanning electron microscopy (EM). Retinal
vascular endothelia were examined for changes
in
mor_phology,
in
particular thickened EC with inireasedcy_
to^.ljlq _otgg"elle s, and plump morpholo gy characteristic
of HEV. The route of leukoiyte passage-ac.oss both the
retinal
endothelia andthe RpE
was investigated Lyexamining
the
structural interactions between thes-ecells. Furthermore,
by
using electron-dense vasculartracers_, the integrity of the two barrier sites and the path through which tracer extravasation occurs was deter_
mined.
RESULTS
AND
DISCUSSIONLlcur
MrcnoscopvToluidine blue-stained sections from the eyes of all
rats were inspected for structural changes. Up io day 10,
all_Tetina appeared histologically
,ror*il
with no.ig";i
infiltrating-leukocytes. By- day-10, a few
inflam.itory
cells were observed particularly in lhe outer nuclear
ani
photorec.eptor layers,
-t!:
t_qttdr being the known turg"iof S-antigen-mediated EAU (19). Fr6m day 10 onwa"rd,
there was increased extravasation of inflammatory cells
within the neuroretina with notable perivascular
*fn"g
particularly of the venules, and,ru*eious cells ."igraii;E
throughout the parenchyma (Fig. 1o). The
irfl";;;;r;
cells were predominantly mononuclear and could be seei tracking from retinal vessels into the outer retina
(Fij
1b). As the disease progressed, destruction of the outei
retinal layers occurred particularly
at
pointsin
closeproximity to retinal vessels (Fig. 1). Focal accumulation
of inflammatory cells in the choiiocapillaris became more
evident with time but with limited e,oiderrce of migraiion
of cells across the RPE. The overall progression of the
9]:"1.^q was. generally similar to that repoited previously (11, 19) with increasing outer retinal destruction and
thl
formation of
vitritis
anda
subretinal..rohu.-orr"gi"
exudate. The disease finally became quiescent in the 4"th
week postimmunization (PI), leavingsevere destructi,on
of the outer layers of the retina, a limited focal inner
layer damage, and little evidence of further
i"nam-aiory
cell infiltration.
ElncrnoN
MrcRoscopy: MoRpnor,ocy oF THERnrrNer,
EC
,qNo RpE_
Th"
injected control animals displayed normal retinalpC m9rylology throughout the uasc.rja. bed during lhe
4-week PIperiod, with no extravasation of inflammitory celts. Similarly, the structure of the RpE was consisteni
with_ normal perfusion-fixed noninjected animals. In
EAU-induced rats, the retinal EC and
RpE
remainedstructurally unchanged during the first I0 to 12 days pI.
.t'rom day 12 onward during
the
active phaseof
thedisease process (11), alterations
in
these cells becamemore evident, although large-scale changes d.id not occur
until there was substantial inflammatory cell infiltration
and parenchymal damage. Occasionally raised, bulbous
endothelia could be identified
with
both transmission (FiS. 2a) and scanning electron microscopy (Fig. 2b),pqrticularly
in
areasof
extensive cellulafinfiltiation.
This phenomenon has previously been described in EAE
(12,.7.3,?-0-,.21), multiple sclerosis (22),EAIJ (1, 11), and
uveitis (23), with the implication that these endoihelia
-{...'..i)
... .*
' ''\ :."..:ij.:;$"-'; .;!' ,!i\r. ,. $ -... i ,.r:' . V
. u.... ..
. i.
\.ss.sil:,ia-.s.r :.s, ,: r
\s:.-', \N:,. '. , ,Stl.,
Vol. 70, No. 1,
la
I.994
BLOOD-RETINAL BARRIER IN EXPERIMENTAL AUTOIMMUNE UVEORETINITISx.b
Ftc. 1. a, Toluidine biue-stained resin section of retina, day 21 PI'
Substantial perivascular cuffing of inflammatory cells (,L) and destruc-tion of underlying outer retina (orrou). Note paucity of leukocytes in choroid. b, Toluidine blue-stained resin section from day 18 postim-munization. Inflammatory cells can be seen migrating into the outer retina from retinal vessel (white atow) causing localized damage' A
few inflammatory cells can be seen in the photoreceptor layer' Adjacent RPE cells contaln numerous cellular inclusions. Figure 1o, x100; b'
x220.
FIc. 2. o, Transmission electron micrograph (?8i14) of retinal
ves-sel, day 21 PI. Raised, bulbous endothelial cell with microvilli (lorge
.'....
\
s:
t-\
arrow) lying over area of large-scale leukocy'te infiltration. HRP
reac-tion product can be seen in BM (srnoll arrow) and' extending into parenchymal extracellular space. b, Scanning electron micrograph (SEM), day 12 PI. Two adherent inflammatory cells can be seen
surrounded by piump, protruding endothelial cells (bloc& arrows). Oc' casional iong microvilli projecting from the EC can be seen (white orrou). Figure 2o, x3,000; b, x2,500.
FIc. 3. o, SEM, day 12 PI. Flattened inflammatory cell adhering to
retinal vessel wall. b, Higher power of o showing microvilli at the junction between ECs (arrorus). Figure 3o, x7,270; b, x3,340'
$Nr,,
| .ri
{$ii-\.x.:r:i:.:.i.l:\l)\\.\:Si\i': ai:,\i:s\$it\\ii\\\ii:\:i.ii:\\
N
\Sl-N' - :-l\
Nr'\\S::'*f\.sx:l N.\$,..\,,1. ',.,.1
42 GREENWOOD, HOWES,
AND LIGHTMAN Llsonerony ItvnsrtclrroN
resemble those of the HEV. Whether these endothelia
also express the addressin adhesion molecules associated with HEV, as has been demonstrated
in
EAE (12, IB),remains to be established.
In
some large vessels of the retina, especially the arteries, raised, ridge-like endothe_lia
appearto
bea
normal feature and- shouldnot
bemistaken for HEV-like endothelia. An additional
poten-tial difficulty in interpreting vessel morphology is derived
from the method of fixation used. In this study, the tissue
was fixed by perfusion through the ascending aorta to
p-reserve vessel tone, obtain rapid fixation, and maintain
the structural interactions belween the retinal EC and
leukocytes (13).
In
previous comparable studies withEAU, immersion fixation was used (10, 11, 14) which does not sustain vessel tone and fixes the vasculature in a 'collapsed' state. Contrary to the state of endothelial preservation,
the
RPE appearsto
be better presewedwith immersion fixation as this method avoids the
vac-uolation of the junctional region between the cells seen
in
perfusion fixed material. Although these differencesin-fixation technique are not critical, they must be con_
sidered when comparisons are made between different
studies.
In those areas where leukoclte adhesion and infiltra_
tion was greatest, the endothelia generally remained flat
but. thickened, displaying increased amounts of cytosol
and a .dramatic upregulation
in
the levelsof
cyttsohcorganelles such as rough endoplasmic reticulum, ribo_
sosmes, and vesicular-like profiles. This activation of the
endothelia has been reported in both EAE (24) and EAU
(10), and
is
likely
to
be
a
consequenceof
increasedendothelial cell metabolism and protei., synthesis.
Cy-tokine activation of retinal endothelia can lead to uprl_ gulation and increased expression
of
moleculesof
im-munologic significance, such intracellular adhesion mol_
99yle-1 (25), major histocompatibility complex class
II
(26), transforming growth
fictor-p
aswell
asthe
in_greeseq production of extracellular matrix proteins (g).
In EAU, the structural changes have been quantified and
have demonstrated a clear correlation between
endothe-Iial cell thickness
in
capillaries, venules, and veins andthe severity of the disease (11).
With
scanning EM, theluminal surface of the endothelial vessels exhii*bited sub-stantial numbers
of
microvilli
that
could be seen todelineate the contact points between cells (Fig. 3)
in
asimilar fashion
to
that
in
EAE
(13, 21). Occisionally, very long processes could also be seen emanating fromthe endothelia (Fig. 2b). As the disease progressedio the
point
of
maximal leukocyte extravasition and tissuedamage, there was evidence of endothelial cell necrosis
and death.
The RPE maintained
its
structure during the earlystages
of the
disease. Duringthis
period,the initial
infiltration of inflammatory cells into the photoreceptor
layer was accompanied
by the
appea.a.rceof
cellularinclusions, such as phagocltosed malerial and large dark
electron-dense bodies, in the adjacent RpE (Fig. tA). R,
the disease progressed, the RPE persisted
u."u
-orro-layer, with only rare focal signs ofhypertrophy in areas
where there was severe infiltration
ofthe
clioioid. Oncedestruction of the photoreceptor layer and formation of
a subretinal exudate had occurred, the RpE remained as
a
qell-presewed monolayerwith
apical microvilli ex_tending
into
the
exudate.At
this
stage, there was areduction in the number of cytosolic org:anelles which is
likely to result from the cessation of ph"agocltosis of rod
outer segments and their subsequent degradation.
PpRiraslsrr,rry oF THE
BRB
ro
ELEcTRoN_DENsETnacnRs
The BRB maintained
its
structural integrity to both HRP and lanthanum during thefirst
few d'ays of onsetofthe disease, being consistent with previous studies (t).
As the numbers of leukocytes interacting with the retinal
E_C and migrating across increased, theie was a detecta_ ble increase in the permeability to tirese tracers. previous
work has demonstrated that this increase in BRB perme_ ability occurs concomitantly
with
leukocytei extravasa_tion
(1).In
both EAU and EAEit
is incieasingly clearthat
thesetwo
events are inextricably linked(Zl_Sl)
despite earlier opinion
that
leakage acrors the barrieroccurs before leukocybe infiltration (92, Ag). Leakage of
tracer was nearly always restricted to areas of inflam_ mation and associated almost exclusively with the retinal vasculature and not the RPE.
HRp
could be seen ex_tending along
the
basement membrane andinto
the extracellular space (Fig. ). Unlike a previous study in which abnormalities of the EC tight junctions were not found (14), we have confirmed ttrai tightlunction disrup_tion does occur (1, 10, 84) as occasionattv
Hnp
*". ."",
alongthe entire length of a'tight' junction (Fig. ad). The
lack
of
finding many junctions hiledwith
t-"raceris
aconsequence of the tortuosity ofjunctions and the diffi_
culty of obtaining, in a single plane, a complete junction from luminal
to
abluminal side. Moreovei,it
is
also afunction of their relative infrequency in that opening of
these junctions is likely to occui in a precise and punciate manner (17) with a substantial capacity to reseal. Junc_
tional disruption remains the most likely route through which
initial
extravasation occurs, although leukocfrepenetration may also carry through very small amounts of tracer (27, 28). Differential permeability to tracers
of
different molecular weight (1), which haj been used to distinguish the route of BBB breakdown after
hyperos-molar disruption (3b), is indirative of small pores foiming
through the junctions. Had extravasation occurred
bi
pinocytosis and vesicular transport, as has previouslj, been suggested
in
EAE
(24, eO;,this
size distinction would not have been apparent. This does not, of course,exclude the possibility that vesicular transport occurs at
a later stage of the disease, although whether
it
plays arole in net transfer remains questionable (37).
Occasion-ally, tracer-filled vesicular-like profiles could be
identi-fi-ed (Fig. 4b), but these were mostly associated with the abluminal plasma membrane
that
have been shown to be a normal featureof
central nervous system (CNS)endothelia (37).
In
addition, vesicular-like profiles, thaiwere largely devoid
of
tracer did become-a
prevalent feature of the activated endothelia during thedevelop-ment of the disease resembling those seen
in
EAE (24),3"{
T.u"V other pathologic conditions of the CNS (aA),including the retina (39).
The factors that are responsible for inducing
VOI. ?0, NO. 1,
1994
BLOOD-RETINAL BARRIER IN EXPERIMENTAL AUTOIMMUNE UVEORETINITISFrc. 4. TEM, day 21 PI. o, Lymphocyte adhering to large retinal vessel EC. HRP reaction product can be seen filling the BM and extracellular space (orror^us). b, Retinal capillary with HRP reaction product inBM (large arrow) and extracelluiar space. HRP-5lled ablu-minal invaginations can be seen (small arrows)- c, Mononuclear cells adhering to wall of large retinal vessel and in perivascular region with extravasated HRP in BM (orror.o)' d, HRP extending the length of a
tight junction (arrow) and filling the BM and abluminal pits
(arroru-h.eads). a, x8,000; b, x10,000; c, x2,500; d, x20,000.
GREENWOOD, HOWES, AND LIGHTMAN LlsoReronv lNvnstrclrtox
those implicated
in
disruptionof
the
BBB
(17, 40).During many disease processes of the CNS, there ls the
release of a wide spectrum of compounds with a potential
for acting upon the cells of the blood-CNS barrieis. Many
ofthese agents have been shown to cause opening
ofth!
barrier, although the cellular mechanisms througi which they operate are poorly defined.
It
has been p6stulatedthat many of these act by altering intracellular calcium
leading to induction of pinocltosis or alterations in the
binding capacity of the tight junctions. Compounds such
as histamine, bradykinin, arachidonic acid and
its
me-tabolites, the eicosinoids, have long been known to bring
about. changes
in
permeabilitybut
more recently,thI
cybokines such as interleukin-1 and tumour neirosis factor, have also been implicated in BRB disruption (41,
42). "Ihe cytokines, that are produced within fhe retina
in EAU (43), are known to induce leukocvte infiltration
(41), but whether the increase
in
vesicuiar activityre-ported is a direct or indirect consequence of this
phenom-enon requires further analysis.
The physical disruption caused
by
leukocltes pene-trating the BRB may also give riseto
smali transientleaks in barrier integrity. In the present study, no tracer
was recorded filling the space between the invading leu_
kocyte and the endothelial membrane as has
been"dem-onstrated
in
EAE (27, 28). This route of extravasation,however, remains
a
distinct
possibility, although theendothelial cell is thought to seal once the leukoclte has
migrated through (13), especially if diapedesis is t-hrough
and not between the EC.
_
At
the RPE,virtually
no leakageof
HRp
could bedetected, even when inflammatory cells were
in
closeproximity
in
both the photoreceptor layer and the cho-roid.In
only
a
single section, where there was total destructionof
the
photoreceptor layer, coulda
small amount of HRP be detectedfilling
the spaces betweenthe apical microvilli.
It
was not cleir, however, whether this had diffused across the RPE or from nearby retinal vessels. The smaller tracer lanthanum was also mostlyexcluded by the RPE.
It
could often be seen extendingthrough the junction between apposing RpE up to, bul not.beyond,,the apical
tight
junctions(Fig.5).
Veryrarely, and
in
minute amounts, lanthanum was seenbetween the apical microvilli beyond the tight junctions
indicating a small but detectable disruption to the RpE
tight junctions. These small permeability changes found
at the RPE in EAU are compatible with previous reports
of minimal damage to the RPE barrier (10).
Lpuxocyrn IxrpnncrroNs wrrH
THE BRBIn_the early phase of the disease, where tissue damage
was localised to occasional perivascular regions and the
photoreceptor layer underlying these areas, infiltrating
cells, predominantly mononuclear, were found
in
th6 surrounding tissue and adhering to the luminal wall ofthe vessel (Fig. 4a and c). The majority of these cells
were lymphocytic in appearance with a smaller
percent-age
of
monocl'tesand
occasional polymorphonuclear cells. These inflammatory cells, especially those that were spherical or ovoid in shape, were often attached tothe EC luminal membrane by what appeared to be fairly
tenuous and infrequent connections (Fig. 4a andc).
Wit[
scanning EM, inflammatory cells could be seen adhering
to the vessel wall of veins and venules in large numberi (Fig. 6o) and occasio,nally within microvessels Gig. 6b).
Mononuclear cells adhering to the vascular endothlehum
could often be seen probing into the endothelial cell in close proximity
tq
but not
into, the tight
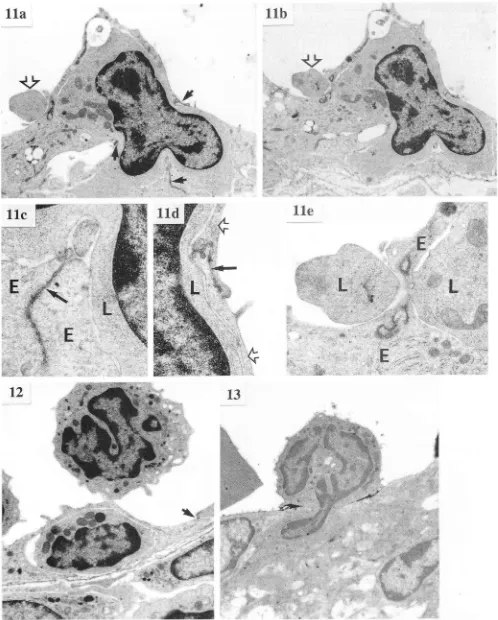
junctionsbetween .apposing ECs (Figs. 7, 8, g, and fb).
thi.
p"rr-etration into the body of the endothelial cell. whicli did
not
disruptthe
endothelial cell membrane. has beenpreviously described for lymphocytes in
Oln
(fS).Sim-ilarly, the direction of penetration sometimes appeared
to bisect- the plane of a junction between two overiapping
endothelial cells in a manner described by Raine
"{ "t.
ii
EAE
(13) and Wekerleet
al.
(aa)witir
myelin basicprotein-specific T cell line lymphocyte migration through brain endothelial cell monolayers in
uitri.
^
Despite the interpretational difficulties imposed by a2-dimensional image,
it
appeared that in-os[
case., lhe migratory route wasnot via
a junction,but
in
closeproximity to
it.
The route that leukocytes take throughthe CNS vascular barrier remains a iontentious issrie.
Despite growing evidence
that
in
CNS inflammation,diapedesis can occur through endothelial cells (11, 13,
27, 44), there remains a degree of scepticism especially
from those working outside the CNS. A reason for this could be
that this
route of passage may be unique toendothelia of the CNS, wheri celli are attached to orr"
another by tight junctions. The strong adhesive
proper-ties
of
these junctions may be suffrciently great thatpenetration is more easily accomplished by the leukocyte
taking an intracellular, rather than an intercellular
paih-way. This would involve invagination of the fluid plasma
membrane of the EC at the point of penetration of the
leukoclte pseudopodium (Fig. T,
8,
and 9), either byphagocytic
type
mechanisms,or by
electrostatic andmechanical forces. The invagination would continue un_
til
the
cell becomes attenuated andthe luminal
andablum-inal plasma membrane abut each other (Fig. Td
arld 8/). Once this has occurred, pore formation betiveen
the two membranes could develop, allowing the
unhin-dered passage of the leukocyte into the periviscular space
(Fig. 10d and e). A further advantage ofthis route wbuld
be the_increased degree
of
control over diapedesisaf-forded by the endothelial cell.
It
is already clear that therecruitment of inflammatory cells by both cerebral and
retinal
endothelia canbe
regulatedby
their
level ofexpression
of
adhesion moleculesthat
can be inducedand upregulated
by
cytokines (E-7,g,
25, 4E). Indeedtreatment of EAE animals with an antibody that blocks
the very late antigen-4/vascular cell adhesibn
molecule-1 T]hesion pairing has been shown to prevent leukocyte
infiltration and paralysis (46). However, the endotheiial
cell may al,so play an additional role by actively
facilitat-ing migration by forming pores at the point of leukocvte
penetration, a process
that is
likelyfo
involve theie-arrangement of the cytoskeleton. This hlpothesis, how_
ever,
will
require careful examinationin
order toestab-lish the route of extravasation and the role of the EC in
diapedesis.
.
In
some cases, mononuclear cells could be seenpar-tially or completely surrounded by endothelial cells (Figs.
VOI.70, NO. 1,
1994
BLOOD'RETINAL BARRIER IN EXPERIMENTAL AUTOIMMUNE UVEORETINITIS,,\$u
Frc. 6. SEM, day 12 PI. a, Large vessel (probablv vein) with
nu-merous inflammatory cells adhering to the vascular wall' Smaller vessel
(probably arteriole) with ridge-like endothelia devoid of inflammatory cells (asierisk). b, Capillary with spread, migrating inflammatory cell in lumen (arrow\. Figure 6o, x1,090; b, x2'500.
Frc. 7. TEM, day 18 PI. o and b, Lymphocytes adhering to retinal
:r!:i*': {: ;l
EC and projecting pseudopodia into the EC at points close to tight junctionJ (imall arrows). Lanthanum coats the surface of the cells
-(open
arrows). c and d, Higher powers of o and b showing lymphocytes probing into EC (arrowheads\ in close association wittr tight junctions Tarrori). Figure 3o, x15,000; b, x20,000; c, x25,000; d,x50,000.
i::' ."
GREENWOOD, HOWES, AND LIGHTMAN
8b
L,leonltonv Invrstrclrron
\i':\l:'.
...$.j..\;. . \4. t. .-. .
';{i\l$i,' .'..,r'.ili
r ia:-::..,.. 1-..-_ :. .'
Sl'i :..:' ..-i.1:\l)..' l.i ':.i;i\...::r-i.l\.i\.s\at:ir:l.)r"i: ;.,:'r'
FIc. 8. TEM of mononuclear cells probing endothelial cells of ret_
inal vessels, day 12 PI. b and.c, Higher power micrographs of o showing mononuclear cell pseudopodia penetrating EC in association with anJ at a distance from_tight junctions (arrowl). An increase in EC rough endoplasmic reticulum, ribosomes, and vesicular profiles (arrowh.eaii
can be seen. d, Mononuclear cell with pseudopod.ia inserted into EC (arrowhea.d,) with no associated tight junction. e and
f, Mononuclli
cell penetrating deep into EC.near tightjuncti on (arrow)' cau*i.rg .euere EC attenuation (arrowhea.ds-). Figure gL, xa,g00; b and c, xZA,}}O;
i,
Vol.70, No. 1,
1994
BLOOD-RETINAL BARRIER IN EXPERIMENTAL AUTOIMMUNE UVEORETINITIS-&
Frc. 9. a and b, TEM of serial sections through migrating mono-nuclear cell at junctional region (arrow). Figure 9o, x7,000; b, x8,000. Day 12 PI.
Ftc. 10. o, b, and c, TEM of serial sections through migrating
mononuclear cell, day 12 PI. Inflammatory cell process is penetrating through the EC close to, but not through, the junction (arrows). d,
Higher power micrograph of b showing a hole in EC close to the
junction (small atow) with intact BM. Increased expression of rough endoplasmic reticulum (atowheads) and vesicular profiles (open
ar-roir.,) in EC. . e, Higher power micrograph of c showing mononuclear cell penetrating EC and BM close to the EC junction (small arrow). Dense accumulation of actin-like filaments can be seen in the leading
edge of migrating ceII (large orroru). Figure 10o to c, x8,000; d, x20,000;
1le
t :. :.. :,. ..' :.'ii''.i.r.y.-1y'11r.:1
\\
\:
l 1a
1.,
W\\\RN\\\N\.S\\\\,\{i' ..is\f:iw
FIc. 11. TEM of a mononuclear cell surrounded by EC, day 12 PI.
o, Part of the inflammatory cell remains within the vessel lumen (open
arrow), but is not close to any EC tight junctions (arrows). b, Serial section from same block as o showing an external part of the mono-nuclear cell in continuity with the enclosed part demonstrating that the path of entry is intraendothelial and is not associated with any tight junction. c and d, Higher powers of o showing EC tight junctions (arrows). EC (E and open atows), inflammatory cell (I). e, Higher power from b of part of mononuclear cell protruding into lumen. The
EC (E) at this point is devoid of tight junctions. Figure 11o and b,
[image:10.595.51.549.37.657.2]x6,000; c and d, x25,000; e, x17,000.
FIG. 12. TEM of mononuclear cell almost completely surrounded by EC at point removed from the tight EC junction (anow). Adherent spherical mononuclear cell attached by tenuous contact point. Day 12
PI. x8,000.
Frc. 13. TEM of inflammatory cell migrating through EC and basement membrane. Some lanthanum can be seen between migrating cell and EC. Day 18 PI. x8,000.
-*s;a*i$''s-*--'r,'if -:r {\.\.:ii\ii.*N$'liaa\i\\-\')'.1l:\\:\\
T
Vol. 70, No. 1,
1994
BLOOD-RETINAL BARRIER IN EXPERIMENTAL AUTOIMMUNE UVEORETINITIS49
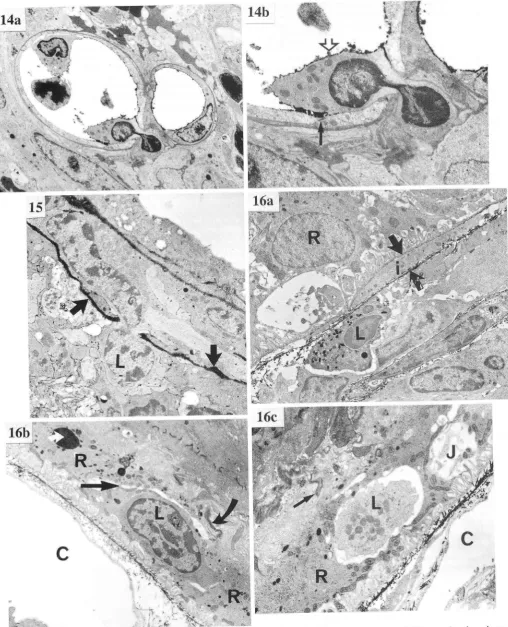
FIG. 14. TEM of lymphocyte in the process of extravasation' day
ra PrI
",
^S".1i;
ilit*gt, two retinal microvessels showing adherent
iii
-igt"ti"c
cells. L-anthanum can be seen coating the luminal;;;;;.ilMigtating
Ivmphocvte near the tight junction (anow)'.o"t"a *itft hnthinum. Figure 14o, x3,900; b' x8'000'
FIc. 15. TEM of inflammatory cell ('L) migratin-g through gap in
tft. gM;iri"h is filled with HRP (arrows)' Dav 21 PI' x4'000'
^
Ftd.- ro. f:Orr'f of RPE. o, Inflammatory cells packed within choroid
""a "ittti"g-g;.tt't t t"-bt"tt.
(l)' Separatio:r-of the layers of Bruch's
[image:11.595.53.562.56.684.2]50 GREENWOOD, HOWES, AND LIGHTMAN LesoRltoRY INVESTIGATI0N
purified bovine retinal S-antigen (54). The antigen was
emul-sified
in
complete Freund's adjuvant (1:1; Gibco, Paisley,United Kingdom), and enriched with 2.5 mg/mI of
mycobacte-rium tuberculosis (strain H37Ra). Each animal was injected
with 100 prl containing 50 pg of S-antigen; 50 pl into the footpad
and 50 pl into the base ofthe tail. In addition, each rat received
5 x 10e killed Bordetella Pertussis organisms given in 300 pl of
phosphate-buffered saline intraperitoneally. Control animals
(N
:
10) were injected intraperitoneallywith
complete Freund's adjuvant alone, with or without killed pertussisorga-nisms.
Ur,rnlsrRucruRAl MoRpHor,ocv
Animals were terminally anaesthetized with pentobarbitone
(50 to 60 mg/kg intraperitoneally, and the thorax opened and
a cannula inserted via the left ventricle into the proximal
ascending aorta. The animals were then killed by perfusing the vasculature with one-half strength Karnovsky's fixative (2%
formaldehyde;2% glutamldehyde; 0.2 u sodium cacodylate; 6.5
mu calcium chloride) at atate of 25 ml/minute. The descending aorta was tied off and afber 3 minutes, the flow rate of fixative
was reduced to 12 ml/minute. After 15 minutes of fixation, the eyes were removed and placed in fixative at
4'
C overnight.Each retina was then prepared for electron microscopy by an
incision through the sclera behind the ciliary body that was
then extended 360 degrees. The cornea and lens were removed,
and the posterior eye cups washed in cacodylate buffer and embedded in 3% agar just before setting. For transmission EM, 100-pm sagittal sections of the embedded posterior eye cup were cut at the level of the optic nerve on a vibroslice (Campden
Instruments, United Kingdom). For scanning EM, 200- to 500-trrm sections were cut from the same eye.
All
sections werepostfixed
in
1% osmium tetroxidefor
t
hour washed anddehydrated through ascending concentrations of ethanol. The
100-1cm sections for TEM were flat embedded in resin between
two aluminium foil-coated glass slides. After the resin had set,
the slides were separated and small blocks of retina cut from clearly defined areas of the flat 100-prm sections, and glued to
larger trimmed resin stubs. Thick sections were cut and stained
with toluidine blue and viewed under the light microscope.
Thin sections were cut from selected areas, placed on copper grids, and counterstained with uranyl acetate and/or lead
cit-rate and observed on an Hitachi H600 transmission electron microscope.
Sections for scanning EM were dehydrated in graded ethanol
and critical point dried with COz. They were then stuck to
stubs and sputter coated with 20 nm of gold before observation on an Hitachi 5520 scanning electron microscope.
PrR[.rnlsrr,rty STUDIES wITH TRAcER
Rats in each group were terminally anaesthetized with
pen-tobarbitone (50 to 60 mg/kg intraperitoneally). For the HRP
study, the anti-histamine, diphenhydramine was injected
intra-peritoneally (0.5 m/kg). After 10 minutes, 50 mg of HRP (Sigma
type
II,
Sigma, Dorset, United Kingdom) in 200 pl of salinewas injected intravenously. After 5 minutes, the thorax was opened and the animals were killed by perfusing the vasculature
with one-half strength Karnovsky's fixative (2% formaldehyde;
2% glutanldehyde; 0.2 tu sodium cacodylate; 6.5 ml,t calcium
chloride) at a rate of 25 ml/minute. The eyes were removed and cut into 100-pm sections as described above. The sections were then incubated with diaminobenzidine for 10 minutes to
produce the electron-dense HRP reaction product. After
thor-and EAE (13, 21). These cells lay between the retinal EC
and basal lamina causing
lifting
of the endothelial cell.This phenomenon may explain the raised appearance of
some
of
the
cellsin
the
SEM micrographs (Fig. 2b).Alternatively, inflammatory cells could also be seen mi-grating through the EC and basal lamina together,
with-out causing separation (Figs. 10, 13 and 14).
Inflamma-tory cells that had already entered the perivascular region
were also detected penetrating the basement membrane
through relatively small gaps and migrating further into
the
parenchyma (Fig. 15).This
processof
migrating throughthe
extracellular spaceis
thoughtto
involve different adhesion molecule pairings to those involved in the binding to, and migration across, the EC (47).Fur-thermore, it often overlooked that activated lymphocytes
are also induced
to
secrete enzymes that are capable ofdegrading the basal lamina and extracellular matrix (48,
49). More recently, the degradation products from these
enzymes have been visualized
in
EAE, generating newdata regarding the mechanisms of lymphocyte migration
(50).
At
the posterior barrier, the choroid was frequently foundto
befull of
inflammatory cellsbut
with
little evidence of migration through the RPE. The presence of inflammatory cells at this site may be due to the releaseof inflammatory and chemotactic factors from the
dam-aged overlying retina inducing leukocyte recruitment and
accumulation in the choroidal extracellular space.
Cyto-kines released from
the
retina couldbring
about theinduction and upregulation
of the
requisite adhesionmolecules on the choroidal endothelia, thus initiating the recruitment of circulating inflammatory cells. This would imply that either the RPE barrier has become permeable
to such factors, or that
it
has been stimulated to secretethem from their basal surface. There is increasing
evi-dence that under certain conditions, RPE cells are
ca-pable of secreting cybokines such as interleukin-6 (51), interleukin-8 (52), and tumour necrosis factor-a (53) which could lead
to
the
recruitmentof
inflammatory cellsin
the absenceof
disruption of the RPE barrier. Migration from the choroid into the retinal parenchymaappears to be limited by the RPE during the early and
mid stages of the disease, but becomes more noticeable
when Iarge scale
retinal
damage and detachment hasoccurred.
In
areas of severeinfiltration
and destructionof the choroid, inflammatory cells could occasionally be
seen between the layers of Bruch's membrane (Fig. 16o),
between the membrane and the RPE, and between
ad-jacent RPE cells. There was also evidence of
inflamma-tory cells apparently
within
RPE cells (Fig. 16b and c)which would suggest an intracellular route.
At
this site,however,
it
wasdifficult
to
ascertainthe
direction ofleukocybe migration.
METHODS ANrulr.s
Female Lewis rats
(N
:
54; 150to
200 gm) were usedthroughout. The EAU group (N
:
44) was immunized withPI. b, Inflammatory cell (.L) engulfed by RPE cell (R) close to junction (straight atow) which has characteristic rows of mitochondria on either
side. The choroid (C) is clear of inflammatory cells. A small amount of
HRP reaction product (curued atow) can be seen between apical microvilli. The photoreceptor layer has been destroyed and a dark
inclusion body (white arrow) can be seen in the RPE. Day 26 PI. c,
Inflammatory cell (.L) apparently within RPE cell (.R) close to junction
Vol.70, No. 1,
1994
BLOOD-RETINAL BARRIER IN EXPERIMENTAL AUTOIMMUNE UVEORETINITIS 51ough washing, they were then osmicated and prepared for transmission EM as described above. For the lanthanum study,
the thorax was opened and the animals were sacrificed by perfusion fixation with one-half strength Karnovsky's fixative
containing 1% lanthanum nitrate for 15 minutes, washed in
distilled water, and the retina prepared for transmission EM
as described above,
Acknowledgenxents: We would like to thank the
Lev-erhulme Trust and Moorfields Eye Hospital Locally
Or-ganized Research Scheme for supporting this work.
Date of acceptance: July 12, 1993.
Address reprint requests to: Dr. J. Greenwood, Department of Clin-ical Science, Institute of Ophthalmology, Bath Street, London ECIV gEL UK.
REFERENCES
1. Lightman S, Greenwood J. Effect of iymphocytic infiltration on the blood-retinal barrier in experimental autoimmune uveoretini-tis. Clin Exp Immunol 1992;88:473-7.
2. de Kozak Y, Sakai J, Thillaye B, Faure JP. S antigen-induced experimental autoimmune uveo-retinitis in rats. Curr Eye Res
1981;1:327-37.
3. Greenwood, J. The blood-retinal barrier in experimental autoim-mune uveoretinitis (EAU): a review. Curr Eye Res 1992;11 (sup-plement):2:5-32.
4. Calder VL, Lightman SL. Experimental autoimmune uveoretinitis
(EAU) verses experimental allergic encephalomyelitis (EAE): a
comparison of T cell-mediated mechanisms. Clin exp Immunol 1992;89:165-9.
5. Greenwood J, Calder V. Lymphocyte migration through cultured endothelial cell monolayers derived from the blood-retinal barrier. Immunoiogy, 1993;80:401-6.
6. Wang Y, Calder VL, Greenwood J, Lightman SL. Lymphocyte adhesion to cultured endothelial cells of the blood-retinal barrier. J Neuroimmunol, 1993;48:161-8.
7. Hughes CCW, Male DK, Lantos PL. Adhesion of lymphocytes to cerebral microvascular cells: effects of interferon-7, tumour necro-sis factor and interleukin-1. Immunology 1988;64:677-81.
8. Mahalak SM, Lin W-L, Essner E, Shichi H. Increased immuno-reactivity of collagen types I, III, and V, fibronectin and TGF-p in retinal vessels of rats with experimental autoimmune uveoretinitis. Curr Eye Res 1991;10:1059-63.
9. Male D, Pryce G, Hughes C, Lantos P. Lymphocyte migration into brain modelled in uitro: control by lymphocyte activation, cyto-kines, and antigen. Cell Immunol 1990;12?:1-11.
10. Lin W-L, Essner E, Shichi H. Breakdown of the blood-retinal barrier in S-antigen-induced uveoretinitis in rats. Graefe's Arch Clin Exp Ophthalmol 199l;229:457 -63.
11. McMenamin PG, Forrester JV, Steptoe RJ, Dua HS. Ultrastruc-tural pathology of experimental autoimmune uveitis. Lab Invest 7992;67:42-55.
12. O'Neill JK, Butter C, Baker D, Gschmeissner SE, Kraal G, Butcher EC, Turk JL. Expression of vascular addressins and ICAM-I by endothelial cells in the spinal cord during chronic relapsing exper-imental allergic encephalomyeiitis in the Biozzi AB/H mouse.
Immunology l99l;7 2:520-5.
13. Raine CS, Cannella B, Duijvestijn AM, Cross AH. Homing to central nervous system vasculature by antigen-specific lympho-cytes. II Lymphocyte/endothelial cell adhesion during the initial
stages of autoimmune demyelination. Lab Invest 1990;63l,476-89.
14. Dua HS, McKinnon A, McMenamin PG, Forrester JV. Ultrastruc-tural pathology of the 'barrier sites' in experimental autoimmune uveitis and experimental autoimmune pinealitis. Br J Ophthalmol 1991;75:391-7.
15. Lightman S. Vascular changes in the posterior segment in clinical and experimental ocular inflammatory disease. Eye lggT;5:432-7. 16. Gratton J, Greenwood J, Luthert P, Lightman S. A quantitative comparison of blood-retinal and blood-brain barrier distribution and density in the rat using image analysis (abstr). J Physiol
1991;446:508.
17. Greenwood J. Experimental manipulation of the biood-brain and blood-retinal barriers. In: Bradbury MWB, editor. Physiology and pharmacology ofthe blood-brain barrier. Handbook Exp
Pharma-col 103: New York; Springer-Verlag, 1992:459-86.
18. Forrester JV, Borthwick GM, McMenamin PG. Ultrastructural pathology of S-antigen uveoretinitis. Invest Ophthalmol Vis Sci 1985;26:7281-92.
19. de Kozak Y, Thillaye B, Renard G, Faure JP. Hyperacute form of experimental autoimmune uveo-retinitis in Lewis rats; electron
microscopic study. Graefe's Arch
Clin Exp
Ophthalmol 7978;208:735-42.20. Cross AH, Raine CS. Central nervous system endothelial cell-polymorphonuclear cell interactions during autoirnmune demyeli-nation. Am J Pathol 1991;139:1401-9.
21. Lossinsky AS, Badmajew V, Robson JA, Moretz RC, Wisniewski HM. Sites of egress of inflammatory cells and horseradish
peroxi-dase transport across the blood-brain barrier in a murine model of chronic relapsing experimental allergic encephalomyelitis. Acta Neuropathol (Berl) 1989;78:359-71.
22. Prineas JW. Multiple sclerosis:presence of lymphatic capillaries and lymphoid tissue in the brain and spinal cord. Science (Wash-ington) 1979;203:1123-5.
23. Charteris DG, Lee WR. Multifocal posterior uveitis: clinical and pathological findings. Br J Ophthalmol 1990;74:688-93.
24. Claudio L, Kress Y, Norton WT, Brosnan CF. Increased vesicular transport and decreased mitochondriai content in blood-brain bar-rier endothelial cells during experimental autoimmune encephalo-myelitis. Am J Pathol 1989;135:115?-68.
25. Liversidge J, Sewell HF, Forrester JV. Interactions between lym-phocytes and cells of the blood-retina barrier: mechanisms of T
Iymphocy'te adhesion to human retinal capillary endothelial cells
and retinal pigment epithelial cells
in
ultro. Immunology1990;?1:390-6.
26. Liversidge JM, Sewell HF, Forrester JV. Human retinal pigment epithelial cells differentially express MHC ciass II (HLA DP, DR and DQ) antigens in response to in uitro stimulation with lympho-kine or purified IFN-7. Clin exp Immunol 1988;73:489-94.
27. Butter C, Baker D, O'Neill JK, Turk JL. Mononuclear cell
traf-ficking and piasma protein extravasation into the CNS during chronic relapsing experimental allergic encephalomyelitis in Biozzi
AB/H mice. J Neurol Sci 1991;104:9-12.
28. Claudio L, Kress Y, Factor J, Brosnan CF. Mechanisms of edema
formation in experimental autoimmune encephalomyelitis. Am J Pathol 1990;137:1033-45.
29. Hawkins CP, Munro PMG, MacKenzie F, Kesselring J, Tofts PS,
du Boulay EPGH, Landon DN, McDonaid WI. Duration and selectivity of blood-brain barrier breakdown in chronic relapsing experimental allergic encephalomyeiitis studied by gadolinium-DTPA and protein markers. Brain 1990;113:365-78.
30. de Rosbo NK, Bernard CCA, Simmands RD, Carnegie PR. Con-comitant detection of changes in myelin basic protein and perme-ability of blood spinal cord barrier in experimental allergic enceph-alomyeiitis by electroimmunoblotting. J Neuroimmunol 1985;
6:349-61.
31. Stoul W, Kaplan MS, Gonatas NK. A quantative assay for exper-imental allergic encephalomyelitis in the rat based on permeability
of the spinal cord to r2sl-human gamma-globulin. J Immunoi
7979;122:920-5.
32. Daniel PM, Lam DKC, Pratt OE. Reiation between the increase
in the diffusional permeability of the blood central-nervous system barrier and other changes during the development of experimental
allergic encephalomyelitis
in
the Lewis rat.J
Neurol Sci1983;60:367-76.
33. Juhler M. Pathophysiological aspects of acute experimental allergic encephalomyelitis. Acta Neurol Scand 1988;78(Suppl 119):1-21. 34. Lightman SL, Caspers-Velu LE, Hirose S, Nussenblatt RB,
Pal-estine AG. Angiography with fluorescein-labeled dextrans in a
primate model of uveitis. Arch Ophthalmol 1987;105:844-8.
35. Robinson PJ, Rapoport SI. Size selectivity of blood-brain barrier permeability at various times after osmotic opening. Am J Physiol 1987;253:R459-R66.
36. Hawkins CP, Munro PMG, Landon DN, McDonald WI. Metabol-ically dependent blood-brain barrier breakdown in chronic
relaps-ing experimental ailergic encephalomyelitis. Acta Neuropathol (Berl) 1991;83:630-6.
37. Broadwell RD. Transcytosis of macromolecules through the blood-brain barrier: a cell biological perspective and critical appraisal. Act Neuropathol (Berl) 1989;79:117-28.
52
GREENWOOD, HOWES, ANDLIGHTMAN
LABoR,AToRY IIvestIGarrOIBradbury MWB, Handbook Exp Pharmacol 103: New York:
Sprin-
matrix. Immunol Today 1991;12:262-6.ger-Verlag,
1992:439-57.
48. Naparstek Y, Cohen IR, Fuks Z, Vlodavsky I. Activated T lympho-39. Essner E. Role of vesicular transport in breakdown of theblood-
cytes produce a matrix-degrading heparin sulfate endoglycosidase.retinal barrier. Lab Invest
1987;56:457-60.
Nature (London) 1984;310:241-4.40. Greenwood J. Mechanisms of blood-brain barrier breakdown.
Neu-
49. Savion N, Vlodavsky I, Fuks Z. Interaction of T lymphocy'tes and roradiology1991;33:95-100.
macrophages with vascular endothelial cells: attachment, invasion 41. Brosnan CF, Claudio L, Tansey FA, Martiney J. Mechanismsof
and subsequent degradatiorr of the subendothelial extracellularautoimmune neuropathies. Ann Neurol 1990;27(Suppl):S75-S9. matrix. J Cell Physiol 1984;118:169-78.
42. Martiney JA, Litwak M, Berman JW, Arezzo JC, Brosnan
CF.
50. MunroPMG,BrennerRE,HawkinsCP,LandonDN.TannicacidPathophysiologic effect of interleukin-lp in the rabbit retina.
Am
visualisation of blood-brain barrier breakdown in chronicexperi-J Pathol
1990;137:1411-23.
mental allergic encephalomyelitis (EAE)(abstr). Neuropathol Appl43. Charteris DG, Lightman S. Interferon gamrna (IFN-r)
production
Neurobiol, 1993;19:449.in uiuo in experimental autoimmune uveoretinitis. Immunology 51. Planck SR, Dang TT, Graves D, Tara D, Ansel JC, Rosenbaum
1992;75:463-7.
JT. Retinal pigment epithelial cells secrete interleukin-6 inre-44. Wekerle H, Engelhardt B, Risau W, Meyermann R. Interaction
of
sponse to interleukin-l. Invest Ophthalmol Vis Sci 1992;33:78-82.T lymphocytes with cerebral endothelial qellS !p ultrc-Brsin
felllol
52. Elner VM, Strieter RM, Elner SG, Bagiolini M, Lindley I, Kunkel1991;1:10?-14.
SL. Neutrophii chemotactic factor (IL-8) gene expression bycy-45. Male D, Pryce G, Linke A, Rahman J. Lymphocyte migration
into
tokine treated retinal pigment epithelial cells. AmJ
Pathol the CNS modelled in uitro. JNeurimmunol1992;40:167-72.
1990;136:?45-50.46. Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman 53. Tanihara H, Yoshida M, Yoshimura N. Tumor necrosis factor-a
L, Karin N. Prevention of experimental autoimmune
encephalo-
gene is expressed in stimulated retinal pigment epithelial cells in myelitis by antibodies against a4131 integrin. Nature(London)
culture. Biochem Biophys Res Commun 1992;187:1029-34.1992;356:63-6.
54. Al-Mahdawi S, Forrester JV, Lee WR. A simplified method for the47. de Sousa M, Tilney NL, Kupiec-Weglinski JW. Recognition of
self
isolation of highly purified bovine retinal S-antigen. J