KLASIFIKASI FRAGMEN METAGENOM MENGGUNAKAN
METODE SVM DAN
FAST CORRELATION BASED FILTER
SEBAGAI PENYELEKSI FITUR
AFDHAL DINILHAK
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM INSTITUT PERTANIAN BOGOR
PERNYATAAN MENGENAI SKRIPSI DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA
Dengan ini saya menyatakan bahwa skripsi berjudul Klasifikasi Fragmen Metagenom Menggunakan Metode SVM dan Fast Correlation Based Filter sebagai Penyeleksi Fitur adalah benar karya saya dengan arahan dari komisi pembimbing dan belum diajukan dalam bentuk apa pun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir skripsi ini.
Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
ABSTRAK
AFDHAL DINILHAK. Klasifikasi Fragmen Metagenom Menggunakan Metode SVM dan Fast Correlation Based Filter sebagai Penyeleksi Fitur. Dibimbing oleh WISNU ANANTA KUSUMA.
Bioinformatika memiliki banyak bidang kajian penting dan terus berkembang, salah satunya adalah analisis metagenom. Metagenom merupakan materi genetik yang diperoleh dari sampel yang langsung diambil dari lingkungan tanpa budidaya di laboratorium. Untuk mengklasifikasikan fragmen metagenom ke dalam tingkat taksonomi yang berbeda perlu dilakukan proses binning. Pada penelitian ini dilakukan proses binning dengan pendekatan komposisi menggunakan metode supervised learning. Proses klasifikasi dilakukan dengan menggunakan Support Vector Machine (SVM) sebagai classifier, perhitungan frekuensi k-mers untuk mengekstraksi fitur, dan Fast Correlation Based Filter (FCBF) sebagai penyeleksi fitur. Pada proses seleksi fitur, jumlah fitur yang terseleksi ditentukan oleh nilai threshold. Dari penelitian ini akurasi hasil klasifikasi SVM dengan seleksi fitur berkisar antara 79.13% sampai 96.68% untuk 3-mers, sedangkan jika menggunakan 4-mers akurasi berkisar antara 83.59% sampai 99.35%.
Kata kunci: metagenom, binning, k-mers, feature selection, fast correlation based filter, threshold, SVM
ABSTRACT
AFDHAL DINILHAK. Metagenome Fragments Binning Using SVM Method and Fast Correlation Based Filter Feature Selection. Supervised by WISNU ANANTA KUSUMA.
Bioinformatics has many fields important and it keeps growing, one of the interesting fields to be studied is metagenome analysis. Metagenome is the genetic material which obtained from samples taken directly from environmental without conducting a laboratory cultivation. Binning is required to classify metagenome fragments into different taxonomic. This research employed the binning based composition approach which is implemented using supervised learning method. Classification process was conducted using Support Vector Machine (SVM) as classifier, counting k-mers frequency as feature extraction, and Fast Correlation Based Filter (FCBF) algorithm as feature selection. In the process of feature selection, the number of selected features was determined by a threshold. The result of this research show that the accuracy of the used method using 3-mers ranged from 79.13% to 96.68%. Moreover, the accuracy increased from 83,59% to 99,35% using 4-mers.
Skripsi
sebagai salah satu syarat untuk memperoleh gelar Sarjana Komputer
pada
Departemen Ilmu Komputer
KLASIFIKASI FRAGMEN METAGENOM MENGGUNAKAN
METODE SVM DAN
FAST CORRELATION BASED FILTER
SEBAGAI PENYELEKSI FITUR
AFDHAL DINILHAK
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM INSTITUT PERTANIAN BOGOR
Penguji: Aziz Kustiyo, SSi MKom Toto Haryanto, SKom MSi
Judul Skripsi : Klasifikasi Fragmen Metagenom Menggunakan Metode SVM dan Fast Correlation Based Filter sebagai Penyeleksi Fitur
Nama : Afdhal Dinilhak NIM : G64124026
Disetujui oleh
Dr Eng Wisnu Ananta Kusuma, ST MT Pembimbing
Diketahui oleh
Dr Ir Agus Buono, MSi MKom Ketua Departemen
PRAKATA
Alhamdulillahi Rabbil ‘alamin, puji dan syukur penulis panjatkan kepada Allah Subhanahu wa ta’ala atas segala karunia-Nya sehingga karya ilmiah ini berhasil diselesaikan. Tema yang dipilih dalam penelitian yang dilaksanakan sejak bulan Juli 2014 ini ialah klasifikasi fragmen metagenom, dengan judul Klasifikasi Fragmen Metagenom Menggunakan Metode SVM dan Fast Correlation Based Filter sebagai Penyeleksi Fitur.
Terima kasih penulis ucapkan kepada seluruh pihak yang telah berperan dalam penelitian ini, yaitu:
1 Ayahanda Apruddin, ibunda Rusmiati, dan keluarga atas doa, semangat, dan dorongan kepada penulis sehingga dapat menyelesaikan penelitian ini.
2 Bapak Dr Eng Wisnu Ananta Kusuma, ST MT selaku pembimbing, yang telah memberikan arahan, ide, masukan, dan dukungan kepada penulis.
3 Bapak Aziz Kustiyo, SSi MKom dan bapak Toto Haryanto, SKom MSi yang telah bersedia menjadi penguji, dan memberikan saran yang berharga sehingga tulisan ini menjadi lebih baik dari sebelumnya.
4 Seluruh staf pengajar Ilmu Komputer IPB yang telah memberikan ilmu semasa perkuliahan.
5 Rekan-rekan Ilmu Komputer IPB yang saling menyemangati selama pengerjaan penelitian di tahun yang sama.
6 Seluruh rekan satu bimbingan yang tidak dapat disebutkan satu persatu dan pihak-pihak lainnya.
Semoga penelitian dan tulisan ini dapat memberikan manfaat.
DAFTAR ISI
DAFTAR TABEL ix
DAFTAR GAMBAR ix
DAFTAR LAMPIRAN ix
PENDAHULUAN 1
Latar Belakang 1
Perumusan Masalah 2
Tujuan Penelitian 2
Manfaat Penelitian 2
Ruang Lingkup Penelitian 2
METODE 3
Pengumpulan Data 4
Pembagian Data 4
Praproses Data 4
Ekstraksi Fitur 4
Seleksi Fitur 4
Support Vector Machine (SVM) 6
Grid Search 8
Pelatihan SVM 8
Pengujian SVM 8
Analisis 8
Implementasi 9
HASIL DAN PEMBAHASAN 9
Pembagian Data 9
Praproses Data 9
Ekstraksi Fitur 10
Seleksi Fitur 10
Grid Search 11
Klasifikasi SVM 12
Analisis 12
SIMPULAN DAN SARAN 18
Simpulan 18
Saran 18
DAFTAR PUSTAKA 19
DAFTAR TABEL
1 Jumlah fitur terseleksi untuk beragam nilai threshold 11 2 Nilai akurasi klasifikasi SVM tanpa seleksi fitur dan dengan seleksi
fitur 14
3 Nilai sensitivity dan specificity hasil klasifikasi SVM dengan ekstraksi fitur menggunakan 3-mers dan panjang fragmen 0.5 Kbp. 16
DAFTAR GAMBAR
1 Tahapan Penelitian 3
2 Ekstraksi fitur k-mers 4
3 Algoritme FCBF (Yu 2003) 6
4 Contoh ilustrasi pemodelan SVM 6
5 Hasil praproses data 10
6 Hasil grid search 11
7 Hasil akurasi klasifikasi SVM dengan 3-mers sebagai ekstraksi fitur dan seleksi fitur menggunakan beragam nilai threshold. 12 8 Hasil akurasi klasifikasi SVM dengan 4-mers sebagai ekstraksi fitur
dan seleksi fitur menggunakan beragam nilai threshold. 13 9 Hasil akurasi klasifikasi SVM dengan menggunakan FCBF dan tanpa
FCBF 14
10 Waktu komputasi klasifikasi SVM dengan seleksi fitur dan tanpa seleksi fitur dan 3-mers untuk ekstraksi fitur. 15
DAFTAR LAMPIRAN
1 Daftar nama organisme data latih 21
2 Daftar nama organisme data uji 23
3 Daftar tingkat taksonomi (genus) 24
4 Daftar hasil grid search 24
5 Daftar waktu komputasi klasifikasi SVM tanpa seleksi fitur dan
dengan seleksi fitur 25
6 Hasil analisis ragam untuk melihat pengaruh seleksi fitur dan panjang
fragmen terhadap akurasi klasifikasi 26
7 Hasil analisis ragam untuk melihat pengaruh seleksi fitur dan panjang fragmen terhadap waktu komputasi klasifikasi 26 8 Confusion matrix hasil dari klasifikasi SVM dengan ekstraksi fitur
menggunakan 3-mers pada data dengan panjang fragmen 0.5 Kbp 27 9 Confusion matrix hasil dari klasifikasi SVM dengan ekstraksi fitur
menggunakan 3-mers dan seleksi fitur (threshold 0) untuk data
dengan panjang fragmen 0.5 Kbp 28
1
PENDAHULUAN
Latar Belakang
Bioinformatika memiliki banyak bidang kajian penting dan terus berkembang, salah satunya adalah analisis metagenom. Metagenomika adalah ilmu yang mempelajari materi genetik yang diperoleh dari sampel yang langsung diambil dari lingkungan tanpa budidaya di laboratorium atau isolasi genom individu (Wu 2008). Di dalam sampel tersebut terdapat berbagai spesies organisme, sehingga perlu dilakukan proses binning untuk mengklasifikasikan scaffolds, contigs dan unassembled contigs ke dalam garis keturunan taksonomi yang berbeda.
Proses binning dapat dilakukan dengan dua pendekatan, yaitu pendekatan homologi dan pendekatan komposisi. Pada pendekatan homologi dilakukan pencarian penjajaran sekuens dengan membandingkan fragmen metagenom dengan basis data sekuens National Centre for Biotechnology Information (NCBI). Hasilnya disimpulkan pada tiap level taksonomi. Metode yang menggunakan pendekatan homologi seperti BLAST (Wu 2008; Zheng dan Wu 2009) dan MEGAN (Huson et al. 2007). Berbeda dengan pendekatan homologi, pendekatan komposisi tidak membandingkan sekuens kueri dengan sekuens referensi. Pada pendekatan komposisi, pasangan basa hasil ekstraksi fitur digunakan sebagai masukan untuk pembelajaran dengan observasi (unsupervised learning) atau pembelajaran dengan contoh (supervised learning). Beberapa contoh penelitian mengenai metagenome binning yang menggunakan pembelajaran dengan observasi, yaitu TETRA (Teeling et al. 2004), Chisel System (Rodriguez et al. 2007), ESTmapper (Wu et al. 2005), Growing Self Organizing Map (Hsu dan Halgamuge 2002), Kohonen Self Organizing Map (Abe et al. 2003), Meta-Clust (Woyke et al. 2006), Self Organizing Clustering (Amano et al. 2003), dan Clustering Metagenome Fragments Using Growing Self Organizing Map (Overbeek et al. 2013). Adapun penelitian yang menggunakan pembelajaran dengan contoh adalah ClaMS (Pati et al.2011), PhyloPythia (McHardy et al. 2007), Naïve Bayessian Classification (Rosen et al. 2008), serta Support Vector Machine dan Characterization Vectors (Kusuma dan Akiyama 2011).
Berkaitan dengan metode supervised learning, McHardy et al. (2007) melakukan penelitian untuk mengklasifikasikan fragmen metagenom dengan menggunakan perhitungan frekuensi k-mers sebagai ekstraksi fitur dan Multiclass Support Vector Machine (SVM) sebagai classifier. Akurasi yang diperoleh cukup baik khususnya untuk panjang fragmen ≥ 5 Kbp yaitu antara 60% sampai ˃ 90% di setiap tingkat takson. Akurasi ini terus menurun secara signifikan jika menggunakan fragmen dengan panjang ≤ 3 Kbp. Pada fragmen dengan panjang 3 Kbp hanya diperoleh akurasi sebesar 40%, sedangkan untuk panjang fragmen 1 Kbp akurasi yang diperoleh < 10%. Ukuran k-mers yang digunakan adalah 5-mers, yang berarti fitur yang dihasilkan memiliki dimensi 45 = 1024 fitur. Dimensi fitur
yang besar dapat membuat kinerja algoritme klasifikasi menjadi tidak efektif dan efisien, misalnya akurasi yang kurang baik dan waktu pemrosesan menjadi lebih lama karena banyak fitur yang harus diproses.
2
salah satu tahap praproses pada klasifikasi dengan memilih fitur-fitur yang relevan terhadap data yang mempengaruhi hasil klasifikasi. Algoritme Fast Correlation Based Filter (FCBF) adalah salah satu algoritme seleksi fitur yang dikembangkan oleh Yu dan Huan (2003). Konsep utama dari algoritme ini adalah menghilangkan fitur-fitur yang tidak relevan serta menyaring fitur-fitur yang redundant terhadap fitur-fitur yang lain. Berdasarkan penelitian yang dilakukan Yu dan Huan (2003) diperoleh hasil bahwa FCBF sangat efisien dalam melakukan seleksi fitur serta memberikan performa yang baik bagi kinerja algoritme klasifikasi. Adapun rataan akurasi yang diperoleh pada penelitian tersebut adalah 89.13% dengan menggunakan C4.5 sebagai classifier dan 86.92% jika menggunakan Naive Bayes Classifier. Oleh karena itu, pada penelitian ini dilakukan klasifikasi fragmen metagenom dengan menggunakan metode SVM dan Fast Correlation Based Filter sebagai penyeleksi fitur, serta perhitungan frekuensi k-mers sebagai metode ekstraksi fitur.
Perumusan Masalah
Permasalahan yang akan menjadi bahan analisis dalam penelitian ini adalah: 1 Bagaimana pengaruh nilai threshold terhadap hasil seleksi fitur dan hasil
klasifikasi?
2 Berapa akurasi yang diperoleh jika menggunakan metode SVM dengan seleksi fitur dan tanpa seleksi fitur?
3 Bagaimana pengaruh panjang fragmen yang digunakan terhadap hasil klasifikasi?
4 Bagaimana pengaruh nilai k-mers yang digunakan terhadap hasil klasifikasi?
Tujuan Penelitian
Tujuan dari penelitian ini adalah:
1 Mengklasifikasikan fragmen metagenom ke dalam tingkat taksonomi genus dengan menggunakan metode SVM dan FCBF sebagai penyeleksi fitur.
2 Mengetahui pengaruh penggunaan beragam nilai threshold terhadap hasil seleksi fitur dan hasil klasifikasi.
3 Mengetahui pengaruh panjang fragmen dan nilai k-mers yang digunakan terhadap hasil klasifikasi.
4 Mengetahui lama waktu komputasi klasifikasi SVM dengan seleksi fitur dan tanpa seleksi fitur.
Manfaat Penelitian
Penelitian ini diharapkan dapat membantu peneliti dalam mengidentifikasi dan mengklasifikasikan fragmen metagenom sesuai dengan tingkat taksonomi, serta dapat memberikan kontribusi untuk mendukung proses analisis metagenome sequence.
Ruang Lingkup Penelitian
Ruang lingkup penelitian ini meliputi:
3 2 Data uji sebanyak 30 organisme yang termasuk dalam taksonomi yang sama
dengan data latih.
3 Fragmen yang digunakan dihasilkan dari perangkat lunak MetaSim yang mensimulasikan Illumina sequencer. Fragmen yang dihasilkan memiliki panjang yang tetap dan tidak mengandung sequencing error.
4 Level taksonomi yang digunakan yaitu genus.
METODE
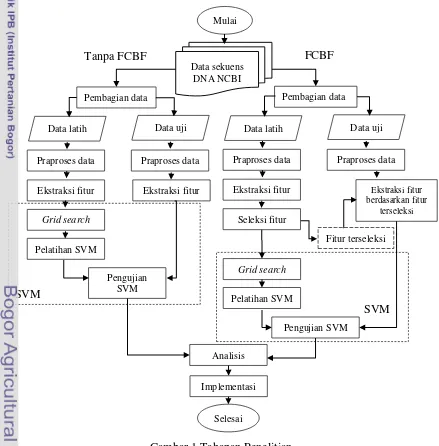
Penelitian ini dilakukan dengan beberapa tahap, yaitu pengumpulan data, pembagian data, praproses data, ektraksi fitur, seleksi fitur menggunakan algoritme FCBF, klasifikasi dengan menggunakan metode SVM, analisis hasil klasifikasi, dan implementasi. Tahapan pada penelitian ini digambarkan pada Gambar 1.
Gambar 1 Tahapan Penelitian
Pembagian data
Data latih Data uji
Praproses data Praproses data
Ekstraksi fitur Ekstraksi fitur
berdasarkan fitur
Praproses data Praproses data
Ekstraksi fitur Ekstraksi fitur
4
Pengumpulan Data
Data yang digunakan dalam penelitian ini adalah data sekuens DNA bakteri yang diambil dari basis data National Centre for Biotechnology Information (NCBI). NCBI adalah sebuah institusi yang fokus di bidang biologi molekuler dan menjadi sumber informasi untuk perkembangan bidang tersebut. Data disimpan dalam fail dengan format FastA. Data dapat diunduh di alamat berikut: ftp://ftp.ncbi.nih.gov/genomes/Bacteria/all.fna.tar.gz.
Pembagian Data
Pada penelitian ini data yang digunakan terbatas pada 50 organisme untuk pelatihan, dan 30 organisme untuk dilakukan pengujian. Pemilihan data uji dilakukan dengan mengambil organisme selain data latih yang juga termasuk ke dalam genus yang sama.
Praproses Data
Pada tahap praproses data, fragmen metagenom dibangkitkan dari data sekuens DNA menggunakan perangkat lunak MetaSim (Richter et al. 2008). MetaSim adalah perangkat lunak untuk mensimulasikan genome sequencer. Data diproses dibaca berulang kali disesuaikan dengan kebutuhan penelitian. Panjang fragmen yang ditetapkan untuk setiap kali pengolahan yaitu 0.5Kbp, 1 Kbp, 3 Kbp, dan 5 Kbp.
Ekstraksi Fitur
Ekstraksi fitur dilakukan dengan menghitung frekuensi dari kombinasi nukleotida yang mungkin terbentuk dengan menggunakan k-mers. Pola kemunculan k adalah pola yang menampilkan k pada suatu waktu dalam suatu sekuens DNA. Pola kemunculan dalam sekuens DNA dihitung menggunakan empat basa utama (A, T, G, dan C) dipangkat dengan rangkaian pasangan basa yang ingin digunakan (pola kemunculan 4 , dengan k >= 1). Dalam penelitian ini akan digunakan beberapa nilai k-mers, yaitu 3-mers dan 4-mers.
Seleksi Fitur
Seleksi fitur bertujuan untuk mengurangi dimensi data, menghilangkan fitur yang tidak relevan dan berlebihan, dan memberikan performa yang baik bagi kinerja algoritme klasifikasi (Yu dan Liu 2003). Algoritme seleksi fitur yang digunakan dalam tahapan ini adalah Fast Correlation Based Filter (FCBF) yang dikembangkan oleh Yu dan Liu (2003). Yu dan Liu melakukan dua pendekatan dengan mengukur korelasi antara dua variabel acak yaitu berdasar pada classical
AAAACCATATGATTACCT 3-mers
5 linear correlation atau linear correlation coefficient dan berdasar pada teori informasi. Pendekatan linear correlation coeficient dirumuskan sebagai berikut:
� = ∑ � − ̅� − ̅� √∑ − ̅ √∑� − ̅�
�
̅adalah rata-rata dari X dan ̅� adalah rata-rata dari Y. Nilai r berada di rentang -1 dan 1. Nilai r adalah 1 atau -1 jika X dan Y memiliki korelasi, dan bernilai 0 jika X dan Y tidak berkorelasi. Pendekatan linear correlation coeficient hanya bisa digunakan pada fitur-fitur yang memiliki nilai-nilai numerik. Untuk fitur-fitur yang tidak bernilai numerik digunakan pendekatan berdasarkan pada information-theoretical concept of entrophy yang mengukur ketidakpastian pada variabel acak. Entrophy dari variabel X didefinisikan sebagai berikut:
= − ∑ � log �
Entrophy dari variabel X jika diketahui variabel Y didefinisikan sebagai berikut:
| = − ∑ � ∑ � | log �( | )
� adalah prior probablities untuk semua nilai X dan � | adalah posterior probabilities dari X jika diketahui Y. Dari nilai entrophy dapat diperoleh Information Gain.
| = − |
Untuk mengukur korelasi antar fitur, digunakan symmetrical uncertainty yang nilainya berkisar pada rentang 0 sampai dengan 1.
�� , = [ +| ]
6
Gambar 3 Algoritme FCBF (Yu 2003) Support Vector Machine (SVM)
SVM merupakan teknik klasifikasi yang masuk ke dalam kelas supervised learning. SVM dikembangkan oleh Vladimir Vapnik tahun 1995, konsep dasar SVM adalah menemukan bidang pemisah (hyperplane) terbaik yang dapat memisahkan d-dimensi data dengan sempurna ke dalam 2 kelas (kelas +1 dan kelas -1). Hyperplane terbaik antara kedua kelas dapat ditemukan dengan mengukur margin hyperplane dan mencari titik maksimalnya. Margin adalah jarak antara hyperplane dengan pattern terdekat dari masing-masing kelas. Pattern yang paling dekat ini disebut sebagai support vector (Boswell 2002). Ilustrasi SVM dapat dilihat pada Gambar 4.
7 berdimensi d, yang didefinisikan seperti berikut:
⃗⃗ . + � =
⃗⃗ . + � − , ∀ ∈ kelas − ⃗⃗ . + � + , ∀ ∈ kelas +
dengan w adalah bidang normal dan b adalah posisi bidang relatif terhadap pusat koordinat. Margin terbesar dapat ditemukan dengan memaksimalkan nilai jarak antara hyperplane dan titik terdekatnya, yaitu 1/||w||. Untuk memisahkan dua buah kelas pada input space yang tidak dapat terpisah secara sempurna SVM dimodifikasi dengan memasukkan slack variable� � > seperti berikut:
. ⃗⃗ + � − � , ∀�
min � ⃗⃗ , � = ‖⃗⃗ ‖ + � ∑ � =
Parameter C dipilih untuk mengontrol tradeoff antara margin dan error klasifikasi �. Nilai C yang besar berarti akan memberikan penalti yang lebih besar terhadap error klasifikasi. Selanjutnya dilakukan penyelesaian dengan formula Lagrangian menggunakan Lagrange multiplier:
⃗⃗ , �, � = ‖⃗⃗ ‖ − ∑ � ( . ⃗⃗ + � − ) =
, � = , , … , �
� adalah Lagrange multipliers, yang bernilai nol atau positif (� ).
Jumlah data yang besar dan beragam dapat mengakibatkan data tersebut tidak dapat dipisahkan secara linear. Untuk mengatasi masalah ini, SVM dimodifikasi dengan memasukkan fungsi kernel yang dapat mentransformasikan data ke dimensi lebih tinggi dengan fungsi transformasi xk → Φ(xk). Data yang sudah berada di dimensi lebih tinggi tersebut dapat dengan mudah dipisahkan dengan hyperplane secara linear (Boswell 2002). Fungsi kernelK(x,xi) = Φ(x) ∙ Φ(xi), memberikan kemudahan dalam proses pembelajaran SVM. Dengan demikian fungsi yang dihasilkan dari pelatihan adalah:
�(Φ ) = ∑ � , + �
�
= ,� �∈��
Kernel yang digunakan dalam SVM pada penelitian ini adalah Gaussian Radial Basis Function (RBF) (Osuna et al. 1997):
x,y = exp -‖ − ‖�
8
( 14)) (14) Pada Multiclass SVM dilakukan N(N-1)/2 pengklasifikasian biner yang berbeda, dengan N adalah banyaknya kelas. Sehingga data baru yang akan ditentukan kelasnya, akan masuk ke dalam kelas yang memiliki nilai fungsi keputusan terbesar. Apabila terdapat dua kelas atau lebih yang memiliki nilai keputusan yang sama besar, maka kelas yang indeksnya lebih kecil dinyatakan sebagai kelas dari data tersebut (Hsu dan Lin 2002).
Grid Search
Akurasi model yang dihasilkan dari proses pelatihan dengan Multiclass SVM dipengaruhi oleh fungsi kernel dan parameter yang digunakan. Untuk mendapatkan parameter terbaik ada beberapa cara yang dapat dilakukan antara lain cross validation, leave-one-out, dan ��- estimator (Quang et al. 2002). Pada penelitian ini metode yang digunakan untuk mendapatkan parameter terbaik adalah cross validation. Pencarian parameter terbaik ini disebut dengan grid search.
Grid search dilakukan dengan menggunakan 10% data latih yaitu sebanyak 6000 fragmen. Pengambilan 10% data latih ini mengacu pada penelitian yang dilakukan oleh McHardy et al. (2007). Melalui k-fold cross validation berbagai nilai parameter akan dicoba sehingga menghasilkan nilai parameter terbaik. Untuk kernel RBF, nilai parameter terbaik yang dihasilkan adalah nilai cost (c) dan gamma (γ). Setelah nilai parameter terbaik ditemukan, pelatihan dilakukan dengan menggunakan keseluruhan data latih yaitu sebanyak 60 ribu fragmen.
Pelatihan SVM
Pelatihan SVM dilakukan untuk data latih yang terdiri dari 60 ribu fragmen. Data latih yang digunakan yaitu data latih yang belum diseleksi fitur dan data latih yang telah diseleksi fitur. Fungsi kernel yang digunakan dalam pelatihan ini adalah fungsi kernel RBF.
Pengujian SVM
Hasil dari pelatihan SVM adalah sebuah model yang akan diuji menggunakan data uji. Pengujian dilakukan pada data uji tanpa seleksi fitur dan data uji dengan seleksi fitur. Pengujian akan mengklasifikasikan data uji yang terdiri dari 30 organisme ke dalam kelas taksonominya. Semua organisme data uji yang telah dikelaskan menghasilkan persentase hasil pengklasifikasiannya.
Analisis
Setelah dilakukan pelatihan dan pengujian SVM, diperoleh hasil untuk kinerja algoritme SVM, baik itu dengan menggunakan seleksi fitur atau tanpa seleksi fitur. Akurasi hasil klasifikasi dihitung dengan menggunakan rumus:
akurasi =∑ data uji benar∑ data uji x %
Setelah perhitungan akurasi selesai dilakukan, beberapa hal yang menjadi bahan analisis adalah:
9 2 Perbandingan nilai akurasi klasifikasi SVM dengan melakukan seleksi fitur dan
tanpa seleksi fitur, serta waktu komputasi klasifikasi.
3 Pengaruh panjang fragmen dan nilai k-mers terhadap hasil klasifikasi. 4 Nilai sensitivity dan specificity.
Implementasi
Implementasi sistem dilakukan ke dalam sebuah aplikasi berbasis website dengan lingkungan pengembangan sebagai berikut:
1 Bahasa pemrograman : PHP
2 Library : LibSVM 3.18
3 DBMS : MySQL.
Sistem ini dirancang untuk melakukan prediksi tingkat taksonomi genus suatu sekuens DNA. Hasil keluaran dari sistem ini adalah nama genus dari sekuens DNA yang dimasukkan.
HASIL DAN PEMBAHASAN
Pembagian Data
Data sekuens DNA yang diunduh dari situs NCBI dibagi menjadi data latih dan data uji. Data latih dipilih 50 organisme dan data uji 30 organisme. Pemilihan data uji dilakukan dengan mengambil organisme selain data latih yang masih berada dalam tingkat taksonomi genus yang sama. Daftar organisme yang digunakan sebagai data latih dapat dilihat pada Lampiran 1 dan organisme data uji pada Lampiran 2. Adapun daftar genus yang digunakan dapat dilihat pada Lampiran 3.
Praproses Data
Pada tahap ini, fragmen metagenom dibangkitkan dari sekuens DNA data latih dan data uji dengan menggunakan perangkat lunak MetaSim. Pada penelitian ini, data latih dipersiapkan sebanyak 6000 dan 60 ribu fragmen yang diurai dari 50 organisme. Adapun untuk data uji sebanyak 24 ribu fragmen yang diurai dari 30 organisme. Setiap genus pada data latih dan data uji memiliki jumlah fragmen yang sama. Jumlah fragmen pada masing-masing genus pada data latih adalah 600 dan 6000, sedangkan jumlah fragmen pada masing-masing genus pada data uji adalah 2400.
10
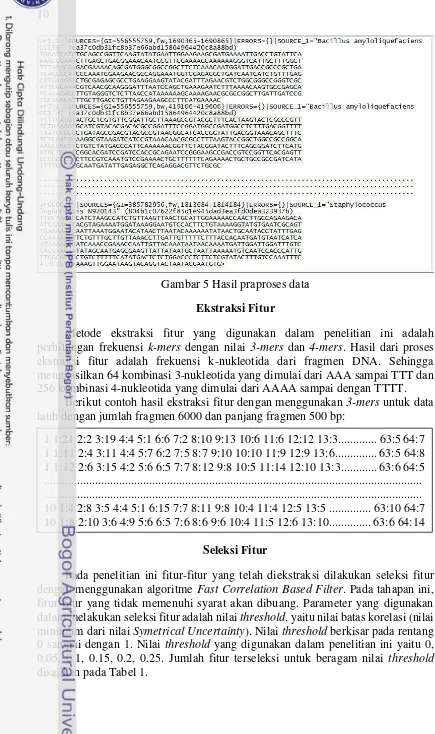
Gambar 5 Hasil praproses data
Ekstraksi Fitur
Metode ekstraksi fitur yang digunakan dalam penelitian ini adalah perhitungan frekuensi k-mers dengan nilai 3-mers dan 4-mers. Hasil dari proses ekstraksi fitur adalah frekuensi k-nukleotida dari fragmen DNA. Sehingga menghasilkan 64 kombinasi 3-nukleotida yang dimulai dari AAA sampai TTT dan 256 kombinasi 4-nukleotida yang dimulai dari AAAA sampai dengan TTTT.
Berikut contoh hasil ekstraksi fitur dengan menggunakan 3-mers untuk data latih dengan jumlah fragmen 6000 dan panjang fragmen 500 bp:
Seleksi Fitur
Pada penelitian ini fitur-fitur yang telah diekstraksi dilakukan seleksi fitur dengan menggunakan algoritme Fast Correlation Based Filter. Pada tahapan ini, fitur-fitur yang tidak memenuhi syarat akan dibuang. Parameter yang digunakan dalam melakukan seleksi fitur adalah nilai threshold, yaitu nilai batas korelasi (nilai minimum dari nilai Symetrical Uncertainty). Nilai threshold berkisar pada rentang 0 sampai dengan 1. Nilai threshold yang digunakan dalam penelitian ini yaitu 0, 0.05, 0.1, 0.15, 0.2, 0.25. Jumlah fitur terseleksi untuk beragam nilai threshold disajikan pada Tabel 1.
11 Tabel 1 Jumlah fitur terseleksi untuk beragam nilai threshold
Nilai threshold
3-mers 4-mers
0.5 Kbp 1 Kbp 3 Kbp 5 Kbp 0.5 Kbp 1 Kbp 3 Kbp 5 Kbp Fullsets 64 64 64 64 256 256 256 256
0 29 34 31 17 82 100 150 156
0.05 29 34 31 17 62 91 143 151
0.1 15 23 31 17 39 68 114 123
0.15 9 13 15 11 20 36 92 99
0.2 8 7 8 8 9 21 42 58
0.25 3 5 4 6 2 5 16 24
Tabel 1 menunjukkan bahwa seleksi fitur dapat mengurangi jumlah fitur hingga mencapai setengah dari jumlah fitur asalnya. Hal ini disebabkan karena fitur-fitur yang terseleksi tersebut adalah fitur yang memiliki nilai Symetrical Uncertainty di atas nilai threshold. Selain itu, dapat dilihat juga bahwa tidak semua fitur relevan dengan kelasnya dan tidak semua fitur mempunyai tingkat korelasi yang tinggi terhadap kelasnya dibandingkan dengan korelasinya terhadap fitur yang lain. Selanjutnya fitur yang telah terseleksi dapat digunakan pada tahapan klasifikasi.
Grid Search
Grid search dilakukan untuk mendapatkan nilai parameter yang dibutuhkan oleh kernel RBF. Parameter tersebut adalah cost (c) dan gamma (γ) yang didapatkan dengan melakukan proses k-fold cross validation dengan k sama dengan 5. Hasil grid search untuk data latih yang telah dilakukan seleksi fitur dengan jumlah fragmen 6000 dan panjang fragmen 500 bp, serta menggunakan 3-mers untuk ekstraksi fitur dapat dilihat pada Gambar 6. Gambar 6 menunjukkan bahwa nilai terbaik c = 4 dan γ = 2 dengan akurasi 5-cross validation = 79.35%. Hasil grid search lainnya untuk setiap data latih yang digunakan dapat dilihat pada Lampiran 4.
12
Klasifikasi SVM
Proses klasifikasi SVM dilakukan terhadap data tanpa seleksi fitur dan data dengan seleksi fitur. Klasifikasi SVM diawali dengan menskalakan (normalisasi) data latih dan data uji terlebih dahulu sebelum melakukan pelatihan dan pengujian. Proses penskalaan diperlukan untuk menghindari atribut atau fitur bernilai besar tidak mendominasi fitur lain yang bernilai kecil. Selain itu penskalaan juga dapat mempermudah perhitungan selama proses pengklasifikasian.
Selanjutnya data latih yang telah diskalakan dilatih dengan menggunakan fungsi kernel (RBF). Hasil pelatihan ini adalah model yang digunakan untuk pengujian data uji. Dari pengujian ini diperoleh hasil klasifikasi berupa akurasi dengan menggunakan Persamaan 14.
Analisis
Analisis yang dilakukan terhadap hasil klasifikasi SVM meliputi bagaimana pengaruh nilai threshold terhadap hasil seleksi fitur dan akurasi klasifikasi, akurasi dan waktu komputasi klasifikasi SVM menggunakan seleksi fitur dan tanpa seleksi fitur, panjang fragmen dan nilai k-mers yang digunakan, serta nilai sensitivity dan specificity.
Pengaruh Nilai Threshold Terhadap Hasil Seleksi Fitur dan Akurasi Klasifikasi
Nilai threshold merupakan parameter yang digunakan untuk menyeleksi fitur. Nilai threshold sangat menentukan jumlah fitur yang terseleksi. Jumlah fitur yang terseleksi berdasarkan beragam nilai threshold yang digunakan disajikan pada Tabel 1. Dari Tabel 1 dapat dilihat bahwa semakin tinggi nilai threshold yang digunakan maka fitur yang terseleksi semakin sedikit. Pemangkasan fitur yang dilakukan pada proses seleksi fitur dengan berbagai nilai threshold memberikan pengaruh terhadap hasil klasifikasi SVM. Pengaruh tersebut dapat dilihat pada Gambar 7 dan Gambar 8.
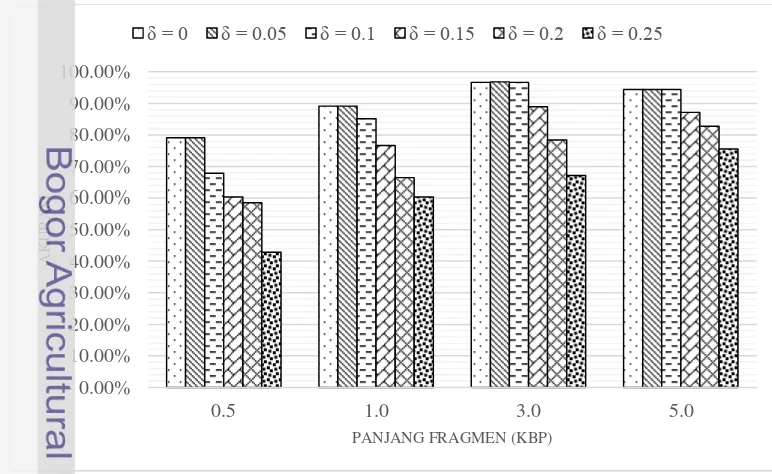
Gambar 7 Hasil akurasi klasifikasi SVM dengan 3-mers sebagai ekstraksi fitur dan seleksi fitur menggunakan beragam nilai threshold.
13 Dari Gambar 7 dapat dilihat bahwa akurasi terbaik dihasilkan ketika menggunakan nilai threshold 0, 0.05, dan 0.1, serta panjang fragmen 3.0 Kbp yaitu sebesar 96.68%. Sedangkan akurasi terkecil didapat saat menggunakan nilai threshold 0.25 dengan panjang fragmen 0.5 Kbp yaitu sebesar 42.81%.
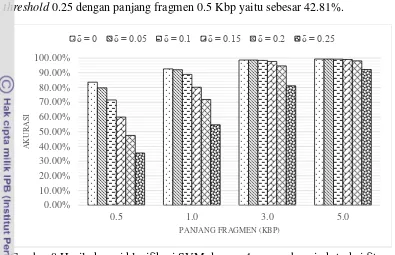
Gambar 8 menunjukkan akurasi terbaik diperoleh saat menggunakan nilai threshold 0.1 dan panjang fragmen 5 Kbp yaitu sebesar 99.36%. Sedangkan akurasi terkecil diperoleh saat menggunakan nilai threshold 0.25 dan panjang fragmen 0.5 Kbp yaitu sebesar 35.5%.
Kecenderungan nilai akurasi pada setiap panjang fragmen untuk setiap nilai threshold tidak dapat diprediksi apakah mengalami kenaikan atau penurunan. Pada klasifikasi SVM dengan menggunakan panjang fragmen 0.5 Kbp dan 4-mers untuk ekstraksi fitur, terjadi penurunan akurasi ketika nilai threshold dinaikkan. Adapun ketika menggunakan panjang fragmen 5 Kbp dan 4-mers untuk ekstraksi fitur, terjadi peningkatan akurasi dari 99.35% (threshold 0.05) menjadi 99.36% (threshold 0.1).
Hasil Klasifikasi SVM dengan Menggunakan FCBF dan Tanpa FCBF
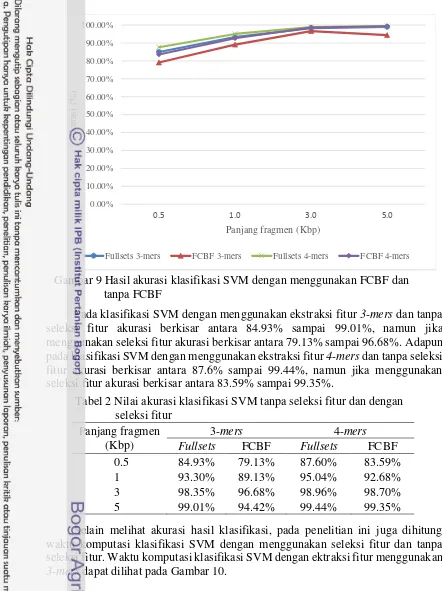
Perbandingan akurasi klasifikasi SVM tanpa seleksi fitur dan dengan seleksi fitur (threshold 0) dapat dilihat pada Gambar 9 dan Tabel 2.
Gambar 8 Hasil akurasi klasifikasi SVM dengan 4-mers sebagai ekstraksi fitur dan seleksi fitur menggunakan beragam nilai threshold.
14
Pada klasifikasi SVM dengan menggunakan ekstraksi fitur 3-mers dan tanpa seleksi fitur akurasi berkisar antara 84.93% sampai 99.01%, namun jika menggunakan seleksi fitur akurasi berkisar antara 79.13% sampai 96.68%. Adapun pada klasifikasi SVM dengan menggunakan ekstraksi fitur 4-mers dan tanpa seleksi fitur akurasi berkisar antara 87.6% sampai 99.44%, namun jika menggunakan seleksi fitur akurasi berkisar antara 83.59% sampai 99.35%.
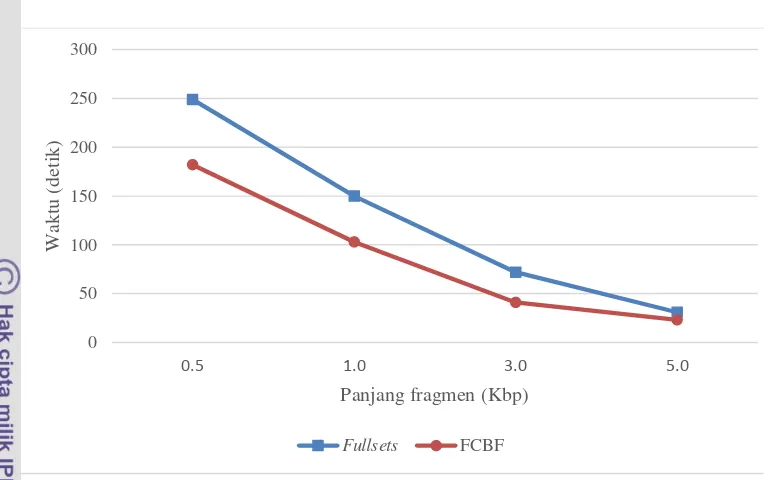
Selain melihat akurasi hasil klasifikasi, pada penelitian ini juga dihitung waktu komputasi klasifikasi SVM dengan menggunakan seleksi fitur dan tanpa seleksi fitur. Waktu komputasi klasifikasi SVM dengan ektraksi fitur menggunakan 3-mers dapat dilihat pada Gambar 10.
Gambar 9 Hasil akurasi klasifikasi SVM dengan menggunakan FCBF dan tanpa FCBF
Fullsets 3-mers FCBF 3-mers Fullsets 4-mers FCBF 4-mers
Tabel 2 Nilai akurasi klasifikasi SVM tanpa seleksi fitur dan dengan seleksi fitur
Panjang fragmen (Kbp)
3-mers 4-mers
Fullsets FCBF Fullsets FCBF
0.5 84.93% 79.13% 87.60% 83.59%
1 93.30% 89.13% 95.04% 92.68%
3 98.35% 96.68% 98.96% 98.70%
15
Gambar 10 menunjukkan bahwa terdapat perbedaan waktu komputasi antara klasifikasi SVM tanpa seleksi fitur dan klasifikasi SVM dengan seleksi fitur. Untuk klasifikasi dengan menggunakan ekstraksi fitur 3-mers, selisih waktu tertinggi terjadi pada panjang fragmen 0.5 Kbp yaitu sebesar 67 detik. Dan selisih terkecil pada panjang fragmen 5 Kbp sebesar 8 detik. Sedangkan waktu komputasi klasifikasi SVM dengan ekstraksi fitur menggunakan 4-mers dapat dilihat pada Lampiran 5. Selisih waktu terbesar berada pada panjang fragmen 1 Kbp sebesar 110 detik, dan terkecil pada panjang fragmen 5 Kbp sebesar 32 detik.
Dari akurasi dan waktu komputasi klasifikasi SVM menggunakan seleksi fitur dan tanpa seleksi fitur, dilakukan analisis ragam untuk melihat apakah seleksi fitur berpengaruh signifikan terhadap hasil akurasi dan waktu komputasi klasifikasi. Hasil analisis ragam untuk mengetahui pengaruh seleksi fitur terhadap akurasi dapat dilihat pada Lampiran 6. Adapun hasil analisis ragam untuk mengetahui pengaruh seleksi fitur terhadap waktu komputasi klasifikasi dapat dilihat pada Lampiran 7. Taraf nyata yang digunakan adalah 0.05. Dari hasil analisis ragam, dapat diperoleh informasi antara lain:
1 Hasil analisis ragam untuk melihat pengaruh seleksi fitur terhadap akurasi klasifikasi SVM dengan ekstraksi fitur menggunakan 3-mers, nilai Prob>F adalah 0.0161. Karena nilai Prob>F (0.0161) lebih kecil dari nilai taraf nyata (0.05), maka tolak H0 yang berarti seleksi fitur mempengaruhi hasil akurasi klasifikasi.
2 Hasil analisis ragam untuk melihat pengaruh seleksi fitur terhadap akurasi klasifikasi SVM dengan ekstraksi fitur menggunakan 4-mers, nilai Prob>F adalah 0.1699. Karena nilai Prob>F (0.1699) lebih besar dari nilai taraf nyata (0.05), maka terima H0 yang berarti seleksi fitur tidak mempengaruhi hasil akurasi klasifikasi.
3 Hasil analisis ragam untuk melihat pengaruh seleksi fitur terhadap waktu komputasi klasifikasi SVM dengan ekstraksi fitur menggunakan 3-mers, nilai Prob>F adalah 0.1217. Karena nilai Prob>F (0.1217) lebih besar dari nilai taraf Gambar 10 Waktu komputasi klasifikasi SVM dengan seleksi fitur dan tanpa
16
nyata (0.05), maka terima H0 yang berarti seleksi fitur tidak mempengaruhi waktu komputasi klasifikasi.
4 Hasil analisis ragam untuk melihat pengaruh seleksi fitur terhadap waktu komputasi klasifikasi SVM dengan ekstraksi fitur menggunakan 4-mers, nilai Prob>F adalah 0.0372. Karena nilai Prob>F (0.0372) lebih kecil dari nilai taraf nyata (0.05), maka tolak H0 yang berarti seleksi fitur mempengaruhi waktu komputasi klasifikasi.
Sensitivity dan specificity
Pada penelitian ini perhitungan sensitivity dan specificity dilakukan pada data yang menghasilkan akurasi klasifikasi SVM terendah, yaitu data dengan panjang fragmen 0.5 Kbp. Nilai sensitivity dan specificity dihitung dengan menggunakan confusion matrix. Adapun confusion matrix hasil dari klasifikasi SVM dengan ekstraksi fitur menggunakan 3-mers dan tanpa seleksi fitur dapat dilihat pada Lampiran 8. Sedangkan confusion matrix hasil dari klasifikasi SVM dengan ekstraksi fitur menggunakan 3-mers dan seleksi fitur (threshold 0) dapat dilihat pada Lampiran 9. Sensitivity dan specificity yang dihasilkan dari klasifikasi SVM dengan ekstraksi fitur menggunakan 3-mers dapat dilihat pada Tabel 3.
Tabel 3 Nilai sensitivity dan specificity hasil klasifikasi SVM dengan ekstraksi fitur menggunakan 3-mers dan panjang fragmen 0.5 Kbp.
No Genus Fullsets FCBF
Sensitivity Specificity Sensitivity Specificity
1 Bacillus 81.63% 98.13% 77.42% 97.48%
17 diprediksi sebagai bagian dari genus lain. Pada genus Corynebacterium terdapat sebanyak 333 organisme diprediksi masuk ke dalam Geobacter dan 183 organisme ke dalam Mycobacterium. Adapun pada genus Lactobacillus terdapat sebanyak 337 organisme diprediksi masuk ke dalam Staphylococcus dan 105 organisme ke dalam Bacillus. Untuk nilai specificity dari klasifikasi SVM dengan ekstraksi fitur menggunakan 3-mers dan tanpa seleksi fitur berkisar antara 97.01% sampai dengan 99.75% dengan rata-rata specificity-nya 98.34%. Adapun nilai specificity dari klasifikasi SVM dengan ekstraksi fitur menggunakan 3-mers dan menggunakan seleksi fitur (threshold 0) berkisar antara 96.20% sampai dengan 99.71% dengan rata-rata specificity-nya sama dengan 97.72%.
Panjang Fragmen
Sebuah fragmen DNA mengandung unsur nukleotida yang merupakan unsur genetik yang dimiliki suatu organisme. Ciri yang dimiliki setiap organisme berbeda satu sama lain, ciri tersebut dapat dilihat dari perbedaan unsur genetik yang dimilikinya. Pada klasifikasi SVM dengan tidak melakukan seleksi fitur, terlihat bahwa panjang fragmen memberikan pengaruh terhadap hasil akurasi. Semakin panjang fragmen yang digunakan, akurasi yang dihasilkan semakin meningkat. Hal ini terjadi karena apabila fragmen yang digunakan untuk proses pengklasifikasian besar, maka perbedaan unsur nukleotida pun semakin besar yang mengakibatkan hasil pengklasifikasian pun lebih baik. Adapun jika menggunakan seleksi fitur, akurasi tidak hanya dipengaruhi oleh panjang fragmen saja tetapi juga dipengaruhi oleh fitur-fitur yang terseleksi. Seperti yang ditunjukkan pada Gambar 9 di atas, hasil akurasi dengan menggunakan panjang fragmen 5 Kbp mengalami penurunan jika dibandingkan dengan akurasi dengan menggunakan panjang fragmen 3 Kbp.
Bila melihat hasil analisis ragam pada Lampiran 6 dan Lampiran 7 dapat diperoleh informasi bahwa panjang fragmen memberikan pengaruh yang signifikan terhadap akurasi dan waktu komputasi klasifikasi SVM.
Penggunaan Nilai k-mers
Nilai k-mers yang digunakan untuk melakukan ekstraksi fitur adalah 3-mers dan 4-mers. Berdasarkan hasil percobaan yang sudah dilakukan dapat diperoleh informasi bahwa penggunaan nilai k-mers memberikan pengaruh terhadap hasil klasifikasi. Pada Gambar 9 di atas dapat dilihat bahwa penggunaan 4-mers memberikan hasil akurasi lebih tinggi dari 3-mers. Pada saat melakukan proses ekstraksi fitur dengan menggunakan 3-mers, jumlah fitur yang dihasilkan ada sebanyak 4 yaitu 64 fitur. Adapun jika menggunakan 4-mers jumlah fitur yang dihasilkan sebanyak 4 yaitu 256 fitur.
Implementasi
18
SIMPULAN DAN SARAN
Simpulan
Pada penelitian ini dilakukan pengklasifikasian fragmen metagenom dengan menggunakan metode Support Vector Machine (SVM) dan Fast Correlation Based Filter sebagai penyeleksi fitur. Seleksi fitur merupakan salah satu tahapan praproses klasifikasi yang dilakukan dengan cara memilih fitur-fitur yang mampu memberikan hasil terbaik pada saat klasifikasi. Salah satu parameter yang digunakan dalam menyeleksi fitur adalah nilai threshold. Nilai threshold menentukan banyaknya fitur yang terseleksi, semakin tinggi nilai threshold jumlah fitur yang terseleksi semakin sedikit. Penggunaan nilai threshold yang berbeda menghasilkan akurasi klasifikasi yang berbeda juga.
Pada klasifikasi SVM dengan menggunakan ekstraksi fitur 3-mers dan tanpa seleksi fitur akurasi berkisar antara 84.93% sampai 99.01%, namun jika menggunakan seleksi fitur akurasi berkisar antara 79.13% sampai 96.68%. Adapun pada klasifikasi SVM dengan menggunakan ekstraksi fitur 4-mers dan tanpa seleksi fitur akurasi berkisar antara 87.6% sampai 99.44%, namun jika menggunakan seleksi fitur akurasi berkisar antara 83.59% sampai 99.35%.
Di sisi lain, klasifikasi SVM dengan menggunakan seleksi fitur dapat mereduksi waktu komputasi. Pada klasifikasi SVM dengan menggunakan panjang fragment 1 Kbp dan 4-mers untuk ekstraksi fitur, waktu komputasi klasifikasi dapat direduksi hingga 110 detik.
Setelah dilakukan analisis ragam dengan menggunakan taraf nyata 0.05, diperoleh informasi bahwa pada klasifikasi SVM dengan ektraksi fitur menggunakan 3-mers, seleksi fitur mempengaruhi nilai akurasi namun tidak mempengaruhi waktu komputasi. Adapun pada klasifikasi SVM dengan ekstraksi fitur menggunakan 4-mers, seleksi fitur tidak mempengaruhi akurasi tetapi berpengaruh secara signifikan terhadap waktu komputasi.
Saran
Beberapa saran untuk penelitian selanjutnya yaitu:
1 Menggunakan kombinasi metode klasifikasi dan algoritme seleksi fitur yang berbeda.
2 Menggunakan data dengan jumlah kelas yang lebih banyak, sehingga dapat melakukan prediksi untuk lebih banyak kelas dan untuk mengetahui bagaimana pengaruh algoritme FCBF jika digunakan pada kelas yang banyak.
19
DAFTAR PUSTAKA
Abe T, Kanaya S, Kinouchi M, Ichiba Y, Kozuku T, Ikemura T. 2003. Informatics for unveiling hidden genome signatures.Genome Research. 179(4):693-701. doi:10.1101/gr.634603.
Amano K, Nakamura H, Ichikawa H. 2003. Self-organizing clustering : a novel non-hierarchical method for clustering large amountof sequece DNAs. Genome Informatics. 14: 575-576.
Boswell D. 2002.Introduction to support vector machine [Internet]. [diunduh 2014 Jun 23]. Tersedia pada: http://www.work.caltech.edu/~boswell/ IntroToSVM.pdf
Hsu AL, Halgamuge SK. 2002. Enhancement of topology preservation and hierarchical dynamic self-organizing maps for data visualisation. International Journal of Approximate Reasoning. 32(2003):259-279
Hsu CW, Lin CJ. 2002. A comparison of methods for multiclass support vector machine. IEEE Transactions on Neural Networks. 13(2):415–425. doi: 10.1109/72.991427.
Huson DH, Auch AF. Qi J, Schuster SC. 2007. MEGAN analysis of metagenomic data.Genome Research.17 : 1 – 11. doi : 10.1101/gr/5969107.
Kusuma WA, Akiyama Y. 2011. Metagenome fragments classification based on characterization vectors. Di dalam: Proceedings of International Conference on Bioinformatics and Biomedical Technology; 2011 Mar; Sanya, China. hlm 50-54
McHardy AC, Martin HG, Tsirigos A, Hugenholtz P, Rigoutsos I. 2007. Accurate phylogenetic classification of variable-lenght dna fragments. Nature Methods. 4(1):63-72. doi: 10.1016/j.mib.2007.08.004
Overbeek MV, Kusuma WA, Buono Agus. 2013. Clustering Metagenome Fragments Using Growing Self Organizing Map. ICACSIS 2013: 5th International Conference on Advanced Computer Science and Information
Systems; 2013 Sept; Bali. hlm 285-289. Doi: 10.1109/ICACSIS.2013.6761590.
Pati A, Heath LS, Kyrpides NC, Ivanova N. 2011.ClaMS : A classifier for metagenomic sequences. Standards in Genomic Science.5 : 248 –253. doi :10.4056/sigs.2075298
Quang Anh, Zhang Qian-Li, Li Xing. 2002.Evolving Support Vector Machine Parameters. Di dalam: Proceedingd of the First International Conference on Machine Learning and Cybernetics; 2002 Nov; Beijing.
Richter DC, Ott F, Auch AF, Schmid R, Huson DH. 2008. MetaSim:a sequencing simulator for genomics and metagenomics. PLoS ONE. 3(10):1–12. doi:10.1371/journal.pone.0003373.
Rodriguez AA, Bompada T, Syed M, Shah PK, Maltsev N. 2007. Evolutionary analysis of enzymes using chisel.Bioinformatics. 23( 22).
Rosen G, Garbarine E, Caseiro D, Polikar R, Sokhansanj B. 2008. Metagenome fragment classification using n-mer frequency profiles. Advances in Boinformatics. doi: 10.1155/2008/205969.
20
tetranucleotide usage pattern in sequence DNAs. BMC Informatics.5(163). doi:10.1186/1471-2105-5-163.
Wooley JC, Godzik A, Friedberg I. 2010. A primer on metagenomics. PLos Computational Biology. 6(2):1–13. doi: 10.1371/journal.pcbi.1000667.
Woyke T, Teeling H, Ivanova NN, Hunteman M, Richter M, Gloeckner FO, Boffelli D, Anderson IJ, Barry KW, Shapiro HJ et al.2006. Symbiosis insights through metagenomic analysis of a microbial consortium. Nature. 443(7114): 950-5.
Wu H. 2008. PCA-based linear combinations of oligonucleotide frequencies for metagenomic DNA fragment binning. Di dalam: Computational Intelligence in Bioinformatics and Computational Biology 2008. hlm 46-53.
Wu X, Lee W, Tseng C. 2005. ESTmapper : efficiently aligning sequence DNAs to genomes. IEEE International Paralel and Distributed Processing Symposium. 204(2005) : 196 – 204. doi : 10.1109/IPDPS.2005.204.
Yu L, H Liu. 2003. Feature Selection for High Dimensional Data: A Fast Correlation-Based Filter Solution.
www.hpl.hp.com/conferences/icml2003/papers/144.pdf.
21 Lampiran 1 Daftar nama organisme data latih
No Nama Organisme No Nama Organisme
1 Bacillus amyloliquefaciens CC178
23 Clostridium botulinum A str. Hall chromosome
2 Bacillus amyloliquefaciens FZB42
24 Clostridium difficile B19 chromosome
3 Bacillus anthracis str. A0248 25 Clostridium kluyveri DSM 555 chromosome
4 Bacillus anthracis str. Ames chromosome
26 Corynebacterium diphtheriae NCTC 13129 chromosome 5 Bacillus cereus AH187
chromosome
27 Corynebacterium glutamicum ATCC 13032
6 Bacillus megaterium DSM 319 chromosome
28 Corynebacterium jeikeium K411 chromosome
7 Bordetella avium 197N chromosome
29 Corynebacterium urealyticum DSM 7109 chromosome 8 Bordetella bronchiseptica RB50
chromosome
30 Geobacter bemidjiensis Bem chromosome
9 Bordetella parapertussis 12822 chromosome
31 Geobacter sp. M18 chromosome 10 Burkholderia ambifaria AMMD
chromosome 1
32 Geobacter metallireducens GS-15 chromosome
11 Burkholderia cenocepacia AU 1054 chromosome 1
33 Helicobacter acinonychis str. Sheeba chromosome
12 Burkholderia cenocepacia HI2424 chromosome 1
34 Helicobacter hepaticus ATCC 51449 chromosome
13 Burkholderia cenocepacia J2315 chromosome 1
35 Helicobacter pylori G27 chromosome
14 Burkholderia cenocepacia MC0-3 chromosome 1
36 Helicobacter pylori HPAG1 chromosome
15 Burkholderia gladioli BSR3 chromosome 1
37 Lactobacillus acidophilus 30SC chromosome
16 Burkholderia mallei ATCC 23344 chromosome 1
38 Lactobacillus amylovorus GRL 1112 chromosome
17 Burkholderia mallei NCTC 10229 chromosome I
39 Lactobacillus buchneri NRRL B-30929 chromosome
18 Burkholderia mallei SAVP1 chromosome I
40 Lactobacillus casei str. Zhang chromosome
19 Burkholderia sp. 383 chromosome 1
41 Lactobacillus delbrueckii subsp. bulgaricus ND02 chromosome 20 Clostridium acetobutylicum
ATCC 824 chromosome
42 Mycobacterium abscessus chromosome
21 Clostridium beijerinckii NCIMB 8052 chromosome
43 Mycobacterium avium 104 chromosome
22 Clostridium botulinum A str. ATCC 19397 chromosome
22
Lampiran 1 Lanjutan
No Nama Organisme No Nama Organisme
45 Mycobacterium leprae TN chromosome
48 Staphylococcus epidermidis ATCC 12228 chromosome 46 Staphylococcus aureus subsp.
aureus COL chromosome
49 Staphylococcus haemolyticus JCSC1435 chromosome 47 Staphylococcus aureus subsp.
aureus JH1 chromosome
23 Lampiran 2 Daftar nama organisme data uji
No Nama Organisme No Nama Organisme
1 Bacillus amyloliquefaciens DSM7 16 Corynebacterium resistens DSM 45100 chromosome
2 Bacillus anthracis str. CDC 684 chromosome
17 Geobacter daltonii FRC-32 chromosome
3 Bacillus cereus ATCC 10987 18 Geobacter sp. M21 chromosome 4 Bacillus cereus Q1 chromosome 19 Helicobacter felis ATCC 49179
chromosome
5 Bordetella bronchiseptica 253 20 Helicobacter pylori B38 chromosome
6 Bordetella parapertussis Bpp5 21 Lactobacillus acidophilus NCFM chromosome
7 Burkholderia ambifaria MC40-6 chromosome 1
22 Lactobacillus brevis ATCC 367 8 Burkholderia cepacia GG4
chromosome 1
23 Lactobacillus delbrueckii subsp. bulgaricus ATCC 11842
chromosome 9 Burkholderia mallei NCTC 10247
chromosome I
24 Lactobacillus gasseri ATCC 33323 chromosome
10 Burkholderia sp. CCGE1001 chromosome 1
25 Mycobacterium africanum GM041182 chromosome 11 Burkholderia sp. YI23
chromosome 1
26 Mycobacterium bovis BCG str. Tokyo 172 chromosome
12 Clostridium botulinum A str. ATCC 3502 chromosome
27 Mycobacterium leprae Br4923 chromosome
13 Clostridium difficile CD196 chromosome
28 Staphylococcus aureus subsp. aureus JH9 chromosome 14 Clostridium kluyveri NBRC
12016
29 Staphylococcus aureus subsp. aureus MSSA476 chromosome 15 Corynebacterium aurimucosum
ATCC 700975 chromosome
24
Lampiran 3 Daftar tingkat taksonomi (genus)
Lampiran 4 Daftar hasil grid search No Genus
Nilai parameter c dan γ terbaik yang didapat pada tahap grid search (3-mers) Fullsets FCBF (threshold = 0)
Panjang fragmen
25 Lampiran 5 Daftar waktu komputasi klasifikasi SVM tanpa seleksi fitur dan
dengan seleksi fitur Panjang
fragmen (Kbp)
3-mers 4-mers
26
Lampiran 6 Hasil analisis ragam untuk melihat pengaruh seleksi fitur dan panjang fragmen terhadap akurasi klasifikasi
Keterangan:
Columns : Fitur (seleksi fitur dan tanpa seleksi fitur) Rows : Panjang fragmen
Lampiran 7 Hasil analisis ragam untuk melihat pengaruh seleksi fitur dan panjang fragmen terhadap waktu komputasi klasifikasi
Keterangan:
Columns : Fitur (seleksi fitur dan tanpa seleksi fitur) Rows : Panjang fragmen
Ekstraksi fitur menggunakan 3-mers
Ekstraksi fitur menggunakan 4-mers
Ekstraksi fitur menggunakan 3-mers
27
Lampiran 8 Confusion matrix hasil dari klasifikasi SVM dengan ekstraksi fitur menggunakan 3-mers pada data dengan panjang fragmen 0.5 Kbp
Genus hasil prediksi
Genus asal Bacillus Bordetella Burkholde ria
Clostridi um
Corynebac
terium Geobacter
Helicobac ter
Lactobacil lus
Mycobac terium
Staphyloco ccus
Bacillus 1959 3 2 102 12 15 18 57 4 228
Bordetella 0 2162 66 0 43 29 2 1 97 0
Burkholderia 3 73 2186 0 36 22 0 1 79 0
Clostridium 59 1 0 2220 4 17 13 21 0 65
Corynebacterium 5 95 15 1 1741 333 0 26 183 1
Geobacter 63 52 3 8 147 1944 4 136 38 5
Helicobacter 6 0 0 7 34 2 2330 12 0 9
Lactobacillus 105 1 0 61 53 24 39 1770 10 337
Mycobacterium 4 85 34 0 207 39 0 6 2025 0
Staphylococcus 160 0 0 88 9 2 19 76 0 2046
28
Lampiran 9 Confusion matrix hasil dari klasifikasi SVM dengan ekstraksi fitur menggunakan 3-mers dan seleksi fitur (threshold 0) untuk data dengan panjang fragmen 0.5 Kbp
Genus asal
Genus hasil prediksi
Bacillus Bordetella Burkholde ria
Clostridi um
Corynebac
terium Geobacter
Helicobac ter
Lactobacil lus
Mycobac terium
Staphylo coccus
Bacillus 1858 1 1 108 13 13 65 98 3 240
Bordetella 2 2042 92 1 68 40 0 1 153 1
Burkholderia 3 141 2116 0 48 16 0 2 74 0
Clostridium 62 0 0 2215 3 14 17 25 0 64
Corynebacterium 8 186 17 1 1455 513 1 32 187 0
Geobacter 71 44 2 7 310 1698 6 189 66 7
Helicobacter 26 0 0 7 36 1 2279 30 0 21
Lactobacillus 186 1 0 87 63 39 62 1458 16 488
Mycobacterium 7 146 43 0 231 53 0 9 1911 0
Staphylococcus 179 0 0 100 3 1 43 115 0 1959
29 Lampiran 10 Tampilan sistem hasil implementasi
1 Tampilan awal sistem
30