i
VALIDASI METODE ANALISIS SPEKTROFOTOMETRI ULTRAVIOLET PADA PENETAPAN KADAR PIRANTEL PAMOAT
DALAM SEDIAAN SUSPENSI MERK X®
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm)
Program Studi Farmasi
Diajukan oleh : Agnes Mutiara Kurniawan
NIM : 098114131
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
iv
HALAMAN PERSEMBAHAN
Kami hanya ingin pendidikan yang layak bagi kaum kami. Bukan semata-mata untuk menjadi pesaing kaum pria. Namun demi kodrat kami sebagai ibu, yaitu
pendidik yang pertama.
-R.A.Kartini-
Karya ini kupersembahkan untuk:
Bapa Yesus Kristus yang telah memberikan kesempatan yang luar biasa dalam hidupku untuk merasakan apa yang disebut pendidikan.
Seluruh keluargaku (Papa,Mama, Kakak, dan Adikku) atas dukungan, doa, dan perhatiannya selama ini.
Almamaterku Fakultas Farmasi Universitas Sanata Dharma,
dan seluruh anak Indonesia yang belum bisa merasakan pendidikan, semoga semangat belajar mereka tetap berkobar meski keadaan yang
vii PRA KATA
Puji dan syukur penulis panjatkan kepada Tuhan Yesus Kristus untuk
anugerah dan penyertaanNya yang begitu besar kepada penulis, selama proses
penelitian dan penyusunan naskah ini.
Skripsi yang berjudul “Validasi Metode Analisis Spektrofotometri
Ultraviolet Pada Penetapan Kadar Pirantel Pamoat Dalam Sediaan Suspensi Merk
X®” ini disusun untuk memperoleh gelar Sarjana Farmasi (S.Farm) Program Studi Ilmu Farmasi Universitas Sanata Dharma.
Selama proses penelitian dan penyusunan naskah ini, tidak terlepas dari
dukungan banyak pihak yang telah memberikan dukungan, semangat, kritik dan
sarannya kepada penulis. Pada kesempatan kali ini, penulis mengucapkan terima
kasih yang sebesar-besarnya kepada:
1. Ipang Djunarko,M.Sc., Apt., selaku dekan Fakultas Farmasi
Universitas Sanata Dharma.
2. Prof. Dr. Sudibyo Martono, M.S., Apt., selaku dosen pembimbing
yang dengan penuh sabar memberikan masukan, nasehat, arahan,
kritik, saran, serta waktu dan tenaga untuk membimbing penulis
selama proses penelitian maupun penyusunan naskah ini.
3. Jeffry Julianus, M.Si., selaku dosen penguji yang telah memberikan
banyak masukan, kritik dan saran kepada penulis sehingga penulisan
viii
4. Lucia Wiwid Wijayanti, M.Si., selaku dosen penguji yang telah
memberikan banyak masukan, kritik dan saran kepada penulis
sehingga penulisan naskah ini menjadi lebih baik.
5. PT.Konimex, yang telah memberikan baku pirantel pamoat yang
sangat bermanfaat selama proses penelitian ini.
6. Semua dosen dan karyawan Fakultas Farmasi Univesitas Santa
Dharma
7. Mas Bimo dan Pak Parlan selaku Laboran Laboratorium Kimia
Analisis Instrumental dan Kimia Organik yang telah banyak
memberikan bantuan kepada penulis selama proses penelitian.
8. Mas Kethul yang telah memberikan kemudahan waktu kepada penulis
dan tim untuk menjalankan penelitian ini.
9. Seluruh keluargaku tercinta, Papa, Mama, Ko Deddy, dan Dicky untuk
segala doa, dukungan, dan perhatiannya selama ini kepada penulis
10.Novia Sarwoning Tyas dan Victor Purnama Agung FanggidaE, selaku
rekan sekelompok, yang selalu memberikan kebersamaan, semangat,
bantuan, dan dukungannya kepada penulis selama proses penelitian ini
berlangsung.
11.Natalia Windari dan Kaleb Franky Limawan yang telah banyak
memberikan masukan, kritik, saran, serta waktu untuk berdiskusi
bersama dengan penulis dan tim, sehingga proses penelitian ini boleh
ix
12.David Chandra Putra, yang telah banyak memberikan dampak positif
kepada penulis. Kebersamaan, kesabaran, dan pengertiannya kepada
penulis, telah banyak membantu penulis selama proses penelitian ini
berlangsung.
13.Phebe Hendra, Ph.D., Apt., atas nasehat, dukungan, dan motivasi
kepada penulis, selama proses penelitian maupun studi S1, sehingga
penulis dapat menyelesaikan tugas akhir ini tepat waktu.
14.Kedua sahabatku tercinta Giesta Artatia dan Auxilliadora P.H.G. yang
telah memberikan banyak perhatian, kasih persaudaraan, serta
kebersamaan kepada penulis untuk bersama-sama meraih cita-cita
masing-masing.
15.Wuri Kinanti, yang dengan setia mendengarkan keluh kesah, serta
memberikan motivasi yang kuat kepada penulis untuk terus berusaha
melakukan yang terbaik.
16.Teman-teman “Konco Dolan” (Sasya, Metri, Shinta, Laras, Eric, Is,
Novia, Anta, dan Nindy) yang telah banyak memberikan kebahagiaan
dan kenangan indah selama proses pembelajaran di S1.
17.Teman-teman seperjuangan lantai 4, Mas Dika, Sasya, Metri, Shinta,
Leo, Ina, Topan, Agus Teti, Jimmy, Rachel, Gunggek, Febrin, Wisnu,
Joe, Netty, Saka, Jati, Felix.Kebersamaan, kebahagian, dan
dukungannya selama ini telah memberikan semangat kepada penulis
selama proses penelitian, sehingga penelitian ini menjadi lebih
x
18. Rekan-rekan KKMG, Ebed Obed, Kak Megya, Ko Henry, Ko Aan,
Ko Lingga, Ko Vino, Ko Sugeng, Fajar, Kak Listo, dan Kak Kitty, atas
pengertian dan doa selama ini kepada penulis, sehingga penelitian ini
boleh berjalan tepat waktu.
19.Teman-teman Kost Dewi 2, Sheilla, Nindy, Adel, Lani, Maria, Silvi,
atas persahabatan dan persaudaraan bersama penulis selama 4 tahun
ini.
20.Semua pihak yang membantu penulis selama proses penelitian dan
penyusunan naskah ini, baik secara langsung maupun tidak, yang tidak
dapat penulis sebutkan satu per satu.
Akhir kata, penulis berharap semoga skripsi ini dapat bermanfaat bagi yang
membacanya. Tuhan memberkati.
Yogyakarta, Juni 2013
xi DAFTAR ISI
Halaman
HALAMAN JUDUL ………... i
HALAMAN PERSETUJUAN PEMBIMBING ...………. ii
HALAMAN PENGESAHAN ...………... iii
HALAMAN PERSEMBAHAN ...……….... iv
PERNYATAAN KEASLIAN KARYA ……… v
PERNYATAAN PERSETUJUAN PUBLIKASI ………. vi
PRAKATA ...……… vii
DAFTAR ISI ...……….. xi
DAFTAR TABEL ...………..…... xv
DAFTAR GAMBAR ...………... xvi
DAFTAR LAMPIRAN ……….... xviii
INTISARI ……….... xx
ABSTRACT ...………...……… xxi
BAB I PENGANTAR ……… 1
A. Latar Belakang ………... 1
xii
2. Keaslian penelitian ………... 4
3. Manfaat penelitian ……… 5
B. Tujuan Penelitian ………... 5
BAB II PENELAHAN PUSTAKA ………... 6
A. Pirantel Pamoat ……….. 6
B. Ekstraksi ………. 7
C. Spektrofotometri UV ...……….………….. 9
1. Instrumentasi ...……….……….. 10
2. Interaksi elektron dengan REM ………. 12
3. Hukum Lambert-Beer ………...………. 15
4. Analisis Kuantitatif ……… 16
D. Validasi Metode Analisis ………. 18
1. Presisi ………. 20
2. Akurasi ………... 21
3. Linieritas ……… 23
4. Kisaran (range) ……….. 23
E. Landasan Teori ………. 24
F. Hipotesis ………... 26
BAB III METODE PENELITIAN …….………... 27
A. Jenis dan Rancangan Penelitian ………... 27
B. Variabel Penelitian ...………....… 27
xiii
2. Variabel Tergantung ………...……… 27
3. Variabel Pengacau Terkendali ...…..……….………. 27
C. Definisi Operasional ...……….………. 28
D. Bahan Penelitian ...……….………... 28
E. Alat Penelitian ...……….……….. 29
F. Tata Cara Penelitian ..……….………….. 29
1. Pembuatan larutan stok baku pirantel pamoat ……….…………...29
2. Penentuan panjang gelombang pengamatan ……….…. 30
3. Pembuatan larutan seri baku dan kurva baku pirantel pamoat ...….... 30
4. Penentuan rentang linieritas baku pirantel pamoat …...………. 31
5. Penentuan akurasi dan presisi baku pirantel pamoat ...………... 31
6. Penentuan akurasi dan presisi baku dalam matrik sampel dengan metode standard adisi ..………..………. 32
a. Pembuatan larutan baku adisi pirantel pamoat ………. 32
b. Pembuatan larutan sampel tanpa adisi baku pirantel pamoat (blank sample) ………. 32
c. Pembuatan larutan sampel dengan penambahan baku pirantel pamoat (addition sample) ……… 33
d. Ekstraksi larutan blank sample dan addition sample dengan metode ekstraksi cair-cair menggunakan ultrasonikator ………... 33
e. Penetapan akurasi presisi baku pirantel pamoat yang diadisi dalam matrik sampel ………... 34
xiv
a. Selektivitas ………... 35
b. Linieritas ……….. 35
c. Presisi ………..………. 36
d. Akurasi ………...…………..……….... 36
BAB IV HASIL DAN PEMBAHASAN ………. 37
A. Pembuatan larutan baku pirantel pamoat ………...………..……… 37
B. Penentuan panjang gelombang pengamatan ……….……... 38
C. Pembuatan kurva baku pirantel pamoat ………..….… 41
D. Validasi metode ……….…... 44
1. Selektivitas (Spesifisitas) ………... 45
2. Linieritas ……… 47
3. Akurasi ………... 49
4. Presisi ………. 54
5. Rentang ……….. 55
BAB V KESIMPULAN DAN SARAN………... 59
DAFTAR PUSTAKA ……….. 60
LAMPIRAN ………. 63
xv
DAFTAR TABEL
Halaman
Tabel 1. Tipe validasi untuk prosedur analisis ………..…. 19
Tabel 2. Kriteria penerimaan nilai RSD ………. 21
Tabel 3. Kriteria penerimaan nilai % recovery ………... 22
Tabel 4. Data replikasi kurva baku pirantel pamoat ………...… 42
Tabel 5. Presisi kurva baku pirantel pamoat ……….. 44
Tabel 6. Data % recovery larutan baku pirantel pamoat ……… 50
Tabel 7. Data % recovery larutan baku pirantel pamoat (metode standar adisi) ……….……… 52
Tabel 8. Nilai CV baku pirantel pamoat tanpa adisi ……….. 55
Tabel 9. Nilai CV baku pirantel pamoat dalam matrik sampel ………….. 55
xvi
DAFTAR GAMBAR
Halaman
Gambar 1. Struktur pirantel pamoat ………... 6
Gambar 2. Instrumentasi spektrofotometri UVsingle beam ……….. 11
Gambar 3. Instrumentasi spektrofotometri UV double beam ……… 12
Gambar 4. Diagram tingkat energi elektronik ………... 13
Gambar 5. Absorpsi cahaya oleh analit ………. 15
Gambar 6. Diagram parameter validasimetodemenurut ICH ……….. 18
Gambar 7. Kromofor dan auksokrom pirantel pamoat ……….……. 38
Gambar 8. Pola spektra baku pirantel pamoat konsentrasi 10, 20, dan 30 ppm dalam pelarut DMSO-metanol .………...… 40
Gambar 9. Pola spektra larutan blangko (DMSO-metanol) .……….. 41
Gambar 10. Grafik kurva baku pirantel pamoat …………..…...…..………... 43
xvii
Gambar 12. Pola spektra larutan baku pirantel pamoat 20 ppm dalam pelarut
DMSO-metanol (a); Panjang gelombang maksimal larutan baku
pirantel pamoat (b) ……….……….. 46
Gambar 13. Linieritas kurva baku pirantel pamoat replikasi I, II, dan III …... 48
Gambar 14. Rentang linieritas kurva baku pirantel pamoat replikasi I, II, dan III
xviii
DAFTAR LAMPIRAN
Halaman
Lampiran 1. Sertifikat analisis pirantel pamoat ……….…... 64
Lampiran 2. Penentuan panjang gelombang maksimum ……….………. 65
Lampiran 3. Scanning panjang gelombang maksimum konsentrasi 10, 20,
dan 30 ppm ………... 66
Lampiran 4. Pembuatan kurva baku pirantel pamoat ………... 66
Lampiran 5. Kurva baku pirantel pamoat ……….… 68
Lampiran 6. Perhitungan Coefficient of Variation (CV) replikasi kurva baku
pirantel pamoat ……….… 69
Lampiran 7. Penimbangan bahan dan seri konsentrasi baku pirantel pamoat
untuk penentuan akurasi dan presisi ……….... 69
Lampiran 8. Akurasi dan presisi baku pirantel pamoat 10 ppm (konsentrasi
rendah) ………..… 70
Lampiran 9. Akurasi dan presisi baku pirantel pamoat 20 ppm (konsentrasi
tengah) ………..… 71
Lampiran 10. Akurasi dan presisi baku pirantel pamoat 30 ppm (konsentrasi
tinggi) ……….….. 71
xix
Lampiran 12. Perhitungan pencuplikan sampel ………... 73 Lampiran 13. Penimbangan bahan dan seri konsentrasi baku pirantel pamoat
untuk penentuan akurasi dan presisi dalam matrik sampel …….. 74
Lampiran 14. Akurasi dan presisi larutan baku adisi pirantel pamoat 5 ppm
(konsentrasi rendah) ………..…... 77
Lampiran 15. Akurasi dan presisi larutan baku adisi pirantel pamoat 10 ppm
(konsentrasi tengah) ………..…... 79
Lampiran 16. Akurasi dan presisi larutan baku adisi pirantel pamoat 15 ppm
(konsentrasi tinggi) ………...…… 81
Lampiran 17. Kurva adisi baku pirantel pamoat dalam matrik sampel ………... 83
Lampiran 18. Rentang linieritas baku pirantel pamoat ……….... 84
xx INTISARI
Pirantel pamoat merupakan senyawa dengan aktivitas farmakologi sebagai antelmintik, yang salah satunya diformulasikan sebagai sediaan suspensi oral. Aktivitas farmakologi pirantel pamoat, tergantung pada ketepatan dosis terapi. Oleh karena itu, diperlukan proses penjaminan mutu sediaan dengan metode yang telah tervalidasi, untuk menjamin bahwa metode tersebut memenuhi persyaratan aplikasi analitik.
Penelitian ini mengikuti jenis dan rancangan penelitian deskriptif non eksperimental. Metode analisis yang divalidasi adalah metode spektrofotometri UV dengan panjang gelombang pengamatan 301 nm. Parameter validasi yang digunakan meliputi: selektivitas, linieritas, akurasi, presisi, dan rentang.
Hasil penelitian menunjukkan nilai koefisien korelasi untuk linieritas adalah 0,9998 pada konsentrasi 10-30 ppm. Rentang nilai recovery adalah 98,09-100,54%; 99,88-100,37%; dan 98,35-100,88% dengan nilai CV pada konsentrasi tersebut adalah 1,30%; 0,25%; dan 1,27% untuk konsentrasi 10, 20, dan 30 ppm. Rentang nilai recovery standard addition method adalah 98,49-99,49%; 100,49-101,24%; dan 100,49-101,82% dengan nilai CV adalah 0,51%; 0,51%; dan 0,69% pada penambahan baku pirantel pamoat 5, 10, dan 15 ppm. Selektivitas metode ditunjukkan dengan tidak adanya absorbansi pelarut pada panjang gelombang pengamatan yang digunakan untuk pengukuran pirantel pamoat dan pola spektra yang sama antara sampel dengan baku pirantel pamoat. Hasil tersebut menunjukkan bahwa metode yang digunakan memenuhi parameter validasi yang baik.
xxi
ABSTRACT
Pyrantel pamoate is a compound with farmacology activity as anthelmintic, which formulated as oral suspension dosage form. Farmacology activity of pyrantel pamoate is depent on the precise of therapy dose. Therefore, the quality control for every single product is required to do with a validated analysis method, to ensure that the analysis method is complied to the requirement of analitic application.
The type and design of this research is non experimental descriptive. The aimof this study was to validated the spectrophotometric UV method with meansurement wavelength 301 nm.The validation parameters are selectivity, linierity, accuracy, presision, and range.
The result of this research show that the coefficient corelation for linierity is 0,9998 at concentration 10-30 ppm. The recovery range are 98,09-100,54%; 99,88-100,37%; and 98,35-100,88% at concentration 10, 20, and 30 ppm. The CV of that recovery are 1,30%; 0,25%; and 1,27%. The recovery range of standard addition method are 98,49-99,49%; 100,49-101,24%; and 100,49-101,82% at concentration 5, 10, and 15 ppm of pyrantel pamoate added. The CV of that recovery are 0,51%; 0,51%; and 0,69%. The selectivity of this method shown by the difference of spectra between pyrantel pamoate reference standard and solvent. The spectra of solvent didn’t show an absorbance at measurement wavelength of pyrantel pamoate. This result showed that the method is complied a good validation parameters.
1 BAB I
PENGANTAR
A. Latar Belakang
Penyakit cacingan merupakan salah satu masalah kesehatan anak di
Indonesia. Sanitasi yang buruk dan pola hidup yang kurang bersih merupakan dua
faktor penyebab utama tingginya prevalensi penyakit ini.
Cacing merupakan salah satu mikroorganisme yang hidup sebagai parasit
dalam tubuh manusia. Proses infeksi cacing dan penularannya pada manusia yang
terjadi dengan mudah, menyebabkan penyakit ini berkembang dengan pesat.
Manifestasi infeksi cacing pada manusia memberikan dampak pada penurunan
kondisi kesehatan orang yang cukup signifikan. Infeksi cacing dapat
menyebabkan penurunan penyerapan zat gizi makanan serta kekurangan darah
atau yang sering disebut dengan anemia, sehingga menurunkan produktivitas kerja
maupun konsentrasi belajar pada anak-anak.
Salah satu obat yang banyak dipasarkan di Indonesia untuk mengobati
penyakit cacingan adalah pirantel pamoat. Berdasarkan kelarutannya, pirantel
pamoat merupakan senyawa yang praktis tidak larut dalam air. Oleh karena itu,
produk pirantel pamoat yang banyak beredar di pasaran diformulasikan dalam
bentuk sediaan tablet dan suspensi. Pada penelitian ini, bentuk sediaan yang
digunakan adalah sediaan suspensi. Hal ini terkait dengan prevalensi penderita
cacingan yang banyak dialami oleh pasien anak-anak. Berdasarkan hasil survey,
konsumen anak-anak, bentuk sediaan suspensi lebih diminati dibandingkan
dengan penggunaan tablet. Hal ini dikarenakan penggunaannya yang lebih mudah
dan kenyamanan penggunaan sediaan suspensi dibandingan sediaan tablet,
khususnya pada pasien anak-anak. Oleh karena itu, sediaan suspensi merupakan
bentuk sediaan yang lebih sering digunakan dibandingkan dengan bentuk sediaan
tablet.
Sediaan suspensi memiliki beberapa kelemahan, khususnya stabilitas dan
homogenitas analit dalam matrik sampel yang rendah. Hal ini dapat menyebabkan
terjadinya penurunan kadar pirantel pamoat pada sediaan suspensi selama proses
distribusi dan penyimpanan.
Aktivitas farmakologis dan efektivitas terapi dapat tercapai ketika dosis
obat yang digunakan tepat. Oleh karena itu, dibutuhkan suatu usaha untuk
menjamin mutu atau kualitas kandungan zat aktif dalam suatu sediaan obat, yang
salah satunya adalah penjaminan kesesuaian dosis sediaan obat terhadap label
klaim pada kemasan. Tujuan penjaminan mutu ini adalah untuk melindungi
konsumen agar tetap mendapatkan obat dengan kualitas zat aktif yang tepat.
Dalam usaha penjaminan mutu suatu sediaan obat, dibutuhkan metode
yang tervalidasi dan memenuhi parameter validitas yang meliputi akurasi, presisi,
selektivitas, linearitas, dan rentang. Validasi metode merupakan suatu usaha
penilaian terhadap karakteristik kinerja suatu metode analisis yang dilakukan
berdasarkan hasil percobaan untuk membuktikan bahwa metode yang digunakan
dimaksudkan. Tujuan validasi metode ini adalah untuk membuktikan dan
memberikan jaminan kebenaran hasil yang didapatkan.
Pada penelitan sebelumnya, pernah dilakukan analisis pirantel pamoat
pada penetapan kadar pirantel pamoat dalam sediaan tablet secara
spektrofotometri ultraviolet (Agustina, 2010). Hal mendasar yang membedakan
penelitian ini dengan penelitian sebelumnya adalah bentuk sediaan pirantel
pamoat yang dianalisis, metode ekstraksi sampel, serta pelarut yang digunakan.
Berdasarkan studi pustaka yang dilakukan oleh peneliti, proses validasi
metode spektrofotometri ultraviolet pada penetapan kadar pirantel pamoat dalam
bentuk sediaan suspensi merk “X®” dengan metode ekstraksi cair-cair, belum pernah dilakukan. Penggunaan metode spektrofometri ultraviolet pada penetapan
kadar pirantel pamoat ini dipilih berdasarkan pada kemampuan dan sensitivitas
metode tersebut untuk mendeteksi senyawa uji yang terkandung dalam sediaan
suspensi. Spektrofotometri ultraviolet merupakan salah satu metode analisis yang
mudah diaplikasikan pada metode penetapan kadar suatu sediaan dengan zat aktif
tunggal.
Pada penelitian ini dilakukan proses validasi metode spektrofometri
ultraviolet dari hasil optimasi yang termasuk dalam satu kesatuan rangkaian
penelitian bersama penetapan kadar pirantel pamoat dalam sediaan suspensi merk
“X®”, yaitu: optimasi, validasi metode, dan penetapan kadar.
Metode spektrofotometri ultraviolet yang akan digunakan oleh peneliti,
linearitas, selektivitas, dan rentang sehingga dapat digunakan untuk menetapkan
kadar pirantel pamoat dalam bentuk sediaan suspensi merk “X®”.
1. Permasalahan
Berdasarkan latar belakang yang telah diuraikan diatas, maka
permasalahan yang muncul adalah: Apakah metode penetapan kadar pirantel
pamoat dalam sediaan suspensi merk “X®” secara spektrofotometri ultraviolet dengan sistem yang telah dioptimasi sebelumnya memenuhi persyaratan validitas
yang meliputi: akurasi, presisi, linearitas, selektivitas, dan rentang ?
2. Keaslian Penelitian
Beberapa penelitan mengenai analisis pirantel pamoat yang pernah
dilakukan sebelumnya antara lain adalah Spectrophotometric Determination of
Pyrantel Pamoate Bulk Samples and Pharmaceutical Formulations (Forcier,
Mushinsky, and Wagner, 1971), High-Performance Liquid Chromatographic
Determination of Oxantel and Pyrantel Pamoate (Allender, 1988), Penetapan
Kadar Pirantel Pamoat Dalam Sediaan Tablet Secara Spektrofotometri Ultraviloet
(Agustina, 2010), Development And Validation of A RP- HPLC Method For The
Quantitation Studies of Praziquantel And Pyrantel Pamoate (Oltean, 2011).
Berdasarkan studi pustaka yang telah dilakukan oleh peneliti, belum
pernah dilakukan penelitian tentang validasi metode spektrofotometri ultraviolet
3. Manfaat Penelitian
a. Manfaat metodologis. Penelitian ini diharapkan dapat memberikan
alternatif metode analisis pirantel pamoat untuk menetapkan kadar pirantel
pamoat dalam sediaan suspensi merk “X®” yang memiliki sistem optimal dan
memenuhi persyaratan validitas yang baik.
b. Manfaat teoritis. Penelitian ini diharapkan dapat memberikan
tambahan informasi ilmiah mengenai validasi metode penetapan kadar pirantel
pamoat dalam sediaan suspensi merk “X®” secara spektrofotometri ultraviolet.
c. Manfaat praktis. Penelitian ini diharapkan dapat digunakan untuk
menetapkan kadar pirantel pamoat dalam sediaan suspensi merk “X®” yang banyak beredar di pasaran.
B. Tujuan Penelitian
Tujuan penelitian ini adalah untuk mengetahui validitas metode penetapan
6 BAB II
PENELAAHAN PUSTAKA
A. Pirantel Pamoat
Gambar 1. Struktur Pirantel Pamoat
Pirantel pamoat (C11H14N2S.C23H16O6) (Gambar 1) memiliki berat
molekul 594,68 g/mol. Pirantel pamoat mengandung tidak kurang dari 97,0% dan
tidak lebih dari 103,0% C34H30N2O6S, dihitung terhadap zat yang telah
dikeringkan. Pemerian pirantel pamoat berupa padatan kuning hingga coklat.
Pirantel pamoat praktis tidak larut dalam air dan dalam metanol; larut dalam
dimetil sulfoksida; dan sukar larut dalam dimetil formamida (Direktorat Jenderal
Pengawas Obat dan Makanan, 1995).
Suspensi oral pirantel pamoat adalah suspensi pirantel pamoat dalam
medium pembawa akuades yang sesuai. Setiap sedian mengandung tidak kurang
90,0% dan tidak lebih dari 110,0% pirantel (C11H14N2S) dari label klaim. Sediaan
suspensi oral pirantel pamoat merupakan sediaan dengan kemasan wadah dosis
tunggal. pH sediaan berkisar antara 4,0-6,5 (Anonima, 2013).
Pirantel memiliki absorbansi pada panjang gelombang 315 nm dalam
pelarut metanol asam dengan nilai % = 920a (Moffat, 2004). Pirantel pamoat
gelombang 300 nm dalam pelarut metanol dengan nilai % = 366 dan nilai ɛ =
21770 M-1.cm-1, dan pada panjang gelombang 288 nm dengan nilai % = 370
dan nilai ɛ = 22000 M-1.cm-1. Pirantel pamoat juga memiliki 2 absorbansi
maksimum dalam pelarut NaOH 0,1 N, yaitu: pada panjang gelombang 301 nm
dengan nilai % = 382 dan nilai ɛ = 22720 M-1.cm-1 dan pada panjang
gelombang 290 nm dengan nilai % = 383 dan nilai ɛ = 22780 M-1.cm-1
(Dibbern, 2002).
B. Ekstraksi
Ekstraksi cair-cair digunakan sebagai cara untuk pra perlakuan sampel
guna memisahkan analit dari komponen-komponen matriks yang mungkin
mengganggu pada saat kuantifikasi atau deteksi analit. Kebanyakan prosedur
ekstraksi cair-cair melibatkan ekstraksi analit dari fase air ke dalam pelarut
organik yang bersifat non polar seperti, heksan, metilbenzen, atau diklorometan.
Meskipun demikian, proses sebaliknya (ekstraksi analit dari pelarut organik non
polar ke dalam air) juga mungkin terjadi (Gandjar dan Rohman, 2007).
Faktor-faktor yang mempengaruhi laju ekstraksi adalah:
1. Tipe persiapan sampel
2. Waktu ekstraksi
3. Kuantitas pelarut
4. Suhu pelarut
5. Tipe pelarut (Direktorat Jenderal Pengawasan Obat dan Makanan POM
Ekstraksi cair-cair ditentukan oleh distribusi Nerst atau hukum partisi yang
menyatakan bahwa “pada konsentrasi dan tekanan yang konstan, analit akan
terdistribusi dalam proporsi yang selalu sama diantara dua pelarut yang saling
tidak campur”. Perbandingan konsentrasi pada keadaan setimbang di dalam 2 fase
disebut dengan koefisien distribusi atau koefisien partisi (KD) dan diekspresikan
dalam rumus berikut:
KD = [ ][ ] ……… (01)
[S]org dan [S]aq masing-masing merupakan konsentrasi analit dalam fase organik dan dalam fase
air.
Analit yang mempunyai rasio distribusi besar (104 atau lebih) akan
mudah terekstraksi ke dalam pelarut organik meskipun proses kesetimbangan
(yang berarti 100% solut terekstraksi atau tertahan) tidak pernah terjadi.
Efisiensi proses ekstraksi tergantung pada nilai distribusinya (D) dan juga
tergantung pada volume relatif kedua fase. Dengan menggunakan ekstraksi,
banyaknya analit yang terekstraksi dapat dihitung dengan rumus berikut:
E =
[ ] ………. (02)
Vorg dan Vaq masing-masing merupakan banyaknya volume fase organik
dan fase air yang digunakan; D merupakan rasio distribusi. Pada analit dengan
nilai D yang kecil, adanya ekstraksi berulang (bertingkat) akan meningkatkan
(Caq)n = Caq [( )] ……… (03)
(Gandjar dan Rohman, 2007)
C. Spektrofotometri Ultraviolet
Spektrofotometri absorbansi merupakan suatu pengukuran terhadap
interaksi antara radiasi elektromagnetik dan molekul atau atom dari suatu zat
kimia. Teknik yang sering digunakan dalam analisis farmasi antara lain;
spektrofotometri ultraviolet, cahaya tampak atau visible, infra merah, dan
absorbansi atom. Jangkauan panjang gelombang daerah ultraviolet (UV) berkisar
antara 190-380 nm, untuk daerah cahaya tampak atau visible berkisar antara
380-780 nm, daerah inframerah (IR) dekat antara 380-780-3000 nm, dan daerah inframerah
berkisar antara 2,5-40 μm atau 4000-250 cm-1 (Direktorat Jenderal Pengawasan
Obat dan Makanan, 1995).
Spektorfotometri UV-Vis adalah anggota teknik analisis spektroskopik
yang memakai sumber radiasi elektromagnetik ultraviolet dekat (190-380 nm) dan
sinar tampak (380-780 nm) dengan memakai instrumen spektrofotometer.
Spektrofotometri UV-Vis melibatkan energi elektronik yang cukup besar pada
molekul yang dianalisis, sehingga spektrofotometri UV-Vis lebih banyak
digunakan untuk analisis kuantitatif dibandingkan kualitatif (Mulja dan
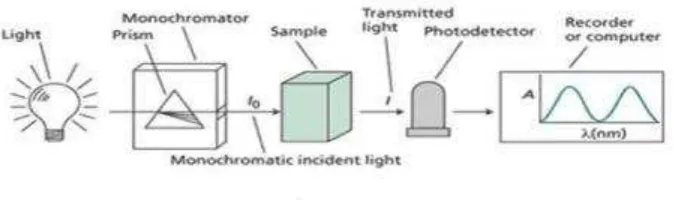
1. Instrumentasi Spektrofotometer Ultraviolet
Spektrofotometer yang sesuai untuk pengukuran di daerah spektrum
ultraviolet dan sinar tampak terdiri atas suatu sistem optik dengan kemampuan
menghasilkan sinar monokromatis dalam jangkauan panajang gelombang 200-800
nm. Komponen-komponennya meliputi sumber-sumber sinar, monokromator, dan
sistem optik.
i. Sumber lampu; lampu deuterium digunakan untuk daerah UV pada panjang gelombang dari 190-350 nm, sementara lampu halogen kuarsa atau
lampu tungsten digunakan untuk daerah visibel (pada daerah panjang gelombang
antara 350-900 nm).
ii. Monokromator; digunakan untuk mendispersikan sinar ke dalam komponen-komponen panjang gelombangnya yang selanjutnya dipilih oleh celah
(slit). Monokromator berputar sedemikian rupa sehingga kisaran panjang
gelombang dilewatkan pada sampel sebagai scan instrumen melewati spektrum.
iii. Optik-optik, dapat dirancang untuk memecah sumber sinar sehingga sumber sinar melewati 2 kompartemen, dan sebagaimana dalam
spektrofotometer berkas ganda (double beam), suatu larutan blangko dapat
digunakan dalam satu kompartemen untuk mengkoreksi pembacaan atau spektrum
sampel. Larutan yang paling sering digunakan sebagai blangko dalam
spektrofotometri adalah semua pelarut yang digunakan untuk melarutkan sampel
Gambar 2. Instrumentasi spektrofotometri UVsingle beam
Spektrofotometer dibagi menjadi dua jenis, yaitu spektrofotometer single
beam dan spektrofotometer double beam. Perbedaan kedua jenis spektrofotometer
tersebut terdapat pada pemberian cahaya. Pada skema spektrofotometer single
beam (Gambar 3), cahaya hanya melewati satu arah dan yang diperoleh hanya
nilai absorbansi dari larutan yang dimasukan. Berbeda dengan spektrofotometer
single beam, nilai blangkopada spektrofotometer double beam dapat langsung
diukur bersamaan dengan nilai absorbansi larutan yang diinginkan dalam satu kali
proses yang sama, sehingga nilai absorbansi larutan yang diukur telah mengalami
pengurangan nilai terhadap nilai absorbansi blangko.Prinsipnya adalah dengan
adanya chopper yang akan membagi sinar menjadi dua, dimana salah satunya
melewati blangko (reference beam) dan yang lainnya melewati larutan (sample
beam). Selain itu, pada skema spektrofotometer double beam (Gambar 4) juga
dapat mengatasi kelemahan pada spektrofotometer single beam seperti adanya
Gambar 3
2. Interaksi elektron dengan REM
Secara umum ada tiga macam distribusi elektron di dalam suatu senyawa
organik yang selanjutnya dikenal sebagai orbital elektron pi (π), sigma (σ), dan
elektron tidak berpasangan (n). Apabila pada molekul tersebut dikenakan rad
elektromagnetik maka akan terjadi eksitasi elektron ke tingkat energi yang lebih
tinggi yang dikenal sebagai orbital elektron “
1995).
Ada empat jenis transisi elektronik yang terjadi diantara tingkat
energi di dalam suatu molekul, yaitu:
a. Transisi sigma
b. Transisi
n-c. Transisi
n-d. Transisi
n-Gambar 3. Instrumentasi spektrofotometri UV double beam
Interaksi elektron dengan REM
Secara umum ada tiga macam distribusi elektron di dalam suatu senyawa
organik yang selanjutnya dikenal sebagai orbital elektron pi (π), sigma (σ), dan
ektron tidak berpasangan (n). Apabila pada molekul tersebut dikenakan rad
elektromagnetik maka akan terjadi eksitasi elektron ke tingkat energi yang lebih
tinggi yang dikenal sebagai orbital elektron “anti bonding” (Mulja dan Suharman,
Ada empat jenis transisi elektronik yang terjadi diantara tingkat
i dalam suatu molekul, yaitu:
Transisi sigma-sigma star (σ σ*) -sigma star (n σ*)
-phi star (n π*)
-phi star (π π*) (Gandjar dan Rohman, 2007)
double beam
Secara umum ada tiga macam distribusi elektron di dalam suatu senyawa
organik yang selanjutnya dikenal sebagai orbital elektron pi (π), sigma (σ), dan
ektron tidak berpasangan (n). Apabila pada molekul tersebut dikenakan radiasi
elektromagnetik maka akan terjadi eksitasi elektron ke tingkat energi yang lebih
(Mulja dan Suharman,
Ada empat jenis transisi elektronik yang terjadi diantara tingkat-tingkat
Gambar 4. Diagram tingkat energi elektronik
a. Transisi elektron sigma sigma star (σ σ*)
Eksitasi elektron (σ σ*) memberikan energi yang terbesar dan terjadi pada daerah ultraviolet jauh yang diberikan oleh ikatan tunggal, contohnya
pada senyawa alkana (Mulja dan Suharman, 1995).
Energi yang diperlukan untuk transisi ini besarnya sesuai dengan energi
sinar yang frekuensinya terletak di antara UV vakum (kurang dari 180 nm).
Jenis transisi ini (σ σ*) terjadi pada daerah ultraviolet vakum sehingga kurang bermanfaat untuk analisis dengan cara spektrofotometri UV-Vis
(Gandjar dan Rohman, 2007).
b. Transisi elektron non bonding sigma star (n σ*)
Jenis transisi ini terjadi pada senyawa organik jenuh yang mengandung
atom-atom yang memiliki elektron non bonding (n). Energi yan diperlukan
untuk transisi jenis ini lebih kecil dibanding transisi (σ σ*), sehingga sinar
yang diserap mempunyai panjang gelombang lebih panjang, yaitu 150-250
nm. Transisi ini banyak terjadi pada panjang gelombang kurang dari 200 nm.
Transisi jenis ini dapat terjadi pada gugus karbonil (dimetil keton dan
asetaldehid) yang terjadi pada daerah UV jauh. Gugus karbonil dapat
memberikan eksitasi elektron (n σ*) yang terjadi pada panjang gelombang
280-290 nm, tetapi eksitasi ini dihindari karena memberikan nilai εmaks =
12-16 (< 1000 M-1.cm-1) (Mulja dan Suharman, 1995).
c. Transisi elektron phi phi star (π π *)
Eksitasi elektron (π π *) terjadi pada ikatan rangkap dua dan rangkap
tiga (alkena dan alkuna) yang terjadi pada daerah ultraviolet jauh. Transisi ini
terjadi pada molekul organik yang mempunyai gugus fungsional yang tidak
jenuh, sehingga ikatan rangkap dalam gugus tersebut memberikan orbital phi
yang dibutuhkan transisi ini. Jenis transisi ini merupakan transisi yang paling
cocok untuk analisis. Hal ini karena transisi ini terjadi pada panjang
gelombang 200-700 nm, yang secara teknis dapat diaplikasikan pada
spektrofotometer (Gandjar dan Rohman, 2007).
d. Transisi elektron non bonding phi star (n π*)
Pada senyawa organik dikenal adanya gugus auksokrom, yaitu gugus
fungsional yang mempunyai elektron bebas seperti: -OH, O- NH2, dan -OCH3
yang dapat memberikan transisi (n σ*). Terikatnya gugus auksokrom pada
gugus kromofor akan mengakibatkan pergeseran pita absorbansi menuju
panjang gelombang yang lebih panjang (pergeseran batokromik) serta disertai
pada dua ikatan rangkap yang terkonjugasi (-C=C-C=C-) (Mulja dan
Suharman, 1995).
3. Hukum Lambert-Beer
Pengukuran absorpsi cahaya oleh molekul analit dalam larutan, diatur oleh
Hukum Lambert-Beer yang dirumuskan dengan persamaan berikut:
log Io/It = A = ε.b.c ………. (04)
Io : intensitas radiasi yang masuk
It : intensitas radiasi yang ditransmisikan
A : absorbansi
ε : konstanta koefisien molar ekstingsi
b : ketebalan kuvet yang dinyatakan dalam cm
c : konsentrasi analit (mol.L-1)
Absorbansi adalah jumlah cahaya yang diabsorpsi oleh larutan sampel
yang diukur. Konstante koefisien molar ekstingsi merupakan absorbansi analit
dalam larutan dengan konsentrasi 1Molar (M).
Gambar 5. Absorpsi cahaya oleh analit
Pada produk farmasi, konsentrasi atau jumlah sering dinyatakan dalam
produk farmasi Hukum Lambert-Beer sering dituliskan dengan persamaan
berikut:
ε = A (1%,1cm). b . c ……….. (05)
% : absorbansi sampel dengan konsentrasi 1% (b/v) dengan ketebalan kuvet 1 cm
b : ketebalan kuvet yang dinyatakan dalam cm
c : konsentrasi sampel yang dinyatakan dalam g/100mL
Monografi dalam British Pharmacope (BP) selalu menulis % untuk
baku pembanding atau standar yang dapat digunakan untuk uji kuantifikasi
(Watson, 2003).
4. Analisis Kuantitatif
Analisis kuantitatif dengan metode spektrofotometri UV dapat
digolongkan atas tiga macam pelaksanaan pekerjaan, yaitu:
a. Analisis kuantitatif zat tunggal (analisis satu komponen).
b. Analisis kuantitatif campuran dua macam zat (analisis dua
komponen).
c. Analisis kuantitatif campuran tiga macam zat atau lebih (analisis multi
komponen).
Analisis kuantitatif zat tunggal dilakukan pengukuran nilai absorbansi
pada panjang gelombang maksimum atau dilakukan pengukuran % transmitan
pada panjang gelombang minimum. Alasan dilakukan pengukuran pada panjang
gelombang tersebut adalah: perubahan absorbansi untuk setiap satuan konsentrasi
adalah paling besar pada panjang gelombang maksimal, sehingga akan diperoleh
panjang gelombang maksimal datar dan pengukuran ulang dengan kesalahan yang
kecil, dengan demikian akan memenuhi hukum Lambert-Beer. Ada empat cara
pelaksanaan analisis kuantitatif zat tunggal, yaitu:
1. Pertama, dengan cara membandingkan absorbansi atau persen transmitan
zat yang dianalisis dengan reference standard pada panjang gelombang
maksimal. Persyaratan pada cara kuantifikasi ini adalah pembacaan nilai
absorbansi sampel dan reference standard tidak jauh berbeda.
2. Kedua, dengan memakai kurva baku dari larutan reference standard
dengan pelarut tertentu pada panjang gelombang maksimum. Dibuat grafik
sistem koordinat Cartesian dimana sebagai ordinat adalah absorbansi dan
sebagai absis adalah konsentrasi.
3. Ketiga, adalah dengan jalan menghitung nilai absorbansi larutan sampel
( % λmaks) pada pelarut tertentu dan dibandingkan dengan absorbansi zat
yang dianalisis yang tertera pada literatur.
4. Keempat, dengan menggunakan perhitungan nilai ekstingsi molar
(absoptivitas molar ε) sama dengan cara yang ketiga hanya saja
perhitungan absorbansi molar lebih tepat karena melibatkan massa
molekul relatif (MR).
ε = % . M
R . 10-1(M-1.cm-1) ……….……… (06)
D. Validasi Metode Analisis
Validasi metode analisis adalah suatu tindakan penilaian terhadap
parameter-parameter tertentu berdasarkan percobaan di laboratorium, untuk
membuktikan bahwa parameter tersebut memenuhi persyaratan untuk penggunaan
(Harmita, 2004). Validasi merupakan suatu persyaratan dasar untuk menjamin
kualitas dan kehandalan hasil dari semua aplikasi metode analisis (Ermer and
Miller, 2005).
Proses validasi dimulai dengan perangkat lunak yang tervalidasi dan
sistem yang terjamin, untuk selanjutnya metode yang divalidasi menggunakan
sistem yang terjamin dikembangkan. Akhirnya, validasi total diperoleh dengan
melakukan kesesuaian sistem. Masing-masing tahap dalam proses validasi ini
merupakan suatu proses yang secara keseluruhan bertujuan untuk mencapai
kesuksesan validasi (Gandjar dan Rohman, 2007).
ICH (International Conference on Harmonization) membagi karakteristik
validasi metode sebagai berikut:
Gambar 6. Diagram parameter validasi metode menurut ICH (Gandjar dan Rohman, 2007)
Validasi Metode
Presisi Akurasi
Batas Deteksi
Batas Kuantifikasi
Spesifisitas
Linieritas
Ketahanan (robustness)
Ada empat tipe prosedur analisis yang perlu divalidasi, yaitu:
a. Uji identifikasi
b. Uji kuantifikasi untuk komponen pengotor (impurities)
c. Uji ambang batasuntuk pengontrolan kandungan pengotor (impurities)
d. Uji kuantifikasi untuk senyawa aktif pada sampel produk obat (Anonim,
2004b).
Berdasarkan pembagian tersebut, masing-masing tipe analisis memiliki
parameter validasi dari yang berbeda-beda. Parameter validasi dari masing-masing
tipe analisis dijabarkan pada tabel 1.
Tabel 1. Tipe Validasi untuk Prosedur Analisis
Karakteristik
* : mungkin dibutuhkan, tergantung pada kondisi pengujian
1. Presisi
Presisi suatu prosedur analisis menunjukkan kedekatan nilai (derajat
penyebaran) antara serangkaian pengukuran yang dilakukan dari proses sampling
ganda (multiple sampling) dari sekumpulan sampel homogen dengan kondisi yang
telah ditentukan. Presisi dapat dipertimbangkan dalam tiga tingkatan, yaitu:
keterulangan (repeatability), intermediate precision, dan reproduciblity.
a. Keterulangan (repeatability) menunjukkan variabilitas analisis
pada kondisi operasional yang sama dengan interval waktu yang pendek.
Sekurang-kurangnya terdapat 9 determinasi yang perlu dilakukan meliputi
range yang spesifik atau 6 determinasi pada konsentrasi test 100%.
b. Presisi antara (intermediate precision) meliputi pengaruh tambahan
efek randomisasi dalam laboratorium, sesuai dengan tujuan penggunaan
prosedur, contohnya pengerjaan pada hari yang berbeda, analis, dan
peralatan yang berbeda.
c. Reproducibility merupakan presisi antar laboratorium (studi
kolaboratif atau antar laboratorium). Reproducibility tidak perlu dilakukan
untuk penyerahan hasil analisis, namun dapat digunakan sebagai
pertimbangan untuk standarisasi prosedur analisis (Ermer and Miller,
2005).
Presisi seringkali diekspresikan dengan SD atau standar deviasi relatif
(RSD) dari serangkaian data. Perhitungan RSD dapat digunakan rumus:
Keterangan : SD = Standar deviasi serangkaian data
= rata-rata data
(Gandjar dan Rohman, 2007)
Kriteria seksama diberikan jika metode memberikan simpangan
baku relatif atau koefisien variasi 2% atau kurang untuk kadar analit
100%. Akan tetapi kriteria tersebut sangat fleksibel tergantung pada
konsentrasi analit yang diperiksa, jumlah sampel, dan kondisi laboratorium
seperti yang ditunjukkan pada tabel 2.
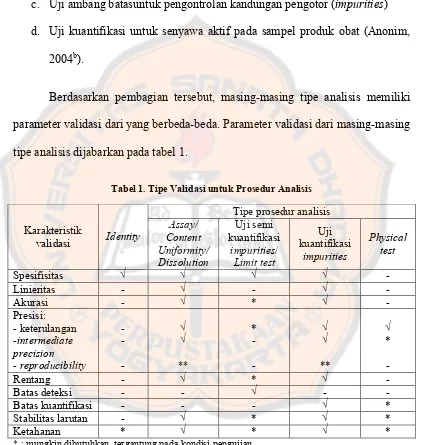
Tabel 2. Kriteria penerimaan nilai RSD
Analit (%) Fraksi analit Konsentrasi analit Nilai RSD (%)
100 1 100% 2
(Horwitz cit. Gonzales, Herrador,and Asuero, 2010)
2. Akurasi
Akurasi pada prosedur analisis menunjukkan kedekatan penerimaan antara
hasil yang diterima sebagai nilai konvensional yang sebenarnya atau hasil
referensi yang diterima, dan hasil yang ditemukan (Ermer and Miller, 2005).
Akurasi pada dapat ditunjukkan melalui hal-hal berikut:
b. Membandingkan hasil dengan karakteristik penerimaan yang baik,
serta prosedur yang bebas.
c. Aplikasi referensi bahan (untuk senyawa obat).
d. Perolehan kembali senyawa obat yang dicampurkan dalam placebo
atau produk obat (untuk produk obat).
e. Perolehan kembali pengotor yang dicampurkan dalam substansi
obat atau produk obat (untuk analisis impurities) (Ermer and
Miller, 2005).
ICH (International Conference on Harmonisation) merekomendasikan
pengumpulan data dari 9 kali penetapan kadar dengan 3 konsentrasi yang berbeda
(misal 3 level konsentrasi dengan masing-masing 3 replikasi) untuk
mendokumentasikan akurasi. Data akurasi dilaporkan dalam nilai persen
perolehan kembali (Gandjar dan Rohman, 2007).
Persen perolehan kembali seharusnya tidak melebihi nilai presisi RSD.
Rentang kesalahan yang diijinkan pada setiap konsentrasi analit pada matriks
dapat dilihat pada tabel 3di bawah ini:
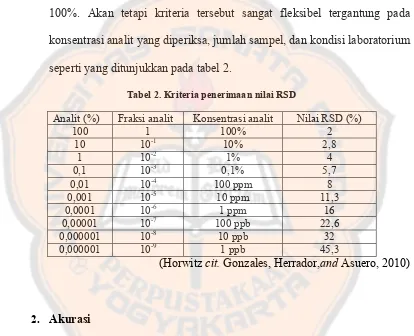
Tabel 3. Kriteria penerimaan nilai %recovery
Analit (%) Fraksi analit Konsentrasi analit Rentang recovery (%)
3. Linieritas
Linieritas merupakan kemampuan suatu metode untuk memperoleh
hasil-hasil uji yang secara langsung proporsional dengan konsentrasi analit pada kisaran
yang diberikan. Linieritas suatu metode merupakan ukuran seberapa baik kurva
kalibrasi yang menghubungkan antara respon (y) dengan konsentrasi (x).
Linieritas dapat diukur dengan melakukan pengukuran tunggal pada konsentrasi
yang berbeda-beda. Data yang diperoleh selanjutnya diproses dengan metode
kuadrat terkecil, untuk selanjutnya dapat ditentukan nilai kemiringan (slope),
intersep, dan koefisien korelasinya (Gandjar dan Rohman, 2007).
Linieritas sebaiknya dievaluasi secara visual dengan memplotkan respon
analit sebagai fungsi konsentrasi analit atau komponen. Untuk pengukuran
linieritas, dianjurkan minimal untuk 5 konsentrasi larutan (Anonim, 2004b).
Syarat suatu metode dikatakan memiliki linearitas yang baik apabila
memiliki nilai koefisien korelasi (r) ≥ 0,999, terutama untuk penetapan kadar
senyawa tunggal (Snyder et al., 1997).
4. Kisaran (range)
Kisaran suatu metode didefinisikan sebagai konsentrasi terendah dan
tertinggi yang mana suatu metode analisis menunjukkan akurasi, presisi, dan
linieritas yang mencukupi. Kisaran-kisaran konsentrasi yang diuji tergantung pada
jenis metode dan kegunaanya. Untuk pengujian komponen utama (mayor), maka
konsentrasi baku harus diukur di dekat atau sama dengan konsentrasi kandungan
Hal yang perlu diperhatikan dalam penentuan rentang:
- Untuk analisis substansi obat atau produk obat akhir, umumnya digunakan
80 sampai 120% dari konsentrasi yang diuji.
- Untuk keseragaman kandungan (content uniformity), minimal melingkupi
70 sampai 130% dari konsentrasi yang diuji. Range yang lebih lebar
diijinkan tergantung bentuk dasar sediaan obat, contohnya sediaan inhalasi
dengan dosis terkontrol (metered dose inhalers) (Anonim, 2004b).
E. Landasan Teori
Pirantel pamoat memiliki sistem kromoforik dan gugus auksokrom yang
memungkinkan untuk dianalisis dengan metode spektrofotometri ultraviolet.
Pirantel pamoat memiliki nilai absorptivitas molar lebih dari 1.000 M-1.cm-1 sehingga pada konsentrasi yang rendah masih mampu untuk dianalisis secara
spektrofotometri ultraviolet.
Pirantel pamoat memiliki dua absorbansi maksimum pada dua panjang
gelombang, karena pirantel pamoat memiliki dua sistem kromoforik, yaitu; pada
struktur asam pamoat dan pada struktur basa pirantel. Panjang gelombang yang
digunakan untuk penetapan kadar pirantel pamoat adalah pada panjang gelombang
300 nm. Hal ini disebabkan karena adanya UV cut off pelarut yang digunakan,
sehingga dipilih panjang gelombang yang lebih jauh.
Suspensi pirantel pamoat mengandung tidak kurang 90,0% dan tidak lebih
dari 110,0% basa pirantel (C11H14N2S) dari label klaim sediaan (Anonima, 2013).
tiap 5 mL larutan. Kadar pirantel pamoat sebagai analit dalam sediaan suspensi
oral merk “X®”memiliki kadar analit 7,21% b/v, namun dilakukan penambahan baku dengan metode standar adisi sehingga kriteria penerimaan nilai RSD adalah
<2,0% (Horwitz cit. Gonzales et al., 2010) dan nilai perolehan kembali sebesar
98-102% (AOAC cit.Gonzales and Herrador, 2007).
Nilai RSD digunakan untuk menyatakan ketelitian metode analisis. Nilai
RSD yang semakin kecil menyatakan bahwa hasil analisis antar replikasi memiliki
kedekatan yang baik. Hal tersebut menunjukkan bahwa prosedur analisis yang
digunakan memiliki ketelitian yang baik.
Nilai perolehan kembali menyatakan akurasi metode analisis, yang
ditunjukkan dengan kedekatan nilai antara hasil pengukuran dengan hasil secara
teoritis. Nilai perolehan kembali dinyatakan dalam persentase. Hasil perolehan
kembali yang mendekati 100%, menunjukkan prosedur analisis yang digunakan
memiliki akurasi yang baik (hasil pengukuran mirip dengan hasil teoritis).
Metode penetapan kadar pirantel pamoat dalam sediaan suspensi merk
“X®” ini digunakan metode analisis kuantitatif dengan menggunakan kurva baku
larutan reference standard. Pada metode ini dilakukan pembuatan grafik sistem
koordinat cartesian dimana sebagai ordinat adalah absorbansi dan sebagai absis
adalah konsentrasi. Kurva baku yang digunakan untuk penetapan kadar harus
menunjukkan hubungan yang linier antara respon absorbansi dengan konsentrasi
larutan. Kriteria penerimaan linieritas kurva baku adalah r ≥ 0,999 (Snyder et al.,
F. Hipotesis
Metode analisis spektrofotometri ultraviolet pada penetapan kadar pirantel
pamoat dalam sampel suspensi oral merk “X®” memenuhi parameter validasi;
27 BAB III
METODE PENELITIAN
A. Jenis dan Rancangan Penelitian
Jenis penelitian yang dilakukan adalah non eksperimental dengan
rancangan penelitian deskriptif. Jenis penelitian non eksperimental karena tidak
dilakukan manipulasi atau pemberian perlakuan pada subyek uji, yaitu metode
ekstraksi dan panjang gelombang pengukuran tetap. Rancangan penelitian
deskriptif karena peneliti hanya mendeskripsikan keadaan yang ada.
B. Variabel Penelitian 1. Variabel bebas
Variabel bebas dalam penelitian ini adalah kondisi optimal dari hasil
optimasi FanggidaE (2013).
2. Variabel tergantung
Variabel tergantung dalam penelitian ini adalah parameter validasi yang
meliputi; rentang, linieritas, akurasi, presisi, dan selektivitas.
3. Variabel pengacau terkendali
Variabel pengacau terkendali dalam penelitian ini adalah kemurnian bahan
baku pirantel pamoat, pengotor dari alat gelas, dan kemurnian pelarut yang
digunakan. Bahan baku pirantel pamoat yang digunakan adalah pharmaceutical
gelas yang digunakan dicuci menggunakan asam pencuci dan metanol sebelum
digunakan. Pelarut yang digunakan adalah pelarut pro analysis yang memiliki
derajat kemurnian tinggi.
C. Definisi Operasional
1. Baku pirantel pamoat yang divalidasi adalah baku pirantel pamoat yang
diperoleh dari PT. Konimex, Indonesia (Certificate of Analysis pada lampiran
1).
2. Metode ekstraksi yang digunakan adalah metode ekstraksi cair-cair
menggunakan ultrasonikator yang merupakan hasil optimasi FanggidaE
(2013).
3. Kadar pirantel pamoat dinyatakan dalam satuan part per million (ppm).
4. Spektrofotometer yang digunakan merupakan seperangkat alat
spektrofotometer UV merk Shimadzu UV-1800 yang dihubungkan dengan
seperangkat komputer merk Advance dan printer merk Hp.
5. Parameter validasi yang digunakan adalah linieritas, rentang, akurasi, presisi,
dan selektivitas.
D. Bahan Penelitian
Bahan-bahan yang digunakan dalam penelitian ini adalah baku pirantel
pamoat (PT. Konimex) dengan kemurnian 102,3% secara HPLC, metanol, heksan,
pirantel pamoat merk “X®” kemasan 10 mL, kertas saring, kapas, akuades
(Laboratorium Kimia Analisis Instrumental, USD).
E. Alat Penelitian
Alat-alat yang digunakan dalam penelitian ini meliputi spektrofotometer
UV merk Shimadzu UV-1800 yang dihubungkan dengan seperangkat komputer
merk Advance dan printer merk Hp, kuvet UV merk Hellma, neraca analitik merk
Ohaus dengan kepekaan 0,1 mg (4 angka di belakang koma, satuan g), hotplate
merk LabTech, mikropipet skala 100-1000 µL merk Socorex, vortex merk Genie,
ultrasonikator merk Retsch UR-275, dan seperangkat alat gelas yang umum
digunakan di laboratorium.
F. Tata Cara Penelitian
1. Pembuatan Larutan Stok Baku Pirantel Pamoat (1023 ppm)
Ditimbang saksama lebih kurang 100,0 mg baku pirantel pamoat,
kemudian dilarutkan dengan DMSO sebanyak 8 mL dalam Beaker glass. Larutan
dimasukkan ke dalam labu ukur 100,0 mL dan ditambahkan metanol hingga tanda
batas.
Keterangan : kemurnian baku pirantel pamoatadalah 102,3% (b/b) dengan metode HPLC,
sehingga penimbangan 100,0 mg baku pirantel pamoat akan menghasilkan konsentrasi
2. Penentuan Panjang Gelombang Pengamatan
Ditimbang saksama lebih kurang 100,0 mg baku pirantel pamoat,
kemudian dilarutkan dengan DMSO sebanyak 8 mL dalam Beaker glass. Larutan
dimasukkan ke dalam labu ukur 100,0 mL dan ditambahkan metanol hingga tanda
batas. Dipipet 100; 200; dan 300 µL larutan stok baku pirantel pamoat, kemudian
dimasukkan ke dalam labu ukur 10,0 mL. Ditambahkan metanol hingga batas
tanda sehingga diperoleh konsentrasi larutan baku pirantel pamoat sebesar 10; 20;
dan 30 ppm. Dilakukan perekaman pola spektra pada panjang gelombang
200-400 nm menggunakan spektrofotometer UV. Panjang gelombang pengamatan
ditentukan dari nilai absorbansi yang paling tinggi, lebih kurang 2 nm dari lamda
teoritis (Direktorat Jenderal Pengawas Obat dan Makanan, 1995).
3. Pembuatan Larutan Seri Baku dan Kurva Baku Pirantel Pamoat
Dipipet 100; 150; 200; 250; dan 300 µL dari larutan stok baku pirantel
pamoat, kemudian dimasukkan ke dalam labu ukur 10,0 mL. Ditambahkan
metanol hingga batas tanda sehingga diperoleh seri konsentrasi larutan baku
pirantel pamoat sebesar 10; 15; 20; 25; dan 30 ppm. Larutan seri baku tersebut
diukur absorbansinya pada panjang gelombang pengamatan 301 nm menggunakan
spektrofotometer UV. Dibuat kurva regresi liniear antara kadar pirantel pamoat
dan absorbansi senyawa, kemudian ditentukan persamaan garis regresi linier dan
nilai koefisien korelasinya. Syarat suatu metode dikatakan memiliki linieritas
yang baik apabila memiliki nilai koefisien korelasi (r) ≥ 0,999, terutama untuk
4. Penentuan Rentang Linieritas Baku Pirantel Pamoat
Ditimbang saksama lebih kurang 100,0 mg baku pirantel pamoat, kemudian
dilarutkan dengan DMSO sebanyak 8,0 mL dalam Beaker glass. Larutan
dimasukkan ke dalam labu ukur 100,0 mL dan ditambahkan metanol hingga tanda
batas. Dipipet 10; 30; 50; 100; 150; 200; 250; 300; 350; 400; 450; dan 500 µL dari
larutan stok baku pirantel pamoat, kemudian dimasukkan ke dalam labu ukur 10,0
mL. Ditambahkan metanol hingga batas tanda sehingga diperoleh seri konsentrasi
larutan baku pirantel pamoat sebesar 1; 3; 5; 10; 15; 20; 25; 30; 35; 40; 45; dan 50
ppm. Larutan seri baku tersebut diukur absorbansinya pada panjang gelombang
pengamatan 301 nm menggunakan spektrofotometer UV. Dibuat kurva regresi
linier antara kadar pirantel pamoat dan absorbansi senyawa, kemudian ditentukan
nilai koefisien korelasinya. Syarat suatu metode dikatakan memiliki linieritas
yang baik apabila nilai koefisien korelasi (r) ≥ 0,999, terutama untuk penetapan
kadar senyawa tunggal (Snyder et al., 1997).
5. Penentuan Akurasi dan Presisi Baku Pirantel Pamoat
Ditimbang saksama lebih kurang 100,0 mg baku pirantel pamoat, kemudian
dilarutkan dengan DMSO sebanyak 8 mL dalam Beaker glass. Larutan
dimasukkan ke dalam labu ukur 100,0 mL dan ditambahkan metanol hingga tanda
batas sehingga diperoleh konsentrasi larutan stok baku pirantel pamoat 1023 ppm.
Dipipet 100; 200; dan 300 µL dari larutan stok baku pirantel pamoat 1023 ppm,
kemudian dimasukkan ke dalam labu ukur 10,0 mL. Ditambahkan metanol hingga
rendah); 20 ppm (konsentrasi tengah); dan 30 ppm (konsentrasi tinggi). Dilakukan
replikasi masing-masing 3 kali untuk setiap konsentrasi, sehingga didapatkan 9
data. Absorbansi larutan seri konsentrasi baku pirantel pamoat diukur pada
panjang gelombang maksimum 301 nm menggunakan spektrofotometer UV. Nilai
absorbansi yang didapat, digunakan untuk menghitung kadar pirantel pamoat
dengan memasukkan nilai absorbansi sebagai nilai y pada persamaan kurva baku
yang telah dibuat.
6. Penentuan Akurasi dan Presisi Baku dalam Matrik Sampel dengan Metode Standar Adisi
a. Pembuatan Larutan Baku Adisi Pirantel Pamoat (2500ppm)
Ditimbang saksama lebih kurang 250,0 mg baku pirantel pamoat,
kemudian dilarutkan dengan 20 mL DMSO dalam Beaker glass hingga tepat larut.
Larutan dimasukkan ke dalam labu ukur 100,0 mL dan ditambahkan metanol
hingga tanda batas.
Keterangan : kemurnian baku pirantel pamoatadalah 102,3% (b/b) dengan metode HPLC,
sehingga untuk mendapatkan kosentrasi larutan stok baku pirantel pamoat 2500 ppm,
ditimbang sebanyak 244,4 mg baku pirantel pamoat.
b. Pembuatan Larutan Sampel Tanpa Adisi Baku Pirantel Pamoat (Blank Sample)
Dipipet sampel suspensi pirantel pamoat merk “X®” yang setara dengan
50,0 mg pirantel pamoat. Sampel yang dicuplik, dimasukkan ke dalam labu ukur
digojog hingga tepat larut. Larutan diencerkan dengan menambahkan metanol
hingga volumenya tepat 100,0 mL. Dilakukan penyaringan larutan stok sampel
tanpa adisi baku pirantel pamoat dengan menggunakan corong kaca, kertas saring,
dan kapas, hingga didapatkan filtrat yang jernih.
c. Pembuatan Larutan Sampel dengan Penambahan Baku Pirantel Pamoat (Addition Sample)
Sepuluh sampel suspensi merk “X®” dengan nomor batch yang sama
dicampur menjadi satu dalam Beaker glass, dan diaduk hingga homogen. Dipipet
sampel suspensi pirantel pamoat merk “X®” yang setara dengan 50,0 mg pirantel
pamoat. Sampel yang dicuplik, dimasukkan dalam labu ukur 100,0 mL,
kemudian dilarutkan dengan 6,0 mL dimetil sulfoksida (DMSO) dan digojog
hingga tepat larut. Ditambahkan 10,0 mL adisi larutan baku pirantel pamoat (2500
ppm) untuk akurasi presisi konsentrasi rendah; 20,0 mL untuk akurasi presisi
konsentrasi tengah; dan 30,0 mL untuk akurasi presisi konsentrasi tinggi, dan
masing-masing labu ukur ditambahkan metanol hingga volumenya tepat 100,0
mL. Dilakukan penyaringan larutan stok sampel adisi dengan menggunakan
corong kaca, kertas saring, dan kapas, hingga didapatkan filtrat yang jernih.
d. Ekstraksi Larutan Blank Sample dan Addition Sample dengan Metode Ekstraksi Cair-Cair Menggunakan Ultrasonikator
Dipipet 10,0 mL larutan stok sampel tanpa adisi baku (larutan 4.b.) dan
sampel adisi baku pirantel pamoat (larutan 4.c.) yang telah disaring. Larutan yang
heksan. Ekstraksi cair-cair dilakukan dengan menggunakan ultrasonikator yang
telah diisi air sebelumnya selama 15 menit. Larutan yang telah diultrasonikasi,
dimasukkan ke dalam corong pisah, sehingga tampak pemisahan antara fase
metanol dengan fase organik heksan. Fase metanol ditampung dalam Beaker glass
dan diuapkan menggunakan hot plate sampai kering.
e. Penetapan Akurasi Presisi Baku Pirantel Pamoat yang Diadisi dalam Matrik Sampel
Larutan sampel tanpa adisi (larutan 4.b.) dan larutan sampel yang diadisi
(larutan 4.c.) yang telah diuapkan, dilarutkan kembali menggunakan metanol
dengan cara dekantir, dan dimasukkan ke dalam labu ukur 25,0 mL. Larutan
diencerkan dengan menambahkan metanol hingga tanda batas, kemudian dipipet
500 μL dan diencerkan dengan metanol dalam labu ukur 10,0 mL hingga tanda
batas. Dilakukan pengukuran absorbansi larutan menggunakan spektrofotometer
UV pada panjang gelombang pengamatan 301 nm. Nilai absorbansi yang didapat,
digunakan untuk menghitung kadar pirantel pamoat dengan memasukkan nilai
absorbansi sebagai nilai y pada persamaan kurva baku yang telah dibuat.
Dilakukan replikasi 3 kali untuk masing-masing sampel dengan 3 konsentrasi,
sehingga diperoleh 18 data (9 data sampel tanpa adisi dan 9 data sampel adisi
dengan 3 konsentrasi: rendah, sedang, dan tinggi). Dihitung persen perolehan
kembali (% recovery) dan nilai Coefficient of Variation (CV) baku pirantel
7. Analisis Hasil
Parameter validasi metode analisis untuk penetapan kadar pirantel pamoat
yang digunakan pada penelitian ini antara lain adalah sebagai berikut:
a. Selektivitas
Parameter selektivitas pada metode ini dapat diperoleh dengan
membandingan panjang gelombang maksimal dari pola spektra larutan baku
pirantel pamoat dengan panjang gelombang maksimal dari pola spektra
larutan blangko (DMSO dan metanol). Metode ini dikatakan selektif jika
terdapat perbedaan panjang gelombang maksimal antara larutan baku pirantel
pamoat dengan larutan blangko (Ermer and Miller, 2005).
b. Linieritas
Parameter linieritas ditentukan dari nilai koefisien korelasi (r) antara
seri konsentrasi larutan baku pirantel pamoat terhadap absorbansi, yang
diplotkan dalam persamaan garis regresi linier, sehingga diperoleh persamaan
garis kurva baku pirantel pamoat (Y=bX+A) yang memiliki nilai r. Suatu
metode dinyatakan memiliki linieritas yang baik jika nilai koefisien korelasi
(r) dari persamaan regresi linier tersebut ≥ 0,999 terutama untuk penetapan
c. Presisi (Ketelitian)
Ketelitian metode analisis yang digunakan, dinyatakan dengan nilai
Coefficient of variation (CV) yang dapat dihitung dengan cara sebagai
berikut:
CV = ̅ x 100 %
Keterangan : CV = Coefficient of Variation
SD = Simpangan deviasi
̅ = Rerata kadar terukur
Kriteria presisi yang diterima untuk kadar zat analit 100 % adalah CV
< 2,0% (Horwitz cit. Gonzales et al., 2010).
d. Akurasi (Ketepatan)
Ketepatan hasil yang diperoleh dengan metode analisis yang
digunakan pada penelitian ini dinyatakan dalam % perolehan kembali (%
recovery) yang dapat dihitung dengan cara sebagai berikut:
% recovery = x 100%
Kriteria akurasi yang diterima untuk kadar zat analit 100% adalah
37 BAB IV
HASIL DAN PEMBAHASAN
A. Pembuatan Larutan Baku Pirantel Pamoat
Baku pirantel pamoat yang digunakan berupa serbuk berwarna kuning, dan
memiliki kelarutan dalam dimetil sulfoksida, sedikit dalam dimetil formamida,
serta praktis tidak larut dalam air maupun metanol. Baku yang digunakan terdiri
dari basa pirantel dan asam pamoat, yang memiliki tingkat kemurnian 102,3%
(b/b) secara HPLC dengan kadar asam pamoat sebesar 66,1%. (Lampiran 1. CoA
Baku Pirantel Pamoat).
Berdasarkan kelarutan tersebut, pada penelitian ini digunakan dimetil
sulfoksida (DMSO) sebagai co-solvent pada pembuatan larutan stok baku pirantel
pamoat maupun pada pembuatan larutan stok sampel suspensi pirantel pamoat
merk “X®”. Dimetil sulfoksida (DMSO) memiliki indeks polaritas 7,2 (Snyder et al., 1997). Penggunaan DMSO sebagai co-solvent dilakukan untuk membantu
kelarutan pirantel pamoat sehingga dihasilkan larutan stok yang jernih, dan
menghindari adanya penghamburan cahaya pada saat pengukuran dengan
spektrofotometer UV.
Pengenceran larutan baku dan sampel pirantel pamoat, digunakan metanol
pro analysis. Pelarut metanol dipilih karena pirantel pamoat memiliki nilai %
dalam pelarut metanol, yaitu 366 pada panjang gelombang 300 nm, dan 270 pada
panjang gelombang 288 nm (Dibbern, 2002). Digunakan pelarut pro analysis
B. Penentuan Panjang Gelombang Pengamatan
Penentuan panjang gelombang pengamatan pada penelitian ini dilakukan
untuk menetapkan panjang gelombang pengamatan yang akan digunakan untuk
pengukuran absorbansi baku maupun sampel. Pirantel pamoat dapat terdeteksi
dengan spektrofotometer UV karena memiliki gugus kromofor dan auksokrom
seperti yang ditunjukkan pada gambar 7. Gugus kromofor merupakan ikatan
rangkap terkonjugasi dimana terdapat ikatan π. Elektron pada ikatan π, dapat
terkesitasi ke tingkat energi yang lebih tinggi yaitu ke π* jika diberi radiasi
elektromagnetik.
Gambar 7. Kromofor dan Auksokrom Pirantel Pamoat
Senyawa pirantel pamoat memiliki absorbansi maksimal pada dua panjang
gelombang, yaitu pada panjang gelombang 288 dan 300 nm. Hal ini disebabkan
karena pirantel pamoat memiliki dua sistem kromoforik, yaitu pada struktur asam
pamoat dan basa pirantel seperti pada gambar 7. Panjang gelombang 300 nm
diprediksikan sebagai absorbansi dari sistem kromoforik asam pamoat, sedangkan
panjang gelombang 288 nm diprediksikan sebagai absorbansi dari panjang
gelombang basa pirantel. Asam pamoat memiliki sistem kromoforik yang lebih
panjang dibandingkan dengan basa pirantel, sehingga dibutuhkan energi yang
lebih kecil agar elektron dapat tereksitasi. Hubungan antara energi yang Auksokrom
dibutuhkan oleh suatu elektron untuk tereksitasi berbanding terbalik dengan
panjang gelombang. Oleh karena itu, semakin kecil energi eksitasi maka panjang
gelombangnya semakin panjang. Perekaman pola spektra (scanning panjang
gelombang) dilakukan pada panjang gelombang 200-400 nm agar dapat
menjangkau kedua panjang gelombang tersebut.
Penggunaan panjang gelombang maksimal dipilih karena beberapa hal
berikut, yaitu:
a. Panjang gelombang maksimal memiliki kepekaan yang maksimal,
sehingga perubahan absorbansi untuk setiap satuan konsentrasi adalah
yang paling besar.
b. Disekitar panjang gelombang maksimal, bentuk kurva absorbansi datar
dan pada kondisi tersebut hukum Lambert-Beer akan terpenuhi.
c. Jika dilakukan pengukuran ulang maka kesalahan yang disebabkan
oleh pemasangan ulang panjang gelombang akan sangat kecil ketika
digunakan panjang gelombang maksimal (Gandjar dan Rohman,
2007).
Pada penentuan panjang gelombang maksimal pirantel pamoat, digunakan
larutan baku pirantel pamoat dengan konsentrasi 10, 20, 30 ppm. Konsentrasi ini
dipilih untuk mewakili konsentrasi rendah, sedang, dan tinggi dari larutan baku,
serta untuk menunjukkan bahwa pada ketiga level konsentrasi, memiliki panjang