ISOLASI DAN KLONING FRAGMEN DNA MIKROSATELIT
DARI TANAMAN GAHARU (
Aquilaria malaccensis
Lamk.
)
ARIE AQMARINA
DEPARTEMEN SILVIKULTUR FAKULTAS KEHUTANAN INSTITUT PERTANIAN BOGOR
PERNYATAAN MENGENAI SKRIPSI DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA
Dengan ini saya menyatakan bahwa skripsi berjudul Isolasi dan Kloning Fragmen DNA Mikrosatelit dari Tanaman Gaharu (Aquilaria malaccensis Lamk.) adalah benar karya saya dengan arahan dari komisi pembimbing dan belum diajukan dalam bentuk apa pun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir skripsi ini.
Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
Bogor, Agustus 2014
ABSTRAK
ARIE AQMARINA. Isolasi dan Kloning Fragmen DNA Mikrosatelit dari Tanaman Gaharu (Aquilaria malaccensis Lamk.). Dibimbing oleh ULFAH JUNIARTI SIREGAR dan UTUT WIDYASTUTI.
Aquilaria malaccensis Lamk. merupakan salah satu jenis tanaman hutan yang menghasilkan resin beraroma wangi terbaik di dunia dan masuk ke dalam kategori Appendix II dalam CITES. Penanda mikrosatelit dapat dikembangkan sebagai bahan pembeda antara tanaman A. malaccensis yang menghasilkan gaharu dengan yang tidak menghasilkan gaharu. Tujuan dari penelitian ini ialah mengisolasi dan mengklon fragmen DNA mikrosatelit tanaman A. malaccensis hasil PCR. Metode yang dilakukan terdiri atas isolasi DNA genom A. malaccensis, PCR dengan primer mikrosatelit 10pa17 dan 16pa17, elusi produk PCR, dan kloning fragmen DNA mikrosatelit. Proses kloning meliputi persiapan fragmen DNA mikrosatelit untuk sisipan, ligasi ke dalam vektor pGemT-Easy, persiapan sel kompeten E.coli strain DH5α, transformasi sel kompeten dengan vektor pembawa DNA sisipan, serta seleksi koloni bakteri biru-putih. Isolasi DNA genom menghasilkan DNA dengan konsentrasi 20 ng/µl. Fragmen DNA mikrosatelit hasil PCR berukuran 150 bp. Proses kloning telah menghasilkan koloni yang berwarna putih yang mengindikasikan bahwa koloni bakteri tersebut telah tersisipi DNA rekombinan.
ABSTRACT
ARIE AQMARINA. Isolation and Cloning of Microsatellite DNA Fragments of Agarwood Plant (Aquilaria malaccensis Lamk.). Supervised by ULFAH JUNIARTI SIREGAR and UTUT WIDYASTUTI.
Aquilaria malaccensis Lamk. is one of forest tree species which produces best aromatic resin in the world and is listed in the Appendix II of CITES. Its microsatellite marker was developed in order to be able to differentiate plants of A. malaccensis which actually could produce agarwood and those which could not. The objective of this research was to isolate and clone microsatellite DNA fragment of A. malaccensis resulted from PCR process. Methods included A. malaccensis genomic DNA isolation, PCR using microsatellite primers, i.e. 10pa17 and 16pa17, PCR products elution, and cloning of microsatellite DNA fragment. The cloning process was conducted as followed, preparation of microsatellite DNA fragment as insert, ligation of insert to pGemT-Easy vector, preparation of competent E.coli cells strain DH5α, transformation of competent cell with vector carrying inserted DNA, and selection of blue-white colonies. Isolation of genomic DNA could obtain DNA having concentration of 20 ng/µl. The size of microsatellite DNA fragment as PCR product was 150 bp. The cloning process has produced white becterial colonies indicating that the bacterial colonies has contained the inserted recombinant DNA.
Skripsi
sebagai salah satu syarat untuk memperoleh gelar Sarjana Kehutanan
pada
Departemen Silvikultur
ISOLASI DAN KLONING FRAGMEN DNA MIKROSATELIT
DARI TANAMAN GAHARU (
Aquilaria malaccensis
Lamk.)
DEPARTEMEN SILVIKULTUR FAKULTAS KEHUTANAN INSTITUT PERTANIAN BOGOR
BOGOR 2014
Judul Skripsi : Isolasi dan Kloning Fragmen DNA Mikrosatelit dari Tanaman Gaharu (Aquilaria malaccensis Lamk.)
Nama : Arie Aqmarina
NIM : E44100015
Disetujui oleh
Dr Ulfah Juniarti Siregar Dr Utut Widyastuti
Pembimbing I Pembimbing II
Diketahui oleh
Prof Dr Ir Nurheni Wijayanto, MS Ketua Departemen
PRAKATA
Puji dan syukur penulis panjatkan kepada Allah subhanahu wa ta’ala atas segala karunia-Nya sehingga karya ilmiah ini berhasil diselesaikan. Tema yang dipilih dalam penelitian yang dilaksanakan sejak bulan Oktober 2013 ini ialah kloning fragmen DNA, dengan judul “Isolasi dan Kloning Fragmen DNA Mikrosatelit dari Tanaman Gaharu (Aquilaria malaccensis Lamk.).
Terima kasih penulis ucapkan kepada Ibu Dr Ulfah Juniarti Siregar dan Dr Utut Widyastuti selaku pembimbing yang telah banyak memberikan arahan, saran, dukungan dan bantuan lainnya sehingga penulis dapat menyelesaikan skripsi ini. Selan itu, penghargaan penulis sampaikan kepada Mbak Laswi Irmayanti, S.Hut selaku teknisi Laboratorium Genetika Hutan, Departemen Silvikultur dan Mbak Anidah, S.Si selaku teknisi Laboratorium Bioteknologi SEAMEO-BIOTROP, dan Mbak Peppy Elvafina selaku teknisi Laboratorium Biorin, PPSHB IPB yang telah membimbing selama pelaksanaan penelitian ini. Ayahanda Alm. Rusli Nasution, Ibu Emma Salamah, Bang Emir Idris Nasution, dan seluruh keluarga lainnya yang
telah memberikan do’a, kasih sayang dan dukungan dalam proses
penyelesaiannya. Di samping itu, pada saudara-saudara, akang-akang, dan teteh-teteh Rimbawan Pecinta Alam (RIMPALA) khususnya R-XVI, teman-teman Silvikultur 47, sahabat-sahabat penulis Hassyati Shabrina, Desi Nurafida, Intan Nurhajah, Mirzha Hanifah, Kumala Fitriyanita, Mira Febianti, M. Nur Alifudin, dan Bayu Winata yang juga memberikan bantuan, dukungan, saran dan semangat yang melimpah dalam proses penyusunan hingga penulisan penelitian ini. Tak lupa semua pihak yang tidak dapat disebutkan satu per satu yang turut membantu dalam penyusunan skripsi ini.
Semoga karya ilmiah ini bermanfaat.
DAFTAR ISI
DAFTAR TABEL viii
DAFTAR GAMBAR viii
DAFTAR LAMPIRAN viii
PENDAHULUAN 1
Latar Belakang 1
Perumusan Masalah 2
Tujuan Penelitian 2
Manfaat Penelitian 2
Ruang Lingkup Penelitian 2
METODE 3
Waktu dan Tempat 3
Bahan dan Alat 3
Prosedur Penelitian 3
HASIL DAN PEMBAHASAN 8
Isolasi DNA Genom Tanaman Gaharu 8
Perbanyakan Fragmen DNA Gaharu dengan PCR 12
Pembentukan DNA Rekombinan 13
SIMPULAN DAN SARAN 20
Simpulan 20
Saran 20
DAFTAR PUSTAKA 20
LAMPIRAN 24
DAFTAR TABEL
1 Komposisi bahan PCR 5
2 Primer mikrosatelit 10pa17 dan 16pa17 (Eurlings et al. 2009) 6
3 Kondisi PCR 6
4 Reaksi ligasi 7
5 Ukuran konsentrasi DNA genom dari empat individu 11
DAFTAR GAMBAR
dengan 1 adalah ladder berukuran 1 kb. 2 dan 3 adalah contoh fragmenDNA hasil amplifikasi. 13
4 Elektroforesis fragmen DNA untuk persiapan elusi dari fragmen DNA dengan primer 10pa17 (pita DNA kiri) dan primer 16pa17 (pita DNA
kanan) 14
5 Elektroforesis hasil elusi fragmen DNA dengan primer 10pa17 (2) dan
primer 16pa17 (1) 15
6 Peta vektor pGemT-Easy (Promega 2010) 16
7 Koloni E.coli yang tumbuh dari fragmen DNA dengan (A) Primer
10pa17, (B) Primer 16pa17, (C) Kontrol positif 19
DAFTAR LAMPIRAN
1 Komposisi pembuatan buffer TAE 50 kali (Tris-Acetate-EDTA)
(Setyari 2014) 24
2 Cara pembuatan loading dye 6 kali (Fermentas 2011) 24 3 Cara pembuatan media LB (Luria bertani) cair (Apriani 2008). 24 4 Cara pembuatan media LB (Luria bertani) agar (Apriani 2008). 24 5 Cara pembuatan TFB (Transformation Buffer) (Suharsono dan
Widyastuti 2010). 25
6 Cara pembuatan media 2xYT (Alimuddin dkk. 2008) 25
7 Cara pembuatan IPTG 0,1 M (Promega 2010) 25
8 Cara pembuatan X-Gal 50 mg/ml (Promega 2010) 25
9 Prosedur isolasi DNA genom menggunakan "DNeasy Plant Mini Kit"
dari Qiagen (2012) 26
10 Proses perbanyakan DNA dengan PCR (Vierstraete 1999) 27 11 Cara perhitungan perbandingan 3:1 antara DNA sisipan dengan DNA
vektor (Promega 2010). 27
PENDAHULUAN
Latar Belakang
Indonesia sekarang ini telah dikenal sebagai negara penghasil gaharu di dunia karena hutan tropis Indonesia dapat ditumbuhi secara luas oleh beberapa jenis gaharu terbaik. Pohon gaharu terutama dikenal karena manfaatnya untuk menghasilkan produk hutan berupa Hasil Hutan Bukan Kayu (HHBK) yang telah memiliki nilai komersial tinggi disamping pemanfaatan kayunya. Salah satu HHBK yang dihasilkan adalah gaharu atau resin yang berasal dari pohon penghasil gaharu. Resin tersebut kemudian dapat dijadikan bahan baku terutama untuk produk yang memerlukan zat wangi-wangian yang dihasilkan minyak gaharu, serta dijadikan sebagai campuran dalam obat-obatan dan kosmetik. Sejalan dengan semakin berkembangnya teknologi, maka semakin banyak pula pemanfaatan untuk produk turunan gaharu, sehingga permintaan akan komoditi gaharu juga semakin meningkat. Hal ini juga didukung pula dengan semakin tingginya harga jual gaharu di pasaran dunia.
Perkembangan pesat gaharu yang dimulai sejak era tahun 1980an membuat perdangangan ekspor gaharu terus mengalami peningkatan. Tercatat pada tahun 1990-1998 ekspor gaharu mencapai 165 ton/tahun yang bernilai US$ 2 juta. Kemudian, tahun 2000 tercatat ekspor hingga 446 ton dengan nilai US$ 2,2 juta. Akan tetapi, memasuki tahun-tahun berikutnya ekspornya mulai menurun seperti pada tahun 2002 dari kuota ekspor 300 ton/tahun tapi hanya terpenuhi sekitar 10-15%. Selanjutnya, sejak tahun 2004 keadaan semakin buruk dengan penurunan ekspor yakni dengan kuota 150 ton bahkan tidak tercatat adanya transaksi ekspor dari hutan alam Indonesia (Sumarna 2009).
Kondisi penurunan jumlah produksi gaharu ini menunjukkan bahwa keberadaan pohon penghasil gaharu di hutan alam semakin sedikit karena perburuan gaharu dimana- mana yang disebabkan peningkatan jumlah permintaan pasar internasional akan gaharu tersebut. Padahal awalnya produksi gaharu hanya berasal dari pemungutan batang yang telah mati dan menangandung gaharu, tetapi sekarang ini yang terjadi adalah perburuan hingga pada batang yang masih hidup. Menurut Balitbanghut (2006) hal ini menyebabkan beberapa pohon penghasil gaharu sejak tahun 2004 mengalami perubahan status menjadi Apendix II dalam CITES (Convention on International in Trade Endangered of Wild Fauna and Flora Species) untuk pohon dari genus Aquilaria spp. dan Gyrinops sp. Gaharu jenis A. malaccensis sejak tahun 1995 sudah masuk dalam kategori Appendix II di CITES (Ditjen PHKA 2005), sehingga perlu adanya pembatasan mengenai kegiatan perdagangan internasional dari jenis gaharu tersebut agar tidak terancam punah.
2
Langkah awal untuk mengembangkan gaharu budidaya adalah dengan seleksi pohon-pohon yang mempunyai gen untuk menghasilkan gaharu tersebut. Oleh sebab itu, diperlukan teknik untuk membedakan antara tanaman penghasil gaharu dengan yang tidak menghasilkan gaharu. Hal ini dapat dilakukan dengan menggunakan mikrosatelit sebagai penanda untuk mengidentifikasi gen penghasil gaharu.
Mikrosatelit atau Simple Sequence Repeats (SSRs) adalah salah satu penanda DNA berdasarkan teknik PCR (Varshney et al. 2005). Penanda ini menggunakan primer (potongan pendek DNA sintetik) yang mengandung motif
mikrosatelit pendek pada ujung 3’ atau 5’ atau mengapit daerah mikrosatelit. Saat
ini mikrosatelit dapat dipakai sebagai alat dalam program pemuliaan karena kemampuan yang tinggi dalam mendeteksi perbedaan urutan basa, sampai satu pasang basa. Penggunaan mikrosatelit relatif mudah karena menggunakan teknik PCR. Hasil studi dengan menggunakan penanda mikrosatelit ini akan dijadikan sebagai petunjuk untuk menentukan tanaman yang menghasilkan gaharu dengan yang tidak menghasilkan gaharu. Oleh karena itu, perlu dikembangkan sebagai bahan pembeda melalui analisis fragmen DN A melalui teknik pembentukan DNA rekombinan atau kloning.
Perumusan Masalah
Fragmen DNA mikrosatelit yang diisolasi dari tanaman gaharu (A. malaccensis Lamk.) digunakan untuk mengidentifikasi gen penginduksi gaharu yang diperbanyak secara invitro. Hasil studi fragmen DNA mikrosatelit ini kemudian perlu dikembangkan sebagai bahan pembeda antara tanaman penghasil gaharu dengan tanaman yang tidak penghasil gaharu. Oleh karena itu, salah satu teknik yang digunakan adalah dengan melakukan analisis fragmen DNA melalui pembentukan DNA rekombinan atau kloning fragmen DNA mikrosatelit tersebut.
Tujuan Penelitian
Tujuan dari penelitian ini yaitu untuk mengisolasi dan mengklon fragmen DNA mikrosatelit pada tanaman gaharu (A. malaccensis Lamk.) untuk mengidentifikasi gen penginduksi gaharu sebagai salah satu cara untuk pemuliaan tanaman dan konservasi sumberdaya genetik tanaman A. malaccensis.
Manfaat Penelitian
Manfaat yang diharapkan dari penelitian ini adalah memberikan informasi mengenai hasil isolasi dan kloning fragmen DNA mikrosatelit dari tanaman gaharu (A. malaccensis Lamk.) yang akan dikembangkan sebagai bahan pembeda antara tanaman yang menghasilkan gaharu dengan yang tidak menghasilkan gaharu. Hal tersebut merupakan penunjang program pemuliaan dan konservasi sumberdaya genetik tanaman gaharu guna pengembangan gaharu hasil budidaya.
Ruang Lingkup Penelitian
3 serta pembentukan DNA rekombinan atau kloning fragmen DNA mikrosatelit dari tanaman gaharu (Aquilaria malaccensis Lamk).
METODE
Waktu dan Tempat
Penelitian ini dilaksanakan pada Januari 2014–Juli 2014. Penelitian ini berlokasi di tiga laboratorium yakni Laboratorium Genetika Hutan, Departemen Silvikultur, Fakultas Kehutanan, IPB, Laboratorium Bioteknologi, SEAMEO-BIOTROP, serta Laboratorium BIORIN, PPSHB IPB.
Bahan dan Alat
Bahan yang digunakan yakni sampel daun yang berasal dari bibit gaharu jenis A. malaccensis usia 3 bulan yang didapatkan dari persemaian Pusat Penelitian dan Pengembangan Kehutanan Gunung Batu, Bogor, nitrogen cair, DNeasy Plant Mini Kit dari Qiagen (2012), PVP 26% (b/v), agarosa, buffer TAE 50 kali (Tris-Acetate-EDTA) (Lampiran 1), gel red, EtBr (Ethidium Bromide), blue juice 6 kali (Lampiran 2), DNA lambda 50 ng/µl, primer mikrosatelit 10pa17 dan 16pa17 dengan konsentrasi 10 µM untuk forward dan reverse, PCR Nucleotide Mix 10 mM, DNA Taq polymerase 5U/µl, MgCl2 25 mM, 5X Go taq
flexi buffer, nuklease free water, Gel/PCR DNA Fragments Extraction Kit dari Geneaid (2008), vektor pGEM-T Easy, T4 DNA ligase, buffer T4 DNA ligase 2X, bakteri E. coli strain DH5α berbentuk gliserol, media LB (Luria bertani) cair (Lampiran 3), LB agar (Lampiran 4), TFB (Tranformation buffer) (Lampiran 5), DMSO (Dimethyl sulfoxide), media cair 2xYT atau recovery media (Lampiran 6), alkohol, antibiotik ampicilin, IPTG (Isopropyl β-D-1-thiogalactopyranoside) 0,1 M (Lampiran 7), X- gal (5-bromo-4-chloro-3-indolyl-beta-D-galacto-pyranoside) 50 mg/ml (Lampiran 8).
Alat yang digunakan terdiri atas autoclave, gunting, mortar, pestle, spatula, tube berukuran 2 ml, 1,5 ml, 0,5 ml, rak tube, pipet mikro, tips, vortex, spatula, gabus, mesin sentrifuge, waterbath, freezer, timbangan analitik, tabung erlenmeyer, gelas ukur, labu takar, gelas piala, sisir, cetakan agar, dan mesin elektroforesis, Kodak Gel Logic 200, mesin PCR thermal cycler Applied Biosystems, pestile, mesin UV, lemari es, tusuk gigi, shaker incubator, cawan Petri, heat shock, inkubator, batang kaca, bunsen, laminar air flow, kertas sil, plastik wrap, sprayer, oven, sarung tangan, masker, jas laboratorium, dan termos.
Prosedur Penelitian
4
Gambar 1 Bagan alur penelitian
Pengambilan Sampel Daun
Daun gaharu (A. malaccensis) yang digunakan berasal dari persemaian Pusat Penelitian dan Pengembangan Kehutanan Gunung Batu, Bogor. Bibit gaharu yang digunakan berusia tiga bulan dengan tinggi 30-40 cm. Daun gaharu yang diutamakan sebagai sampel adalah daun muda seberat 100 g yang berada pada bagian pucuk.
Isolasi DNA Genom
Protokol ekstraksi DNA yang digunakan adalah dengan DNeasy Plant Mini Kit dari Qiagen (2012). Sampel daun basah seberat 100 g disiapkan sebagai bahan untuk proses disrupsi jaringan tanaman. Sampel daun tersebut ditambahkan nitrogen cair, lalu digerus pada mortar hingga sampel menjadi serbuk yang halus. Proses selanjutnya adalah lisis organel tanaman. Serbuk halus tersebut dimasukkan ke dalam mikrotube yang telah berisi campuran 400 µl buffer AP1, 40 µl PVP 26%, dan 4 µl RNase A stock (100 mg/ml) dan divortex selama 2 menit. Selanjutnya, campuran diinkubasi ke dalam waterbath pada suhu 65o C
selama 30 menit sambil membolak-balikkan tube sebanyak 2-3 kali.
Selanjutnya adalah pemisahan DNA dengan kotorannya, seperti protein, polisakarida, dan polifenol. 150 µl buffer P3 ditambahkan pada lysate dan diinkubasi kembali selama 10 menit pada suhu -20o C. Sentrifuge lysate dengan
kecepatan 14 000 rpm selama 5 menit dan tranfer lysate ke dalam QIAshredder spin colomn. Setelah itu, sentrifuge kembali lysate dengan kecepatan 14 000 rpm selama 2 menit hingga terbentuk pellet. Cairan yang lewat membran tersebut ditransfer ke dalam mikrotube baru (ukuran 2 ml) tanpa mengganggu pellet yang telah terbentuk. Setelah pellet terbentuk, maka buffer AW1 sebanyak 1,5 volume ditambahkan, lalu dicampur dengan teknik pipetting.
5 dan diinkubasi pada suhu ruang selama 10 menit. Selanjutnya, sentrifuge dengan kecepatan 8 000 rpm selama 1 menit. Ulangi langkah penambahan 100 µl buffer AE, hingga didapatkan volume DNA sebanyak 200 µl. Prosedur isolasi DNA genom dengan menggunakan DNeasy Plant Mini Kit dari Qiagen (2012) (Lampiran 9).
Uji Kualitas DNA dengan Elektroforesis
Elektroforesis dilakukan berdasarkan metode Sambrook dan Russell (2001). Gel agarosa 1% (b/v) disiapkan dari 0,3 g agarosa yang dilarutkan dalam 30 ml buffer TAE 1 kali pada tabung erlenmeyer. Larutan dipanaskan di dalam microwave selama 1 menit hingga didapatkan larutan yang bening. Setelah itu, 1 µl gel red dimasukkan ke dalam larutan bening tersebut. Kemudian larutan dituangkan ke dalam cetakan yang telah disiapkan bersama sisirnya. Biarkan hingga agarosa telah menjadi gel yang sudah mengeras. Selanjutnya, gel tersebut dimasukkan ke dalam mesin eletroforesis yang telah berisi buffer TAE 1 kali yang dibuat dari komposisi bahan TAE 50 kali (Lampiran 1).
10 ml TAE 50 kali ditambahkan ke dalam 490 ml aquadest, sehingga TAE 50 kali akan menjadi TAE 1 kali. 5 µl DNA genom hasil ekstraksi dicampurkan dengan 2 µl blue juice 6 kali dan dicampurkan dengan teknik pipetting. Campuran tersebut dimasukkan ke dalam sumur yang terdapat pada cetakan. Buat agar seluruh gel agarosa telah terendam buffer TAE 1 kali pada mesin elektroforesis. Setelah semua sampel dimasukkan nyalakan mesin elektroforesis dengan tegangan 75 Volt selama 60 menit. Hasil proses running kemudian divisualisasi dengan mesin Kodak Gel Logic 200.
Polymerase Chain Reaction (PCR)
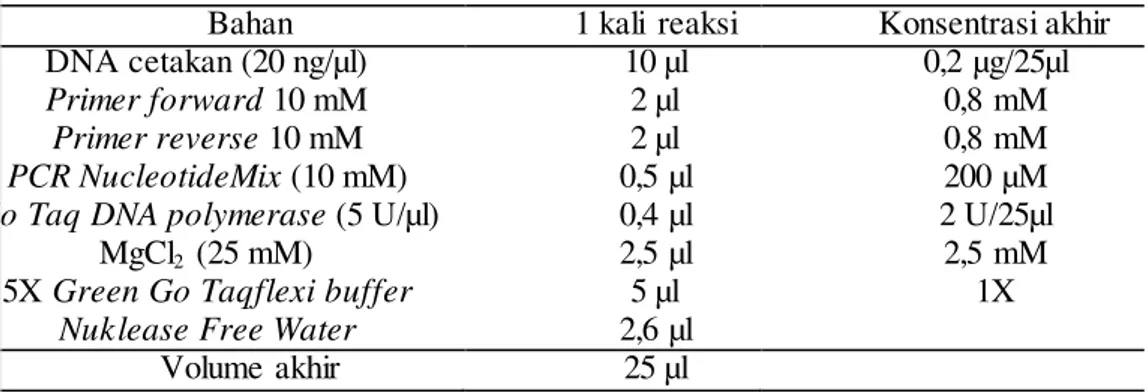
1 µl DNA cetakan diencerkan 10 kali yang akan digunakan sebagai salah satu komponen bahan PCR. Komposisi reaksi PCR ini dibuat berdasarkan GoTaq PCR Core Systems dari Promega (2009) dengan volume akhir 25 µl/reaksi, seperti yang terdapat pada Tabel 1.
Tabel 1 Komposisi bahan PCR (Promega 2009)
Bahan 1 kali reaksi Konsentrasi akhir DNA cetakan (20 ng/µl) 10 µl 0,2 µg/25µl
6
Tabel 2 Primer mikrosatelit 10pa17 dan 16pa17 (Eurlings et al. 2009)
Locus Primer Sequence Repeat Size Range
(bp)
pada Tabel 2. Campuran tersebut dimasukkan ke dalam mikrotube 0,5 ml. Setelah itu, campuran divortex selama 3-5 detik dan dispin. Selanjutnya, dimasukkan ke dalam mesin PCR thermal cycler Applied Biosystems. Proses di dalam mesin PCR diatur sesuai dengan tiap tahapnya seperti yang ditampilkan pada Tabel 3. dilakukan PCR, maka DNA tersebut akan dielektroforesis kembali pada gel agarosa 2% (b/v). Kondisi PCR yang digunakan didapat dari proses optimasi waktu dan suhu annealing PCR.
Purifikasi Hasil PCR
Purifikasi hasil PCR dilakukan dengan Gel/PCR DNA Fragments Extraction Kit dari Geneaid (2008) pada bagian PCR Clean Up Protocol yang dilakukan melalui empat proses. Proses pertama adalah p emisahan/penguraian gel. Gel agarose yang mengandung pita-pita DNA produk PCR hasil elektroforesis sebanyak 100 µl dipotong-potong pada mesin UV.
Gel tersebut dicacah di atas cawan petri dan dimasukkan ke dalam tube 1,5 ml dan ditambahkan dengan 500 µl DF buffer, lalu divortex. Selanjutnya, diinkubasi pada suhu 55-60 oC selama 10-15 menit sambil dibolak-balikkan
selama 2-3 kali. Jika sudah selesai, dinginkan pada suhu ruang. Proses yang kedua adalah DNA binding/pengikatan DNA. 800 µl campuran gel tersebut ditransfer ke dalam DF colomn yang telah ditempatkan pada collection tube 2 ml. Sentrifuge selama 2 menit pada kecepatan penuh dan cairan dibuang, lalu DF column ditempatkan kembali pada collection tube (jika > 800 µl campuran gel, maka ulangi tahap ini).
7 penuh. Setelah itu, cairan dibuang dan DF colomn ditempatkan kembali pada collection tube dan sentrifuge kembali selama 10 menit pada kecepatan penuh untuk mengeringkan matriks. Proses yang keempat adalah elusi DNA. DF colomn kering dipindahkan ke dalam tube 1,5 ml baru, lalu 30 µl elution buffer ditambahkan pada DF colomn kering. Biarkan selama beberapa saat hingga elution buffer terserap seluruhnya ke dalam matriks dan sentrifuge kembali selama 10 menit pada kecepatan penuh untuk mendapatk an produk PCR yang telah murni. Ligasi DNA Plasmid dengan Fragmen DNA Target.
Proses ligasi dilakukan dengan vektor pGemT-Easy (Promega 2010). perbandingan ukuran dan konsentrasi antara stock DNA sisipan dengan DNA vektor sebesar 3:1 (Lampiran 11).
Pembuatan Sel Kompeten
Pembuatan sel kompeten dilakukan dengan metode Zhiming (2005). Bakteri E.coli strain DH5α yang berbentuk gliserol disiapkan sebanyak 100 µl untuk dikulturkan ke dalam media 2 ml LB cair. Campuran gliserol+LB cair diinkubasikan pada shaker incubator pada suhu 37 oC dengan kecepatan 150 rpm
selama 16-20 jam. Setelah itu, hasil kultur tersebut digoreskan ke dalam media LB agar dan diinkubasi pada inkubator pada suhu 37 oC selama 16-20 jam untuk
peremajaan bakteri. Langkah selanjutnya, koloni tunggal bakteri yang terbentuk pada media LB agar diambil dan dimasukkan ke dalam media 2 ml LB cair. Lalu, diinkubasi di shaker incubator pada suhu 37 oC dengan kecepatan 150 rpm selama
16-20 jam.
1 ml kultur bakteri E.coli strain DH5α tersebut dimasukkan ke dalam 10 ml LB cair untuk pembuatan subkultur. Kemudian, diinkubasi pada shaker incubator pada suhu 37 oC dengan kecepatan 150 rpm selama 2,5 jam. Setelah itu, 1,5 ml
subkultur diambil dan dimasukkan pada masing- masing tube 1,5 ml sesuai dengan kebutuhan sampel untuk transformasi. Tube-tube yang telah berisi hasil subkultur tersebut disentrifuge pada suhu 4 oC selama 10 menit dengan kecepatan 5 000 rpm
8
yang berisi subkultur tanpa adanya kontak antara mulut tube dengan mulut wadah subkultur untuk menghindari kontaminasi.
Selanjutnya, 495 µl transformation buffer ditambahkan sambil disuspensikan, lalu diinkubasi di es selama 10 menit. Selanjutnya sentrifuge pada suhu 4°C selama 10 menit dengan kecepatan 5 000 rpm. Cairan hasil sentrifuge dibuang kembali pada wadah yang berisi subkultur dengan hati- hati. 125 µl transformation buffer dan 8,8 µl DMSO ditambahkan, lalu disuspensikan dengan endapan (sambil diaduk dengan mengetuk- ngetukkan dasar tube dengan jari secara perlahan) dan diinkubasi dalam es selama 10 menit. Tiap 50 µl suspensi sel kompeten disimpan ke dalam tiap tube 1,5 ml dan siap untuk digunakan.
Transformasi dan Seleksi DNA Rekombinan
Transformasi dilakukan dengan metode kejut panas (Suharsono dan Widyastuti 2010). Prosedur transformasi (Lampiran 12). 10 µl reaksi ligase dimasukkan ke dalam tube yang telah berisi 50 µl sel bakteri kompeten (sambil diaduk dengan mengetuk- ngetukkan dasar tube dengan jari secara perlahan) dan diinkubasi di es selama 20 menit. Kemudian, campuran sel kompeten dan ligasi dipanaskan pada mesin heat shock pada suhu 42°C, selama 45 detik. Setelah itu, langsung diangkat dan diinkubasikan kembali ke dalam es selama 5 menit. Setelah melakukan langkah heatshock, tube dipindahkan ke suhu ruang dan 100 µl recovery media yakni media cair 2xYT ditambahkan pada campuran reaksi tersebut dan diinkubasi pada shaker incubator dengan kecepatan 250 rpm, suhu 37°C selama 1 jam.
Selanjutnya adalah tahap seleksi DNA rekombinan. 50 µl X- gal dan 10 µl IPTG ditambahkan pada masing- masing campuran reaksi. Setelah tercampur, maka bakteri ditebar di atas media LB agar dengan menggunakan batang kaca steril hingga mengering (telah dicelupkan dalam alkohol dan dibakar oleh api bunsen dan biarkan mendingin). Media LB agar tersebut ada yang mengandung antibiotik ampisilin ataupun yang tidak mengandung ampicilin sebagai kontrol.
Apabila substrat sudah disiapkan, proses seleksi sisipan plasmid dapat diduga melalui marka seleksi dari gen lacZ. Bakteri yang tersisipi fragmen DNA MCS (Multiple Cloning Sites) pada daerah gen lacZ di dalam plasmidnya akan berwarna putih, sedangkan yang tidak tersisipi akan berwarna biru dan mati. Setelah itu, cawan diinkubasi pada suhu 37 °C selama 16 jam dengan posisi cawan terbalik. DNA rekombinan yang terbentuk akan berukuran putih, sedangkan nonrekombinan berwarna biru (Suharsono dan Widyastuti 2010).
HASIL DAN PEMBAHASAN
Isolasi DNA Genom Tanaman Gaharu
9 DNA yakni DNA inti, DNA kloroplas, dan DNA mitokondira. DNeasy Plant Mini Kit dari Qiagen (2012) ini memiliki cara kerja untuk mengektrak total DNA.
DNA (Deoxyribose Nucleid Acid) merupakan susunan sistematis dari polimer asam nukleat yang membawa informasi genetik kepada keturunannya (Yuwono 2009). Watson dan Crick pada tahun 1953 menemukan bahwa struktur asam nukleat DNA adalah double helix yang memutar ke arah kanan (Gaffar 2007). Struktur seperti ini dikarenakan DNA tersusun atas dua rantai nukleotida yang dihubungkan oleh ikatan hidrogen, sedangkan antar nukleotida dalam satu rantai linier tersebut dihubungkan oleh ikatan phospodiester (Sriati 2011). DNA genom adalah satu set lengkap atau keseluruhan informasi genetik yang terdapat pada suatu organisme (Weaver dan Hedrick 1997). Isolasi DNA genom ada lah suatu cara untuk memisahkan molekul DNA yang mengandung keseluruhan informasi genetik dari molekul lainnya pada suatu organisme. Isolasi DNA genom merupakan langkah awal yang digunakan untuk menyalurkan informasi genetik.
Isolasi DNA genom diawali dengan disrupsi jaringan tanaman melalui penghancuran dinding sel tanaman dengan cara menggerus daun hingga menjadi seperti bubuk dengan bantuan nitrogen cair. Sampel daun basah yang digunakan seberat 100 mg yang sebelumnya dipotong-potong terlebih dahulu karena sifat daun gaharu yang berserat (Weising et al. 2005).
Disrupsi ini dilakukan secara fisik menggunakan metode freezing-thawing yakni dengan penggerusan sampel yang telah ditambahkan nitogen cair dengan menggunakan mortar dan pestle. Kehadiran nitrogen cair juga berfungsi untuk mempermudah degradasi dinding sel tanaman yang akan diekstrak sehingga mengeluarkan seluruh isi sel. Tahapan awal dilakukan dengan melisiskan dinding sel yang bertujuan untuk mengeluarkan seluruh isi di dalam sel (Holme dan Hazel 1998).
Potongan-potongan daun yang telah menjadi bubuk dimasukkan ke dalam campuran buffer AP1, PVP, dan RNase yang ada pada mikrotube. Buffer AP1 merupakan buffer lisis yang terdiri atas detergen seperti CTAB, buffer system seperti Tris-HCl, garam seperti NaCl, serta agen pereduksi seperti β -merkaptoetanol. Detergent CTAB (cethyl trimethylammonium bromide) berfungsi untuk melisiskan membran sel tanaman (Bettelheim dan Landesberg 2007) serta mendenaturasikan dan memisahkan protein dari DNA (Weising et al. 2005).
Tris-HCl berguna untuk mengatur kondisi keasaman atau pH agar enzim pendegradasi tidak dapat berkerja secara optimal (Weising et al. 2005). Garam NaCl yang digunakan harus memiliki konsentrasi > 1 M karena fungsi utamanya adalah untuk mendegradasi polisakarida dan memisahkan protein yang terdapat pada inti sel dari DNA (Weising et al. 2005).
10
untuk melisiskan membran sel dan organel-organel yang telah keluar dari dalam sel.
Dimodifikasi dengan penambahan PVP (polyvinyl pyrrolidon) 26% yang berfungsi untuk mereduksi polisakarida dan penjerat polifenol yang banyak di sel tanaman (Weising et al. 2005). RNase A adalah enzim yang berfungsi untuk mendegradasi RNA agar tidak mengkonta minasi DNA. proses melisiskan membran dan organel sel ini juga dibantu dengan menginkubasi sampel pada suhu 65o C.
Tahap selanjutnya adalah presipitasi dari agen-agen pengotor dengan memberikan buffer P3 dan mengikubasinya ke dalam suhu -20o C. Buffer P3 ini
berisi asam asetat yang berfungsi sebagai neutralization buffer. Penambahan buffer P3 ini dimaksudkan untuk menetralkan reaksi dalam tube dan menjaga agar pH reaksi tersebut tidak basa. Hasilnya dapat dilihat melalui sentrifuge dengan kecepatan 14 000 rpm sehingga terbentuklah dua lapisan padat dan cair. Pada akhir tahap ini DNA akan terpisah dengan komponen lainnya setelah mengalami sentrifuge sehingga terbentuk dua lapisan yakni fase organik pada lapisan bawah yang berisi protein dan lipid, serta lapisan aquoeus (cairan) yang merupakan hasil presipitasi yang berisi DNA (Weising et al. 2005).
Lapisan atas kemudian dimasukkan dalam QIAshredder tube untuk disentrifuge sehingga dihasilkan pellet yang merupakan lanjutan proses presipitasi protein dan polisakarida dari tahap sebelumnya. Cairan yang melewati membran QIAshredder tersebut yang digunakan untuk tahap presipitas DNA. Presipitasi DNA dari molekul- molekul lain seperti protein dengan cara menambahkan buffer AW1 yang berisi guanidine hydrochloride yang telah dicampur ethanol. Guanidine-HCl ini merupakan garam chaotropic yang sangat berguna untuk mendegradasi protein-protein sel ataupun jaringan (Novel 2010), sedangkan ethanol yang berfungsi untuk mengendapkan atau presipitasi DNA (Walker dan Rapley 2002).
Molekul- molekul DNA akan tertahan pada membran mini spin colomn setelah disentrifuge. Tahap selanjutnya adalah pemurnian DNA dengan cara penambahan buffer AW2 yang merupakan wash buffer. Wash buffer ini digunakan dengan tujuan untuk mencuci molekul DNA. Tahap akhir adalah elusi DNA dengan menggunakan buffer AE yang merupakan campuran buffer TE dan ethanol pada tube baru.
Fungsi buffer ini adalah untuk melarutkan DNA dengan mengurangi daya ikat atau afinitas molekul DNA dari membran mini spin colomn sehingga molekul DNA tercampur dengan larutan buffer AE pada tube baru (Novel 2010). Buffer TE yang ditambahkan peda pellet DNA berfungsi untuk memisahkan RNA dari DNA (Keller and Mark 1989) serta membuat DNA dapat disimpan dalam waktu berminggu- minggu (Verkuil et al. 2008). Proses ini dilakukan dua kali dengan masing- masing volume buffer AE adalah 100 µl, sehingga volume akhir template DNA adalah 200 µl. Setelah itu, sampel DNA dapat disimpan pada freezer bersuhu -20o C.
11 bermuatan negatif, sehingga ketika dialiri oleh arus listrik, maka akan mengalami migrasi dari kutub negatif (anoda) ke positif (katoda). Media yang digunakan adalah gel agarosa. Pemilihan gel agarosa dikarenakan agarosa tidak bersifat karsinogenik, seperti halnya acrylamide (Larasati 2001).
Sampel DNA yang digunakan kemudian dicampur terlebih dahulu dengan loading dye atau blue juice yang berisi dua zat warna yaitu bromophenol biru dan xilena cyanol FF yang berfungsi untuk melacak migrasi DNA secara visual. Selan itu, loading dye juga mengandung gliserol yang berfungsi sebagai pemberat, sehingga DNA akan membentuk suatu lapisan pada bagia n bawah cetakan gel (Fermentas 2011). Gambar 2 merupakan hasil visualisasi DNA yang telah diisolasi menggunakan DNeasy Plant Mini Kit dari Qiagen (2012).
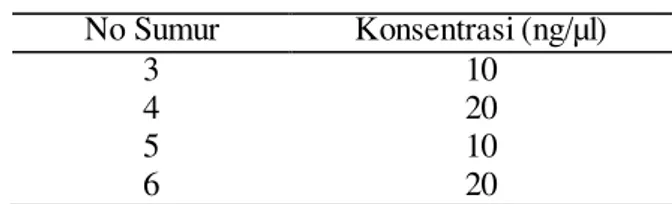
Gambar 2 Elektroforesis hasil isolasi DNA genom pada gel agarosa 1% (b/v). 1 dan 2 adalah DNA lambda dengan konsentrasi 5 ng/µl dan 10 ng/µl. 3, 4, 5, dan 6 adalah DNA genom dengan konsentrasi 10 ng/ µl, 20 ng/µl, 10 ng/µl, 20 ng/µl.
Empat pita DNA tanaman gaharu yang berasal dari individu- individu yang berbeda-beda yang diperlihatkan pada Tabel 5 berikut ini.
Tabel 5 Ukuran konsentrasi DNA genom dari empat individu
No Sumur Konsentrasi (ng/µl)
3 10
4 20
5 10
6 20
Pengukuran konsentrasi hasil isolasi DNA genom dari empat individu tanaman gaharu didasarkan pada perbandingan dengan konsentrasi marker DNA lambda yang digunakan. Marker DNA lambda merupakan DNA yang telah diketahui konsentrasinya sehingga dapat digunakan sebagai acuan dalam menentukan konsentrasi DNA dari sampel yang digunakan. Nomor 1 adalah marker DNA lambda dengan konsentrasi 5 ng/µl, sedangkan nomor 2 berukuran 10 ng/µl. Sampel DNA nomor 3, 4, 5, dan 6 dapat diketahui konsentrasinya dengan cara membandingkan ketebalan pita DNA dari keempat individu tersebut dengan pita DNA lambda, sehingga mendapatkan hasil pengukuran konsentrasi seperti yang terdapat pada Tabel 5. Selanjutnya untuk tahap perbanyakan fragmen DNA gaharu dengan PCR hanya menggunakan satu sampel yakni dari individu nomor 6 yang berkonsentrasi 20 ng/µl, sedangkan yang lainnya dijadikan sebagai persediaan DNA.
12
Perbanyakan Fragmen DNA Gaharu dengan PCR
Amplifikasi fragmen DNA merupakan reaksi rantai polimerase yakni metode melipatgandakan fragmen DNA berupa sekuen nukleotida atau oligonukleotida secara eksponensial yang dilakukan dengan bantuan enzimatis dan secara invitro (Yuwono 2006). Terdapat 5 komponen penting dalam melakukan reaksi rantai polimerase (PCR) yakni : template DNA; oligonukleotida primer; deoksinukleotide triphosphate (dNTP) yang terdiri atas dATP, dTTP, dGTP, dCTP; enzim Taq polymerase, serta buffer PCR (Yuwono 2006). Kelima komponen ini akan sangat mempengaruhi produk PCR.
Template DNA berfungsi menyediakan cetakan utas ganda DNA yang mengandung DNA target yang akan diamplifikasi. DNA target ini dapat di PCR langsung tanpa melalui proses pemurnian terlebih dahulu. Konsentrasi yang digunakan untuk melakukan reaksi PCR harus diencerka terlebih dahulu 10X untuk meningkatkan efisiensi proses amplifikasi. Template DNA yang digunakan untuk proses PCR memiliki konsentrasi stock sebesar 20 ng/ µl, sehingga setelah diencerkan akan memiliki konsentrasi akhir 2 ng.
Primer merupakan sekuen basa-basa nukleotida atau oligonukleotida yang berperan dalam memulai proses penggandaan DNA dengan cara menempel pada bagian template DNA tertentu yang merupakan komplemen dari sekuen basa-basa primer. Terdapat dua jenis primer yakni primer yang berada sebelum daerah target disebut dengan primer forward dan primer yang berada setelah daerah target yang disebut dengan primer reverse (Muladno 2010). Hal yang harus diperhatikan dalam sebuah primer adalah panjang primer, komposisi primer, melting temperature, dan interaksi antar primer-primer (Handoyo dan Rudiretna 2010).
Panjang primer secara umum adalah 18-30 nukleotida. Apabila sekuen primer yang kurang atau lebih dari range tersebut dikhawatirkan dapat menimbulkan ketidakspesifikan proses annealing. Komposisi primer yang dapat mempengaruhi hasil PCR adalah G+C contens (%) yakni dikatakan cukup baik apabila primer tersebut memiliki % G+C sebesar 50-60%. Hal ini berfungsi untuk mencegah terjadinya mispriming (kesalahan penempelan) terutama pada daerah-daerah yang kaya akan G+C (Gelfand dan White 1990).
Temperature melting (Tm) adalah suhu ketika setengah dari rantai template DNA sudah mengalami denaturasi. Tm ini sangat dipengaruhi oleh panjang dan komposisi primer dan sangat mempengaruhi proses annealing DNA-primer. Secara umum suhu annealing berada 5o C dibawah Tm (Muladno 2010).
Interaksi primer-primer yang dimaksud adalah yang dapat menimbulkan mispriming. Penelitian ini menggunakan penanda mikrosatelit sehingga memiliki panjang fragmen 23-25 nukleotida dan persentase G+C sebesar 32-50%.
13 Komponen berikutnya adalah dNTP yang berfungsi sebagai bahan utama amplifikasi DNA. Enzim Taq polymerase merupakan enzim yang digunakan untuk mengkatalisis saat proses polimerisasi terjadi antara template DNA-primer. Buffer PCR berisi Tris-HCl dan KCl yang berfungsi untuk menjaga pH dalam membantu kerja enzim Taq polymerase.
Tahapan PCR yakni denaturasi, annealing (penempelan), dan ekstensi (pemanjangan) juga sangat mempengaruhi keberhasilan proses PCR dari aspek suhu dan waktu amplifikasi. Perbedaan suhu pada tahap annealing akan sangat memengaruhi produk PCR karena sangat disesuaikan dengan tiap primer yang digunakan. Gambar 3 berikut adalah hasil amplifikasi untuk primer mikrosatelit gaharu.
Gambar 3 Elektroforesis hasil PCR fragmen DNA menggunakan (A) Primer 10pa17 dengan 1 adalah ladder berukuran 100 bp, (B) Primer 16pa17 dengan 1 adalah ladder berukuran 1 kb. 2 dan 3 adalah contoh fragmen DNA hasil amplifikasi.
Dua primer yakni 10pa17 dan 16pa17 merupakan suatu pita DNA tunggal yang merupakan persyaratan untuk proses kloning dari produk PCR. Nomor 1 adalah ladder berukuran 100 bp untuk Gambar 3(A) dan ladder berukuran 1 kb untuk Gambar 3(B). Hasil Gambar 3 tersebut menunjukkan panjang rantai DNA yang telah terbentuk sebesar 150 bp. Hal ini dikarenakan pita DNA hasil amplifikasi primer 10pa17 (sampel nomor 3) berada pada ladder yang berukuran 100 bp dan 200 bp (Gambar 3A). Begitu pula dengan pita DNA hasil amplifikasi primer 16pa17 (sampel nomor 3) yang berada di bawah ladder yang berukuran 250 bp (Gambar 3B).
Pita tunggal hasil PCR ini didapatkan melalui hasil optimasi suhu PCR. Suhu annealing yang digunakan untuk amplifikasi primer 10pa17 adalah 50 oC,
sedangkan untuk primer 16pa17 adalah 59 oC. Selain optimasi suhu juga
dilakukan optimasi pada waktu tahap annealing. Primer dengan panjang 18-22 nukleotida cukup dengan waktu 30 detik, sedangkan primer dengan panjang lebih dari 22 nukleotida membutuhkan waktu 60 detik (Handoyo dan Rudiretna 2010). Hal ini dikarenakan primer yang memiliki basa-basa nukleotida lebih banyak akan mengalami proses polimerisasi yang lebih panjang sehingga akan membutuhkan
waktu yang lebih lama saat tahap penempelan “annealing”.
Pembentukan DNA Rekombinan
14
menyatakan bahwa vektor rekombinan yang merupakan agen yang tersisipi oleh DNA target akan ikut terbawa ketika sel inang yang telah mengalami proses transformasi melakukan pembelahan, sehingga koloni sel inang akan menghasilkan salinan identik dari DNA target.
Penyiapan DNA Sisipan
DNA sisipan disiapkan dengan melakukan e lusi yakni suatu proses pemurnian atau isolasi produk PCR yang dilakukan melalui kegiatan preparasi gel agarose. Penelitian kloning fragmen DNA membutuhkan fragmen DNA dengan kualitas yang baik, sehingga tahap pemurnian melalui teknik elusi ini sangat dibutuhkan. Hasil proses running untuk persiapan pemurnian DNA diperlihatkan pada Gambar 4 berikut ini.
Gambar 4 Elektroforesis fragmen DNA untuk persiapan elusi dengan primer 10pa17 (pita DNA kiri) dan primer 16pa17 (pita DNA kanan)
Hasil proses running dari dua primer yang berukuran 150 bp, seperti yang terlihat pada Gambar 4 merupakan pita DNA produk PCR. Pita DNA produk PCR tersebut terlihat tunggal dan tebal sehingga dapat dilakukan proses elusi. Tahap elusi dilakukan dengan menggunakan Gel/PCR DNA Fragments Extraction Kitdari Geneaid (2008) pada bagian PCR Clean Up Protocol.
Terdapat empat kegiatan pada proses ini yang meliputi penguraian gel, pengikatan DNA, pencucian DNA, serta elusi DNA. Penguraian gel dilakukan dengan menambahkan DF buffer ke dalam tube yang telah berisi cacahan gel. Lalu, diinkubasi pada suhu 55-60 oC hingga gel meluruh seluruhnya. Setelah itu,
binding DNA atau pengikatan DNA dilakukan dengan melakukan sentrifuge selama 2 menit pada kecepatan penuh sehingga molekul- molekul DNA akan tersaring pada DF column.
Molekul- molekul DNA yang telah tersaring dimurnikan dengan W1 buffer yang berisi campuran NaOH dan ethanol yang berfungsi untuk memekatkan DNA setelah sentrifuge selama 2 menit pada kecepatan penuh dan dilanjutkan dengan penambahan Wash buffer yang berisi ethanol untuk mencuci molekul- molekul DNA melalui sentrifuge selama 2 menit pada kecepatan penuh dan dikeringkan.
Kegiatan terakhir adalah elusi DNA yang dilakukan dengan menambahkan elution buffer yang berisi 10 mM Tris-HCl pH 8,5 pada DF column yang berisi molekul DNA kering. Elution buffer ini berfungsi untuk menjaga keseimbangan reaksi agar enzim dapat bekerja lebih optimal dan mencegah terjadinya proses lisis sel (Lodish et al. 1995). Kemudian, lakukan sentrifuge kembali selama 2 menit pada kecepatan penuh untuk mendapatkan produk PCR ya ng telah murni. Hasil elusi tersebut kemudian dapat divisualisasi dengan melakukkan elektoforesis pada gel agarose seperti yang terdapat pada Gambar 5.
250 bp
15
Gambar 5 Elektroforesis hasil elusi fragmen DNA dengan primer 10pa17 (2) dan primer 16pa17 (1)
Proses running hasil elusi untuk persiapan DNA sisipan dari kedua primer digunakan dalam memastikan keberhasilan proses pemurnian produk PCR dari kedua primer tersebut. Hal ini terlihat pada ukuran DNA yang sama ketika proses running persiapan elusi (Gambar 4) dengan proses running hasil elusi yakni 150 bp (Gambar 5) karena berada pada posisi yang sama yakni di bawah ladder 250 bp. Hasil pemurnian pada Gambar 5 siap digunakan sebagai sisipan pada tahap kloning produk PCR. Selain itu, fragmen DNA hasil PCR menggunakan primer 10pa17 dan 16pa17 menunjukkan ukuran yang sama antara Gambar 4 dan Gambar 5.
Ligasi
Ligasi merupakan proses pertama pada tahap kloning DNA rekombinan. Secara umum ligasi adalah suatu proses penggabungan fragmen suatu DNA dengan fragmen DNA lainnya yang menghasilkan suatu DNA rekombinan (Anam 2009). Hal- hal yang dibutuhkan pada proses ligasi meliputi DNA target sebagai sisipan, DNA vektor, enzim ligase, dan buffer ligasi. DNA sisipan dalam penelitian ini adalah produk PCR dari primer 10pa17 dan 16pa17 berukuran 150 bp yang telah dimurnikan melalui tahap elusi.
Vektor adalah biotransport yakni sebagai pembawa fragmen DNA target yang kemudian akan disisipkan dan digandakan pada sel inang. Pemilihan vektor didasarkan pada ukuran fragmen DNA target, dan yang paling sering digunakan sebagai vektor adalah plasmid (Brown 1991). Plasmid merupakan DNA untai ganda yang berasal dari sel bakteri, berbentuk sirkuler, yang berukuran lebih dari 500 kb, terdapat pada sitoplasma dan dapat bereplikasi secara autonom (Suharsono dan Widyastuti 2006). Kemampuan yang dimiliki plasmid seperti inilah merupakan keuntungan untuk menggunakannya sebagai vektor kloning.
Vektor yang digunakan adalah plasmid pGemT- Easy yang berukuran 3015 bp. Kriteria terpenting yang harus dimiliki plasmid adalah harus mengandung situs ORI (Origin of Replication), marker seleksi (selectable marker), dan situs kloning (multiple cloning site). Promega (2010) menjelaskan beberapa kelebihan pGemT-Easy yakni ukuran relatif kecil sehingga dapat membawa fragmen DNA target yang berukuran besar, mengandung situs ORI (Origin of Replication) untuk direplikasi pada bakteri E.coli, membawa gen ketahanan ampisilin sebagai marka seleksi, dan mempunyai multiple cloning site (MCS) dengan 19 sisi unik, seperti yang terdapat pada Gambar 6.
1 2 3
16
Gambar 6 Peta vektor pGemT-Easy (Promega 2010)
MCS merupakan suatu partisi dari DNA vektor yang merupakan tempat dimasukkannya fragmen DNA asing. Selain itu, plasmid ini juga memiliki gen lacZ yang dapat digunakan untuk seleksi koloni biru putih. Gen lacZ ini mengkode enzim β-galaktosidase yang mengubah isopropil- β galaktopiranosida (IPTG) dan substrat 5-bromo-4-kloro-3-indol-β-galaktosidase (X-gal) menjadi biru apabila plasmid tidak tersisipi atau termasuki DNA target, sebaliknya jika tersisipi maka gen lacZ tidak akan terekspresi sehingga koloni tetap berwarna putih (Promega 2010).
Plasmid ini juga dapat langsung diligasikan ke DNA target tanpa melalui proses restriksi sehingga kriteria terpenting sudah terpenuhi sebagai vektor plasmid. Plasmid pGemT-Easy merupakan teknologi baru dari plasmid yang didesain khusus untuk mengkloning gen dari hasil produk PCR.
Enzim yang digunakan untuk proses ligasi adalah enzim T4 DNA Ligase yang merupakan enzim ligase yang berasal dari E.coli yang telah terinfeksi virus T4. Enzim ini berfungsi untuk mensistesis ikatan fosfodiester antar basa-basa nukleotida antara DNA sisipan dan DNA vektor. Enzim ini juga meligasikan ujung- ujung DNA sisipan yang sticky end karena memiliki “overhang” dari basa adenin pada ujung 3’, dengan ujung 5’ yang memiliki basa timin pada vektor plasmid pGemT-Easy (Hartati 2007).
Tahap ligasi yang dilakukan menggunakan perbandingan antara DNA sisipan dengan DNA vektor sebesar 3:1. Cranenburhg (2003) menyatakan bahwa perbandingan antara molar DNA sisipan dengan vektor menjadi hal penting dalam proses pembentukan DNA rekombinan dan juga dapat mencegah pembentukan fragmen- fragmen DNA yang tidak diinginkan. Konsentrasi DNA sisipan yang digunakan pada reaksi ligasi adalah 2,5 ng, sedangkan vektor pGemT-Easy berukuran 50 ng. Panjang fragmen DNA sisipan adalah 150 bp, sedangkan panjang vektornya adalah 3.015 bp, sehingga menghasilkan plasmid rekombinan berukuran 3.165 bp.
17 Kemudian, kondisi ligasi yang optimum dilakukan pada suhu 4-15 oC dengan
waktu 16 jam. Hal ini digunakan untuk meningkatkan kestabilan pembentukan ikatan- ikatan hidrogen yang menghubungkan antar rantai nukleotida pada proses ligasi (Rifai 2011).
Reaksi ligasi dari kedua primer memiliki perbandingan volume DNA sisipan dan DNA vektor yang berbeda. Primer 10pa17 menggunakan perbandingan volume DNA sisipan dan DNA vektor sebesar 1:1, sedangkan primer 16pa17 menggunakan perbandingan volume DNA sisipan dan DNA vektor sebesar 0,5:1. Kontrol positif dan negatif dari reaksi ligasi memiliki reaksi yang sama, tetapi perbedaannya terdapat pada saat transformasi. Kontrol positif menggunakan media LB agar yang tidak mengandung ampicilin, sedangkan kontrol negatif menggunakan media LB agar yang mengandung ampicilin. Setelah itu, reaksi ligasi ini dipersiapkan untuk tahap transformasi selanjutnya.
Transformasi
Setelah proses ligasi dilakukan, maka tahap selanjutnya adalah mempersiapkan sel inang yang digunakan untuk tempat memperbanyak DNA rekombinan tersebut. Sel inang tersebut adalah bakteri E.coli strain DH5α. Sel inang inilah yang perlu ditingkatkan kemampuannya untuk dapat menyerap DNA rekombinan sebagai DNA asing yang kemudian akan digunakan dalam proses transformasi, sehingga disebut sebagai sel kompeten (Muladno 2010).
Sel inang bakteri E.coli menjadi kompeten karena melalui perlakuan khusus dengan diberi perlakuan secara kimia yakni dengan penambahan TFB (transformation buffer) yang berisi CaCl2 (Zhiming 2005). Hal ini dikarenakan
E.coli memiliki kemampuan alami untuk menyerap DNA asing yang rendah (Tsen et al. 2002), sehingga diperlukan perlakuan khusus untuk meningkatkan kemampuannya agar menjadi kompeten melalui CaCl2. Fungsi buffer ini adalah
untuk mengganggu keseimbangan kalsium yang terdapat pada membran sel bakteri, sehingga membran sel bakteri menjadi terbuka dan dapat dimasuki oleh DNA rekombinan.
Di samping itu, garam CaCl2 ini juga menyebabkan terganggunya struktur
dinding sel bakteri sehingga dapat meningkatkan kapasitasnya untuk mengikat DNA asing pada bagian luar sel (Old dan Primrose 2003). Mekanismenya adalah molekul lipopolisakarida (LPS) yang terdapat pada dinding sel bakteri kompeten akan mengikat DNA asing dan memasukkannya ke dalam sitosol sel kompeten karena adanya disintegrasi membran sel akibat CaCl2 (Sarkar et al. 2002).
Ditambahkan pula DMSO yang berguna untuk meningkatkan efisiensi transformasi hingga 100-300 kali lipat (Tu et al. 2005).
Keberhasilan preparasi sel kompeten juga dipengaruhi oleh kondisi bakteri itu sendiri. Bakteri E.coli yang dipilih juga berada pada awal fase logaritmik ya ng ditumbuhkan pada suhu 37 oC (Ausubel et al. 1995). Hal ini dikarenakan agar
pertumbuhan bakteri tersebut dapat memperbanyak diri secara optimal dan juga berada pada kondisi yang sangat aktif membelah sehingga mudah untuk diintroduksi oleh DNA asing (Ausubel et al. 1995).
18
karena kelebihan yang dimilikinya, yakni pertumbuhannya cepat dan mudah dalam penanganannya. Selain itu, secara umum merupakan organisme yang mudah dipahami pada tingkat molekular, sehingga lebih mudah dipelajari dan biasa digunakan untuk menyisipkan gen- gen tertentu yang ingin dikembangkan (Pelczar 1986). Strain DH5α dipilih pada penelitian ini karena telah digunakan secara ekstensif untuk penelitian teknologi DNA rekombinan (Griffith 2001).
Kemudian, strain ini juga dapat bermutasi menjadi lacZΔM15 sehingga menonaktifkan aktivitas gen lacZ yang memproduksi β-galactosidase (Yannish-Perron et al. 1985). Oleh karena itu, strain ini juga tidak dapat memecah X-gal dan menghasilkan warna putih jika gen lacZ tersebut tersisipi, sehingga β -galactosidase tidak aktif. Sebaliknya, strain ini dapat memecah X-gal dan menghasilkan warna biru jika gen lacZ tersebut tidak tersisipi, sehingga β -galactosidase menjadi aktif.
Tahap selanjutnya ketika reaksi ligasi sebagai bahan untuk sisipan DNA rekombinan dan sel kompeten sebagai inang sudah siap, maka dilanjutkan dengan tahapan transformasi. Proses penyisipan fragmen DNA ke dalam suatu sel inang yang dapat mengubah fenotip sel itu sendiri sehingga disebut dengan transformasi (Muladno 2010).
Teknik transformasi yang digunakan pada penelitian ini adalah teknik kejut panas (heatshock) yakni dari suhu 0 oC hingga pada suhu 42 oC. Ketika suhu
panas (42 oC), maka membran sel bakteri akan terbuka, sehingga DNA
rekombinan dapat terinduksi ke dalam sel bakteri. Setelah dilakukan perlakuan panas tersebut, maka campuran reaksi ligasi dan sel ko mpeten tersebut langsung diinkubasi lagi di dalam es agar membran ataupun dinding sel bakteri tersebut dapat langsung tertutup.
Brown (2006) juga menambahkan bahwa pada tahap transformasi proses kejut panas merupakan proses yang paling penting yang menentukan kemungkinan masuknya DNA asing yang telah menempel pada dinding sel ke dalam sitoplasma sel kompeten saat pemberian perlakuan panas tersebut. Selanjutnya, diberi recovery media agar sel dapat cepat pulih dan mengekspresikan gen resisten antibiotiknya (Ausubel et al. 1995). Proses transformasi juga melibatkan penambahan dimetil sulfoksida (DMSO) yang berfungsi untuk meningkatkan efisiensi transformasi (Zhiming et al. 2005).
19
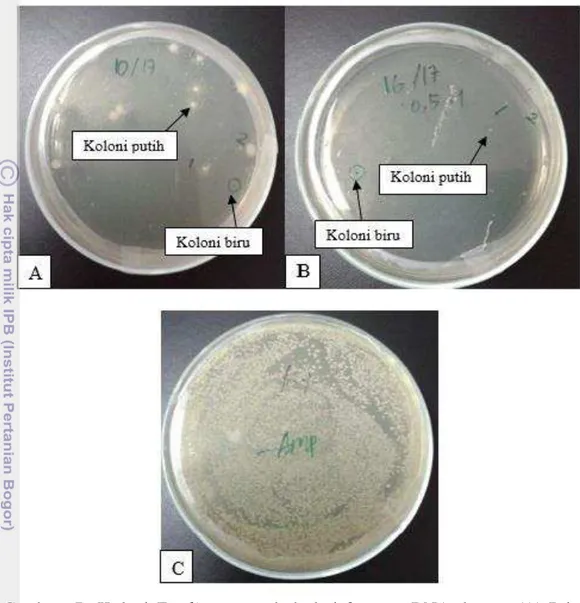
Gambar 7 Koloni E.coli yang tumbuh dari fragmen DNA dengan (A) Primer 10pa17, (B) Primer 16pa17, (C) Kontrol positif
E.coli sebagai inang yang membawa vektor pGemT-Easy yang telah tersisipi fragmen DNA mikrosatelit dari tanaman gaharu pada MCS (Multiple Cloning Sites), maka gen lacZ akan terganggu sehingga IPTG sebagai inducer tidak membantu mengekspresikan enzim β-galaktoside dan mengakibatkan X-gal sebagai substrat tidak terurai, sehingga koloni tetap berwarna putih seperti yang terlihat pada Gambar 7(A) dan 7(B).
Koloni yang berwarna biru memperlihatkan terjadinya ligasi DNA rekombinan pada vektor pGemT-Easy. Koloni bakteri E.coli tetap tumbuh karena plasmid tersebut mengandung gen resisten terhadap antibiotik ampicilin. Akan tetapi, MCS tidak tersisipi oleh DNA target, sehingga IPTG sebagai inducer dapat membantu gen lacZ untuk mengekspresikan enzim β-galaktosidase, sehingga mengurai substrat X-gal menjadi berwarna biru, seperti yang terlihat pada Gambar 7(A) dan 7(B).
20
tersisipi. Oleh karena itu, terbentuklah koloni-koloni baik putih ataupun biru dari sel inang tersebut. Koloni yang terbentuk tersebut adalah sekumpulan sel yang bersifat identik karena tumbuh dari hasil perkembangbiakka n satu sel. Kontrol positif dan negatif juga disebar pada media LB agar tanpa menambahkan DNA sisipan. Kontrol positif yang terdapat pada Gambar 7(C) ditebar pada media LB agar yang tidak mengandung ampicilin sehingga pertumbuhan bakteri tersebut menjadi optimal. Namun, kontol negatif disebar pada media LB agar yang mengandung ampicilin.
SIMPULAN DAN SARAN
Simpulan
Fragmen DNA mikrosatelit sudah berhasil diperoleh dari amplifikasi tanaman gaharu menggunakan primer 10pa17 dan primer 16pa17 dengan ukuran 150 bp. Selanjutnya, fragmen DNA mikrosatelit tersebut telah diisolasi dari tanaman gaharu dengan konsentrasi 20 ng/µl. Proses kloning yang dilakukan dari hasil isolasi telah sampai pada tahap seleksi biru-putih dan menghasilkan koloni transforman E.coli strain DH5α yang berwarna putih yang mengandung DNA rekombinan dan koloni transforman E.coli strain DH5α yang berwarna biru yang tidak mengandung DNA rekombinan.
Saran
1. Perlu dilakukan penelitian lanjut hingga tahap verifikasi isolasi DNA plasmid.
2. Perlu dilakukan penelitian hingga pada tahap sequencing untuk menganalisis hingga pada analisis basa-basa nukleotida.
3. Perlu dilakukan penelitian kloning gaharu dengan primer spesifik yang menyandikan sifat-sifat tertentu untuk mengetahui pengaruh ukuran fragmen DNA.
DAFTAR PUSTAKA
[Balitbanghut] Badan Penelitian dan Pengembangan Kehutanan. 2006. Budidaya gaharu dan rekayasa produksinya [Leaflet]. Bogor (ID): Pusat Penelitian dan Pengembangan Hutan dan Konservasi Alam.
[Ditjen PHKA] Direktorat Jenderal Perlindungan Hutan dan Konservasi Alam. 2005. Peran management authority dalam ekspor gaharu Indonesia. Peluang dan Tantangan Pengembangan Gaharu di Indonesia. Prosiding Seminar Nasional Gaharu; 2005 Desember 1-2; Bogor, Indonesia. Bogor (ID): SEAMEO-BIOTROP.
Alimuddin, Octavera A, Arifin OZ, Sumantadinata K. 2008. Karakterisasi
21 Apriani L. 2008. Seleksi bakteri penghasil enzim kitinolitik serta penguraian beberapa variasi suhu dan pH untuk produksi enzim. [Skripsi]. Depok (ID): Universitas Indonesia.
Anam K. 2009. DNA Rekombinasi. Bogor : Bioteknologi, Sekolah Pascasarjana, Institut Pertanian Bogor.
Anam K. 2010. Isolasi dan Pemetaan DNA Plasmid. Bogor : Bioteknologi, Sekolah Pascasarjana, Institut Pertanian Bogor.
Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Sturhl K, Albright LM, Coen DM, Varki A, Jamsen K. 1995. Current Protocols in Molecular Biology. 2nd ed. Massachusetts (US): John Wiley & Sons. Inc. Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Sturhl K.
1998. Current Protocol in Molecular Biology 1. Canada (US): John Wiley & Sons Inc.
Brown TA. 1991. Pengantar Kloning Gen. Yogyakarta (ID): Yayasan Essential Mediaca.
Brown TA. 2006. Gene Cloning and DNA Analysis : An Introduction. 5th ed. Oxford (GB): Blackwell publishing.
Clark MS. 1997. Plant Molecular Biology. A Laboratory Manual. Berlin (DE): Springer.
Cranenburgh RM. 2003. An equation for calculating the volumetric ratios requires in a ligation reaction. Applied Genetics and Molecular Biotechnology 65:200-202.
Corkill G, Rapley R. 2008. The Manipulation of Nucleic Acids: Basic Tools and Techniques. In: Molecular Biomethods Handbook . 2nd ed. Walker JM, Rapley R, editor. (US): Humana Press, NJ.
Eurlings MCM, Van Beek HH, Gravendeel B. 2009. Polymorphic microsatellites for forensic identification of agarwood (Aquilaria crassna). FSI 197(30):34. Fermentas. 2011. 6X DNA loading dye [Internet]. [dinduh 2014 Agu 9].
Tersedia pada: http://www.fermentas.com/en/products/all/dna-electrophoresis/buffers-reagents/r061-dna-loading-dye.
Gaffar S. 2007. Buku Ajar Bioteknologi Molekul. Bandung (ID): Universitas Padjajaran.
Gelfand DH, White TJ. 1990. Thermostable DNA Polymerase. Di dalam : Innis MA, Glefand DH., Sninsky JJ, White TJ, editor. PCR Protocols A Guide to Methods and Applications. San Diego (US): Academic Press.
Geneaid. 2008. Gel/PCR DNA fragments extraction kit [Internet]. [diunduh
2014 Agu 9]. Tersedia pada
http://www.geneaid.com/sites/default/files/DF12_0.pdf .
Glick BR, Pasternak JJ 1998. Molecular Biotechnology. 2nd ed. Washington DC (US): ASM Press.
22
Handoyo D, Rudiretna A. 2001. Prinsip umum dan pelaksanaan polymerase chain reaction (PCR) [general principles and implementation of polymerase chain reaction]. Unitas. 9(1):17-29.
Hartati. 2007. Kloning gen penyandi sukrosa sintase dari tanaman sengon (Paraserianthes falcataria). [tesis]. Bogor (ID): Sekolah Pascasarjana, Institut Pertanian Bogor.
Holme DJ, Hazel P. 1998. Analytical biochemistry. 3rd ed. London (GB): Addison Wesley Longman.
Keller GH. Mark MM. 1989. DNA Probe. Macmilan (US): University Michigan. Larasati P. 2011. Isolasi Plasmid dan Elektroforesis pada Gel Agarosa. Bogor
(ID): Institut Pertanian Bogor.
Lodish et al. 1995. Molecular Cell Biology. New York (ID): Scientific American Books.
Mizawarti. 2003. Penerapan teknik-teknik kloning gen dalam kehidupan manusia. [tesis]. Medan (ID): Universitas Sumatera Utara.
Muladno. 2010. Teknologi Rekayasa Genetika. Bogor (ID): IPB Press.
Novel SS., Firstianty F, Safitri R, Nuswantara S. 2010. Whatman flinders technology associates (FTA) card untuk uji diagnostik molekular bakteri Mycobacterium tuberculosis berbasis metode polymerase chain reaction. Bandung (ID): Jurusan Biologi, Fakultas Metematika dan Ilmu Pengetahuan Alam, Universitas Padjajaran.
Old RW, Primrose SB. 2003. Prinsip-prinsip manipulasi gen : pengantar rekayasa genetik. Ed ke-4. Susilo H, Corebima AD, editor. Jakarta (ID): UI-Press. Terjemahan dari: Principles of gene manipulation : An Introduction to Genetic Engineering. 4th ed,.
Pelczar MJ, Chan ECS. 1986. Dasar-dasar Mikrobiologi. Hadioetomo RS, Imas T, Tjitrosomo SS, Angka SL, penerjemah. Jakarta (ID): Universitas Indonesia. Porebski S, Bailey LG. Baum BR. 1997. Modification of CTAB DNA extraction
protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 15:8-15.
Promega. 2010. Technical Manual pGemT and pGemT-Easy Vector System. USA : Promega Corporation.
Promega. 2009. Technical Bulletin GoTaq PCR Core Systems. Madison (US): Promega Corporation.
Sarkar S, Chaudhuri S, Basu T. 2002. Mechanism of artificial transformation of E.coli with plasmid DNA-clues from the influence of ethanol. Current Science 83:1376-1380.
Setyari PS, Wirasutha IG, Junitha IK. 2014. Metode analisis kualitatif dan kuantitatif LDL-C menggunakan elektroforesisagarose dapar TAE (Tris-Asam asetat-EDTA). Bali (ID): Universitas Udayana.
23 Suharsono, Widyastuti U. 2006. Penuntun Praktikum Pelatihan Teknik Pengklonan. Bogor (ID): Departemen Biologi, Fakultas matematika dan Ilmu Pengetahuan Alam, Institut Pertanian Bogor.
Suharsono, Widyastuti U. 2010. Penuntun Praktikum Pengantar Genetika Molekular. Bogor (ID): Departemen Biologi, Fakultas matematika dan Ilmu Pengetahuan Alam, Institut Pertanian Bogor.
Sumarna Y 2009. Gaharu Budidaya dan Rekayasa Produksi. Bogor (ID): Penebar Swadaya.
Tsen et al. 2002. Natural plasmid transformatio n in Escherichia coli. Journal of Biomedical Science 9:246-252.
Tu et al. 2005. An improved system for competent cell preparation and high efiiciency plasmid transformation using different Escherichia coli strains. Electronical Journal of Biotechnology 8.
Varshney RK,Graner A, Sorrells ME. 2005. Genic Microsatellite Markers in Plants: Feature and Applications. Trends in Biotechnology. 23:48-56.
Verkuil PVE. 2008. Principles and Technical Aspects of PCR Amplification, 141 Springer Science + Business Media B.V.
Vierstraete A. 1999. Principles of PCR [Internet]. [diunduh 2014 Agu 9]. Tersedia pada: http://users.ugent.be/~avierstr/principles/pcr.html.
Walker JM, Rapley R. 2002. Molecular Biology and Biotechnology. 4th ed. Great Britain : The Royal Society of Chemistry.
Weaver NA, Hedrick PW. 1997. Genetics. 3rd ed. Dubuque (US): Wm. C. Brown Publisher.
Weising K, Nybom H, Wolff K, Kahl G. 2005. DNA Fingerprinting in Plants Principles, Methods, and Applications. 2nd ed. Francis : CRC Press.
Wong DWS. 1997. The ABC of Gene Cloning. New York (US): International Thompson Publishing.
Yuwono T. 2006. Teori dan Aplikasi Polymerase Chain Reaction. Yogyakarta (ID): Penerbit ANDI.
Yuwono T. 2009. Biologi Molekular. Jakarta (ID): Erlangga.
24
LAMPIRAN
Lampiran 1 Komposisi pembuatan buffer TAE 50 kali (Tris-Acetate-EDTA) (Setyari 2014)
Buffer TAE 50 kali dibuat dari komponen:
Tris-base 48,4 g
Acetic acid glacial 11,4 g EDTA 0,5 M pH 8,0 20 ml
Komponen tersebut kemudian dilarutkan dengan menggunakan aquadest hingga volumenya 1000 ml dan dihomogenkan dengan menggunakan magnetic strirrer. Setelah itu, simpan pada suhu ruang.
Lampiran 2 Cara pembuatan loading dye 6 kali (Fermentas 2011) Loading dye 6 kali dapat dibuat dari komponen :
Bromophenol blue 0,25 %
Xylene cyanol 0,25 %
Ficoll tipe 15 %
EDTA 120 mM
Kemudian, komponen tersebut dihomogenkan dan disimpan pada suhu ruang. Lampiran 3 Cara pembuatan media LB (Luria bertani) cair (Apriani 2008).
Media luria bertani cair dapat dibuat dengan komposisi dari: Ekstrak khamir 0,5 g
NaCl 1 g
Pepton 1 g
Komponen tersebut dilarutkan ke dalam 100 ml aquadest. Setelah itu, dihomogenkan pada mesin magnetic stirrer hingga larutan berwarna kekuningan. Kemudian, larutan LB cair tersebut disterilisasi pada autoclave dengan suhu 121
oC selama 1 jam.
Lampiran 4 Cara pembuatan media LB (Luria bertani) agar (Apriani 2008). Media luria bertani agar dapat dibuat dengan komposisi dari :
Ekstrak khamir 0,5 g
NaCl 1 g
Pepton 1 g
Agar 2 g
Komponen tersebut dilarutkan ke dalam 100 ml aquadest. Setelah itu, dihomogenkan pada mesin magnetic stirrer hingga larutan berwarna kekuningan. Kemudian, larutan LB cair tersebut disterilisasi pada autoclave dengan suhu 121
25 Lampiran 5 Cara pembuatan TFB (Transformation Buffer) (Suharsono dan
Widyastuti 2010).
Pembuatan transformation buffer terdiri atas komponen :
PIPES 10 mM 3 g
CaCl2.2H2O 15 mM 2,2 g
KCl 250 mM 18,2 g
MnCl2.4H2O 55 mM 10,9 g
Aquadest 950 ml
Komponen tersebut diaduk dengan menggunakan magnetic strirrer dan tambahkan KOH hingga pH 6,7 dan tambahkan aquadest hingga volume 1 L. Lampiran 6 Cara pembuatan media 2xYT (Alimuddin et al. 2008)
Recovery media atau media 2xYT dapat dibuat dari komponen :
Polypeptone 1,6 %
Yeast extract 1 %
NaCl 0,5 %
Agarosa 1,5 %
Komponen di campur dalam SDW.
Lampiran 7 Cara pembuatan IPTG 0,1 M (Promega 2010)
IPTG sebanyak 1,2 g ditambahkan aquadest sampai volume akhir 50 ml. Kemudian, dilakukan sterilisasi dengan alat filtrasi dan disimpan pada 4 oC.
Lampiran 8 Cara pembuatan X-Gal 50 mg/ml (Promega 2010)
26
27 Lampiran 10 Proses perbanyakan DNA dengan PCR (Vierstraete 1999)
Lampiran 11 Cara perhitungan perbandingan 3:1 antara DNA sisipan dengan DNA vektor (Promega 2010).
Rumus untuk menghitung perbandingan antara DNA sisipan dengan DNA vektor sebesar 3:1 adalah:
Jumlah insert (ng) = vektor (ng) x ukuran insert (kb) x 3 Ukuran vektor (kb) 1 Jumlah insert (ng) = 50 ng x 0,15 kb x 3 = 7,5 ng
3 1
28