BAB II
TINJAUAN PUSTAKA
2.1. THALASSEMIA
2.1.1. Definisi
Thalassemia berasal dari kata Yunani yaitu Thalassa (laut) dan Haema (darah) yang mengacu pada adanya gangguan sintesis dari rantai globin-α dan rantai globin-β, merupakan subunit dari hemoglobin Hb A yaitu (α2; β2).16,18, Gen untuk sintesis rantai globin terletak di kromosom 11 rantai (β) dan kromosom 16 rantai (α).20 Dimana sindrom Thalassemia diklasifikasikan berdasarkan adanya gangguan dari rantai globin-α atau rantai globin-β.21Thalassemia adalah kelainan herediter yang terkait dengan endemisitas malaria ditandai dengan tidak adekuatnya sintesis dari satu atau lebih rantai dari globin
2.1.2. Struktur dan Sintesis Hemoglobin
4,22.
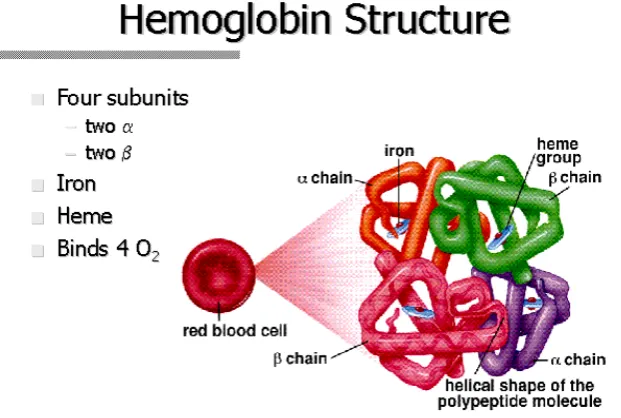
Heme sangat penting untuk transportasi oksigen sedangkan globin berfungsi untuk melindungi heme dari oksidasi. Struktur molekul hemoglobin menghasilkan lingkungan internal hidrofobik yang melindungi besi pada heme dari air, dan juga dari oksidasi. Rantai Globin polipeptida akan mengikat heme, yang nantinya hemoglobin di eritrosit berfungsi untuk mengangkut oksigen dan sebagai transportasi oksigen dari paru-paru ke jaringan 23,24,25
Gambar2.2. Struktur normal dan regulasi pada gen globin manusia. (sumber: Disorder Hemoglobin)
24
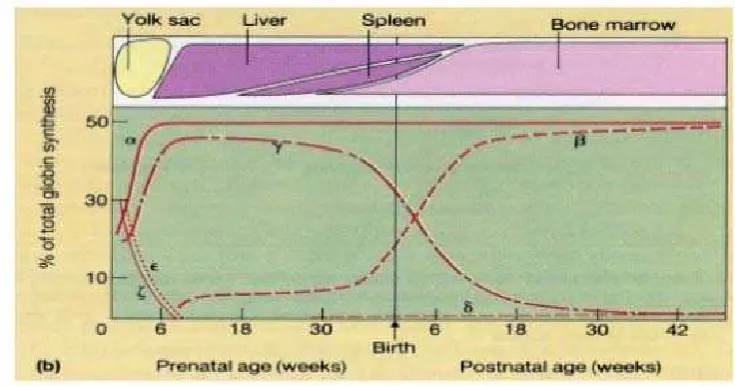
Lokus gen globin pada rantai globin-β terletak pada kromosom 11, dan lokus gen rantai globin-α terletak pada kromosom 16.6 Urutan
aktivasi dimulai dari masa embrional sampai dewasa dari gen ζ ke α dari
genƐ ke ϒᴳ,ϒᴬ,δ dan β. Maka hemoglobin utama pada masa embrional
adalah Hb Gower 1 (ζ₂Ɛ₂), Hb Gower 2 (α₂Ɛ₂), dan Hb Portland (ζ₂ϒ₂).
Pada masa janin sampai perinatal adalah HbF(α₂ϒ₂), dan pada anak
yang berumur lebih dari 1 tahun sampai dewasa normal terdiri dari HbA
(α₂β₂) dan HbA2 (α₂δ₂).22,24,27 Pada 6 bulan pertama perkembangan janin
kehidupan neonatal, terjadi pola yang kompleks dari ekspresi gen globin yang disebut hemoglobin switch.
Pada awal kehidupan embrional sampai delapan minggu sintesis rantai globin akan disintesis yolk sac dan hati yaitu rantai ζ yang berkombinasi dengan rantai Ɛ akan membentuk Hb Gower 1, Hb Gower 2 dan Hb Portland. Ekspresi yang singkat dari gen globin pada masa embrio, maka pada akhir kehamilan akan dibentuk hemoglobin utama
pada janin yaitu Hemoglobin F (α2γ2) dan organ yang terlibat dalam
sintesis rantai globin tersebut adalah hati, limpa dan sumsum tulang, kemudian akan digantikan oleh rantai globin-β dewasa yaitu hemoglobin
A (α2β2), hemoglobin A2 (α₂δ₂) dan Hemoglobin F (α₂ϒ₂) yang kadarnya
<0,5%.7,24,28
Gambar 2.3.Sintesis rantai globin. (sumber: hoffbrand,2001)
24
2.1.2.1. Eritropoiesis di sumsum tulang
sideroblastik, laju eritropoiesis inefektif ini meningkat jauh dibanding laju eritropoiesis yang sangat rendah pada keadaan normal.
Eritropoiesis mencakup tahapankomitmen turunan sel stem pluripoten untuk berdiferensiasi, fase awal eritropoiesis yang independen terhadap eritropoietin (EPO), dan phase lanjut eritropoiesis yang dependen terhadap EPO. Tahapan diferensiasi mulai dari sel stem (hematopoietic stem cell, HSC),burst forming unit-erythroid (BFU-E), colony forming unit-erythroid (CFU-E), pronormoblast/proerythroblast, basophilic normoblast/basophilic erythroblast, polychromatophilic normoblast/ polichromatic erythroblast, orthochromatic normoblast/ orthochromatic erythroblast, retikulosit dan eritrosit matur.
29
Sintesis reseptor transferin dan sintesis hemoglobin sudah dimulai sejak tahap CFU-E.Tahap awal proliferasi dan diferensiasi BFU-E adalah independen terhadap EPO, namun sangat dependen terhadap interleukin-3 (IL-interleukin-3). Dependensi terhadap EPO berkembang pada tahapan antara BFU-E dan CFU-E.
29,30
Berbagai parameter dapat digunakan untuk menilai status eritropoiesis. Secara keseluruhan, status eritropoiesis dapat diperkirakan dari rasio prekursor mieloid: prekursor eritroid (ME ratio) di sumsum tulangyang normalnya sekitar 4:1. Eritropoiesis total antara lain dapat diukur dengan sTfR, karena sumber utama sTfR adalah eritroblast.Kadar sTfR merupakan parameter yang baik untuk eritropoiesis total apabila cadangaan besi adekuat.
29
2.1.3. Klasifikasi
Klasifikasi dari thalassemia berdasarkan jenis subunit globin yang mengalami defek, dan secara garis besar terdiri dari:
2.1.3.1.Thalassemia-α
Pada thalassemia-αproduksi globin-α bergantung jumlah gen-α yang mengalami kerusakan, baik berupa delesi gen maupun non-delesi (mutasi titik). Suatu studi molekul yang menggunakan teknik hibrid telah
mengidentifikasi hilangnya fungsi gen α yang terkait delesi atau non
-delesi dari mutasi gen menyebabkan berkurangnya fungsi gen sehingga menyebabkan mutasi pada kodon yang bertanggung jawab terjadinya syndrom α-thalassemia.33,34
1. Silent thalassemia-α (-α/αα) yang mengalami kerusakan atau delesi 1 gen-α sehingga ratio rantai globin α/β mendekati normal. Selalunya disebut thalassemia-α⁺. Pada keadaan ini tidak terjadi kelainan hematologi . Kelainan ini ditemukan sekitar 15-20% dari populasi keturunan Afrika.
Secara klinis thalassemia-α dapat terbagi menjadi 4 kelompok yaitu:
3. Hemoglobin H disease (--/-α) terjadi kerusakan atau delesi dari 3
gen-α dan hanya terdapat 1 gen-α yang berfungsi. Ciri hematologis
ditandai adanya akumulasi dari rantai globin-β yang mudah larut
membentuk tetramer β₄yang disebut Hb-H yang pada pemeriksaan
pewarnaan supravital dijumpai adanya badan inklusi. Diagnosis penyakit Hb-H dengan menggunakan pemeriksaan hemoglobin Elektroforesis. Penyakit Hb-H memiliki gejala anemia hipokromik mikrositik dengan Hb 8-10 g/dL. Pada pemeriksaan fisik dijumpai adanya pembesaran Hepar dan spleen. Adanya anemia yang berat dapat disebabkan oleh kekurangan asam folat, infeksi akut, paparan stres oksidatif, dan kehamilan. Pengobatan terdiri dari asam folat suplemen (5 mg/hari) dan transfusi darah.
4. Hydrops Fetalis(--/--) disebabkan oleh kerusakan semua gen globin-α. Janin yang terkena akan meninggal di dalam kandungan pada trimester kedua atau trimester ketiga kehamilan atau tidak lama setelah lahir.
35
7,31
Keadaan ini terjadi pada thalassemia-α homozigot, tidak terbentuknya ke empat rantai globin-α. Pada keadaan ini
hemoglobin fetus (HbF atau α2γ2) tidak terbentuk pada masa janin
immature red cell , hipokrom, mikrositer, gambaran sel darah merah anisopoikilositosis.3
2.1.3.2. Thalassemia-β
Thalassemia-β umumnya terdapat di daerah Mediterania, di anak benua India di Asia Tenggara dan umumnya pada orang-orang keturunan Afrika.36,37,38Dimana mutasi thalassemia-β dibagi menjadi dua Kategori: Thalassemia-β (beta zero) dan Thlassemia-β⁺ (beta plus).
Thalassemia-β dapat terjadi dikarenakan hilangnya atau berkurangnya produksi dari rantai globin-β, dapat dibagi menjadi :
36
1. β-Thalassemia minor (trait)terjadi oleh karena ketidakseimbangan sintesa rantai globin-β. Dimana pada β-thalassemia minor (trait) tidak mengalami anemia yang berat, tetapi pada pemeriksaan darah lengkap di jumpai (MCV≤80 fl) dan (MCH≤27 pg) anemia mikrositer hipokrom. Pemeriksaan hemoglobin elektroforesis di jumpai adanyapeningkatan HbA2 (>3,5%), namun pada α-thalassemia trait nilai HbA2 dapat normal atau menurun.39,40Dalam membuat diagnosis
β-thalassemia trait, harus mengesampingkan adanya penyakit
kadang-kadang dilaporkan adanya splenomegaly, perubahan tulang ringan, ulkus pada kaki atau cholelithiasis.18 Kedua orang tua yang memiliki pembawa sifat β-thalassemia, maka akan melahirkan anak-anak 25% normal, 25% β-thalassemia mayor dan 50% β-thalassemia trait.18,36
Gambar 2.4. Skema Penurunan gen β-thalassemia (sumber: D.J.Weatherall)28
umur 2-4 tahun dengan gejala berupa anemia, hiperbilirubinemia, dan hepatospleenomegali. Memiliki pertumbuhan yang lebih baik. Pada beberapa anak thalassemia intermedia, walaupun Hb>7g/dl dapat mengalami kegagalan dalam pertumbuhan ,kurus yang tidak dapat kembali seperti semula kecuali apabila dilakukan transfusi reguler sebelum umur 6 atau 7 tahun.
3. Thalassemia Mayorselalu disebut anemia Cooley, anemia Mediterranean dan anemia Jaksch menunjukkan bentuk penyakit yang homozigot ataupun yang heterozigot dengan gejala anemia berat (1-7 g/dL), hemolisis dan inefektif eritropoesis yang berat. Manifestasi yang muncul pada masa anak-anak dapat terjadi anemia yang berat, ikterus, pertumbuhan terhambat, aktivitas menurun dan sering tidur. Hepatosplenomegaly dengan tanda awal dari wajah thalassemia biasanya ditemukan.
18,24
18
Pada pemeriksaan hapusan darah tepi dijumpai poikilositosis, mikrositosis, hipokrom, target sel, basophilic stipling dan retikulositosis dengan peningkatan Nucleated Red cells.37
2.1.3. Epidemiologi
Timur Tengah melalui Iran, Pakistan, India, Bangladesh, Thailand, Malaysia, Indonesia dan selatan Cina, serta negara-negara di pantai utara Afrika dan Amerika Selatan.
Adanya migrasi penduduk dan perkawinan campuran antara berbagai kelompok etnis telah mengembangkan thalassemia di hampir setiap negara di dunia, termasuk Eropa Utara di mana sebelumnya thalassemia ternyata tidak ada dan sekarang thalassemia menjadi masalah kesehatan umum utama. Diperkirakan 1.5% populasi dunia atau sekitar 80–90 juta orang carrier β-thalassemia, dengan sekitar 60.000 anak lahir pertahun memiliki kasus thalassemia, yang sebagian besar terjadi di dunia yang sedang berkembang.
41
Hemoglobin E-β-thalassemia salah satu hemoglobinopati paling sering dijumpai diseluruh dunia. Insiden HbE banyak terjadi pada 60 populasi di daerah Asia Tenggara. Di daerah pantai Amerika Utara, prevalensi berkembang pesat. Penyakit α- thalassemia sekarang juga
sudah banyak dilaporkan. HbH, Hb Constants Spring , dan homozigot α
-thalassemia mempengaruhi sekitar satu juta orang di seluruh dunia. 3% dari populasi di dunia (sekitar 150 juta orang ) memiliki genβ-thalassemia trait.
41
The dist ribut ion and t he frequency ( % ) of t halassemia bet a carriers in I ndonesia
1
A.S. Sofro & F. Lanni, University of Gajah Mada 4
Gambar 2.5. Distribusi dan frekuensi (%) β-Thalassemia traitdi Indonesia (sumber: Thalassemia International Federation)
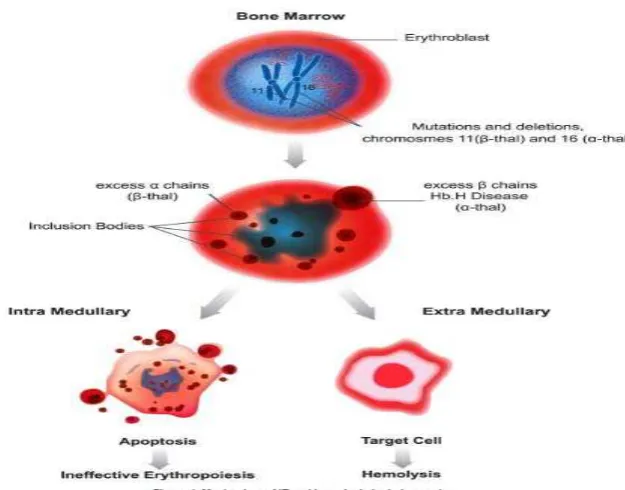
2.1.5. Patofisiologi
Patofisiologi yang mendasari antara jenis thalassemia hampir sama, dengan ditandai penurunan produksi hemoglobin dan sel darah merah (RBC), dan adanya kelebihan rantai globin yang tidak efektif menyebabkan bentuk homotetramers yang tidak stabil sehingga memicu terjadinya heinz body. Alfa homotetramers pada β-thalassemia lebih tidak
stabil daripada β-homotetramers di α-thalassemia dan sebelumnya akan
Pada β-thalassemia patofisiologinya berdasarkan karena berkurang atau hilangnya rantai globin-β yang akan mengakibatkan berlebihnya rantai globin-α. Maka akan terjadi penurunan produksi hemoglobin dan ketidak seimbangan rantai globin. Ini akan mengarah pada penurunan dari volume hemoglobin (MCH) dan volume eritrosit (MCV).42 Pada thalassemia-β yang berat, eritropoesis yang tidak efektif terjadi di sum-sum tulang akan meluas ke tulang-tulang normal dan menyebabkan distorsi dari tengkorak kepala, tulang wajah dan tulang panjang.
Tidak efektifnya eritropoesis yang berat pada anemia kronis dan hipoksia dapat menyebabkan peningkatan absorbsi besi pada saluran pencernaan. Penderita thalassemia homozigot atau pun thalassemia-β heterozygot akan meninggal pada usia 5 tahun karena anemia yang berat. Namun transfusi menyebabkan penumpukan besi yang progressif oleh karena ekskresi yang tidak baik.
18,43
Gambar 2.6. Patofisiologi
2.1.6. Komplikasi
18
dapat terjadi tertundanya pertumbuhan pubertas dan perkembangan seksual. Hampir 90% dari pasien thalassemia mayor memiliki massa tulang yang rendah yang dikaitkan dengan tingginya kejadian fraktur.¹8 Dapat terjadi peningkatan resiko thromboembolik oleh karena adanya berbagai kelainan trombosit dan faktor-faktor pembekuan.2 Telah banyak di laporkan komplikasi thromboembolik pada pasien thalassemia, menggambarkan adanya thrombotik di otak.43,44
2.2.Hepcidin
Hepcidin adalah protein 25-amino-acid dengan 8 residu sistein dan 4 ikatan disulfida, yang terdapat pada banyak spesies. 45Ekspresi hepcidin ini dikode oleh gen HAMP, yang dapat memproduksi 84-amino-acid preprohormon yang akan menjadi hormone matur hepcidin-25.
Hepcidin pertama kali ditemukan pada sampel darah ultrafiltrat plasma dan sampel urin sebagai peptide bakterisida kecil (defensing dan cathelidin ) dan dinamakan liver – expresses antimicrobial peptide (LEAP-1). Hepcidin berasal dari tempat sintesis di dalam hepatosit (hep-) dan aktivitas antimicrobial (-cidin). Gen encoding Hepcidin (HAMP, 19q13) ditemukan di liver , jantung, otak, spinal cord, usus, lambung, pancreas, adiposity, otot skeletal, testis dan makrofag.
45
2.2.1. Struktur dan Sintesa Hepcidin
Gen hepcidin manusia (HAMP) berada di lokasi kromosom 19q13. Dimana Hepcidin mempunyai panjang 2637 base pairs dan terdiri dari 3 ekson dan 2 intron. Ekspresi gen HAMP terutama terjadi di liver, terdapat juga pada jantung, otak, paru, kelenjar prostat, tonsil, kelenjar ludah, dan trachea. HAMP mengkode precursor Hepcidin – Prohepcidin, yang mana merupakan 84 asam amino protein, terdiri dari 24 asam amino pada N-terminal, 35 asam amino pada proregion dan pada C- terminal 20 atau 25 asam amino matang. Hepcidin terdiri dari tiga bentuk, yaitu : 25 asam amino, 20 asam amino dan 22 asam amino peptide. Ketiga bentuk ini terdeteksi dalam urin, tetapi hanya hepsidin 25 dan hepcidin 20 yang terdapat dalam serum manusia.Struktur hepcidin 25, yang merupakan bentuk utama dari hepcidin, yang berisi delapan residu sistein yang dihubungkan oleh ikatan disulfide. Molekul hepcidin berbentuk seperti jepitan rambut (hairpin) dimana kedua lengannya silang oleh gugusan disulfide.44,47
2.2.2. Hepcidin dan metabolisme Besi
Zat besi adalah elemen yang penting bagi tubuh. Walaupun hanya dibutuhkan dalam jumlah sedikit, zat besi berperan penting dalam banyak proses metabolisme tubuh, salah satunya adalah proses hematopoiesis. Hepcidin adalah salah satu protein yang berperan meregulasi kadar zat besi di dalam darah. Kadar hepcidin dalam tubuh dapat dipengaruhi oleh beberapa kondisi, salah satunya adalah rendahnya kadar besi di dalam tubuh.
Hepcidin adalah hormon peptida yang berfungsi sebagai regulator homeostatis metabolisme sistemik besi dan mediator pertahanan tubuh. Sensor sirkulasi besi dan kelebihan besi yang diperkirakan terjadi di liver merupakan tempat utama produksi dan sekresi daripada hepcidin. Beberapa kondisi fisiologis dan patologis dapat mempengaruhi sintesis hepcidin. Situasi yang memerlukan jumlah zat besi yang besar yaitu aktivitas eritropoiesis akan menurunkan sintesis hepcidin. Sedangkan pada kondisi seperti kurangnya cadangan zat besi, inflamasi, infeksi akan meningkatkan sintesis hepcidin.
48
Padaβ-thalassemia traitnilai ferritin lebih tinggi daripada anemia defisiensi besi. Dimana pada proses eritropoiesis menekan hepcidin oleh karena sinyal eritropoiesis memiliki pengaruh fisiologis yang besar. Kadar hepcidin rendah mengindikasikan suatu resiko iron-overload yang signifikan.
14,48
Pada suatu populasi survey dari anak usia sekolah di Sri lanka, β-thalassemia (tetapi bukan HbE) memiliki hubungan dengan peningkatan
eritropoeisis dan penghambatan hepcidin secara ringan. Hal ini mengarahkan kemungkinan terjadinya akumulasi besi. Sebagai kesimpulan pengaruh dari eritropoeisis pada penghambatan hepcidin berhubungan dengan variasi fenotip penyakit dan patogenesa HbE-β thalassemia dan mengidentifikasikan bahwa epidemologi dari β -thalassemia trait perlu ditimbangkan ketika merencanakan intervensi besi pada masyarakat.
Iron-overload menyebabkan cardiomyopathy yang paling sering menyebabkan kematian pada pasien thalassemia yang tergantung pada transfusi tetapi pada thalassemia yang tidak tergantung pada transfusi juga memiliki resiko yang tinggi terhadap iron-overload dan komplikasinya khususnya penyakit hepatic dan endokrin. Hepcidin meregulasi status besi sistemik dengan mengontrol absorbsi besi dengan pelepasan besi dari makrofag. Sintesa hepcidin ditekan oleh eritropoeisis tetapi ditingkatkan oleh akumulasi besi dan inflamasi. Normalnya signal – signal ini seimbang untuk menghasilkan kadar besi yang baik untuk memenuhi kebutuhan eritpoeisis tetapi pada thalassemia eritropoeisis yang berlebihan menekan hepcidin sehingga meningkatkan absorbsi besi. Besi dari transfusi memperbanyak cadangan besi.
49
Ineffective erythropoiesis Chronic anemia / hipoxia
NTDT
↑Erytropoietin Hepcidin↓
↓Ferroportin ↑Release of recycled iron
↑intestinal iron absorption from RES
Iron-overload
Gambar. 2.7.Mekanisme terjadi iron-overload pada NTDT12
2.3.Soluble transferrin reseptor(sTfR)
Reseptor transferin adalah bagianyang terlepas dari permukaan sel berupa bentuk terlarut dalam serum, disebut soluble transferrin receptor(sTfR), dengan berat molekul sekitar 95 kDa. Secara strukturalsTfR merupakan bagian yang menonjol dari reseptor transferin yang kehilangan domain trans-membran dan sitoplasmiknya.
Pada anemia chronic desease, sTfRakan meningkat bila disertai dengan anemia defisiensi besi.
17,57
Beberapa studi menunjukkan retikulosit domba yang dikultur in-vitro memproduksi exosome yang berisi reseptor transferin. Exosome ini berasal dari pembentukan gelembung atau kantong (bleb) internal di dalam vesikel, kemudian membentuk endosom multivesikuler, lalu dikeluarkan dari dalam sel secara eksositosis. Retikulosit melepaskan exosome yang utuh (intact) dan sTfR. Ini menunjukkan bahwa reseptor transferin sudah dipecah secara proteolitik di dalam exosome dan exosome ini merupakan sumber utama sTfR. Ada yang menduga bahwa protease dari granulosit sirkulasi berperan dalam proteolisis reseptor transferin yang dilepaskan dari exosome. Retikulosit meningkat setelah suplementasi Fe pada penderita defisiensi Fe lalu kembali ke nilai normal. Puncak nilai hitung retikulosit tampak pada 2 hari setelah puncak kadar sTfR. Defisiensi Fe akan meningkatkan secara nyata sTfR yang dilepaskan dari dalam sel.50
antara pre dan post menopause. Orang yang tinggal di ketinggian mempunyai sTfR 9% lebih tinggi dibanding yang tinggal di tempat dengan ketinggian permukaan laut. Kadar feritin serum menggambarkan status cadangan Fe sedangkan kadar sTfR menggambarkan status Fe fungsional. Kadar sTfR berhubungan langsung dengan peningkatan massa prekursor eritroid dibandingkan dengan ambilan (uptake) transferin eritron. Ini menunjukkan bahwa sTfR dapat dipakai sebagai ukuran kuantitatif eritropoiesis total.52
2.4. DiagnosisThalassemia
2.4.1. Anamnese
Dalam mendiagnosa thalassemia sangat penting mengetahui tentang riwayat penderita dan keluarga, karena ada beberapa populasi dengan ras etnik tertentu memiliki frekuensi yang tinggi untuk jenis gen abnormalthalassemia.
2.4.2. Pemeriksaan fisik
2.4.3. Pemeriksaan Hematologi 2.4.3.1. Full Blood Count (FBC)
Dengan pemeriksaanFBC (Full Blood Count)dapat dilihat nilai eritrosit rerata seperti Mean Corpuscular Volume (MCV), Mean corpuscular hemoglobin (MCH), Red Blood Cell Distribution Width (RDW). Pada penderitaβ-thalassemia trait biasanya nilai MCV dan MCH rendah (mikrositer hipokrom) dan mengalami anemia ringan
2.4.3.2. Sedian hapus darah tepi
,40,54,55
Pemeriksaan laboratorium pada penderitaβ-thalassemia trait diperlukan juga evaluasi sediaan hapusan darah tepi dengan tujuan untuk melihat morfologi eritrosit, biasanya mikrositer hipokrom.
2.4.3.3. Analisa hemoglobin
7,56
mampu membedakan hemoglobin E (HbE) dari HbA2, sehingga dapat membedakan dengan baik kuantifikasi HbA2 pada pasien dengan HbE.
Manusia dewasa memiliki darah normal yang terdiri dari fraksi hemoglobin HbA, HbF, dan HbA2. HbA merupakan komponen mayor dari
fraksi hemoglobin α2β2, dengan kadarnya 96,8%-97,8%, sedangkan
komponen minor terdiri dari rantai globin α2ϒ2 (HbF) dengan kadar
<0,5%, dan α2δ2 (HbA2) dengan kadarnya 2,2%-3,2%.
43
31
Peningkatan Hemoglobin A2 merupakan parameter yang paling signifikan dalam
mengidentifikasi β-thalassemia trait.39 Distribusi HbA2 pada 200 orang
sehat dengan menggunakan alat elekroforesis dengan media celulosa acetate diperoleh HbA2 berkisar 1,5-3,5%. Cutoff yang banyak digunakan peneliti sebagai batas atas HbA2 pada populasi sehat adalah 3,5%, dan digunakan juga selama penelitian ini untuk studi perbandingan.
Analisa hemoglobin dengan pemeriksaan hemoglobin elektroforesis dengan menilai kadar HbA2 dan kadar HbF. Kuantitasi HbA2 yang meningkat >3,5% mengidentifikasi suatu β-thalassemia trait.
43
10
2.5. Kerangka Konsep
β
-thalassemia trait
Eritropoesis yang tidak efektif
Hepcidin