II.
TINJAUAN PUSTAKA
2.1.
Karbohidrat
Kebanyakan ahli kimia kesulitan dalam mengelompokkan bahan apa saja yang termasuk ke dalam karbohidrat. Definisi klasik karbohidrat berdasarkan asal katanya yaitu carbo dari bahasa Latin dan hydros dari bahasa Yunani adalah ‘hidrat dari karbon’ yang mengandung hidrogen dan oksigen dengan perbandingan 2:1 (Southgate 1978) atau elemen yang terdiri dari air dan karbon dengan perbandingan 1:1 (Kennedy dan White 1988). Karbohidrat adalah senyawa organik yang mengandung karbon, hidrogen dan oksigen baik dalam bentuk molekul sederhana maupun kompleks (Christian dan Vaclavik 2003).
Karbohidrat telah menjadi sumber energi utama untuk metabolisme pada manusia dan sarana untuk memelihara kesehatan saluran pencernaaan manusia. Karbohidrat adalah penyumbang utama dari komponen yang membentuk produk pangan baik sebagai komponen alami maupun bahan yang ditambahkan. Karbohidrat meliputi lebih dari 90% dari berat kering tanaman. Karbohidrat banyak tersedia dan murah. Penggunaannya sangat luas dan jumlah penggunaannya cukup besar (Fennema 1996) baik untuk pemanis, pengental, penstabil, gelling agents dan fat
replacer (Christian dan Vaclavik 2003). Karbohidrat dapat dimodifikasi baik secara kimia dan
biokimia dan modifikasi itu digunakan untuk memperbaiki sifat dan memperluas penggunaannya.
2.1.1 Struktur karbohidrat
Karbohidrat digunakan dalam kimia untuk senyawa dengan formula Cm(H2O)n, tetapi kini rumus molekul itu tidak secara kaku digunakan untuk mendefinisikan karbohidrat (Kennedy dan White 1988). Sebelumnya beberapa ahli kimia memasukkan formaldehid dan glikoaldehid sebagai karbohidrat, namun sekarang istilah karbohidrat dalam biokimia, tidak mengikutsertakan senyawa yang kurang dari tiga atom karbon. Southgate (1978) menggunakan definisi karbohidrat sebagai senyawa yang tersusun oleh polihidroksi aldehid, keton, alkohol, asam dan turunan sederhananya serta polimernya yang memiliki ikatan polimer tipe asetal.
Menurut strukturnya karbohidrat dapat dibagi menjadi kelompok sakarida: monosakarida, oligosakarida dan polisakarida. Monosakarida adalah gula sederhana yang tidak dapat dipecah lagi menjadi molekul yang lebih kecil dan monosakarida inilah yang menjadi unit penyusun dari oligosakarida dan polisakarida. Oligosakarida dan polisakarida tersusun dari monosakarida yang dihubungkan dengan ikatan glikosidik.
2.1.2. Monosakarida
Monosakarida terdiri dari tiga sampai delapan karbon atom, tetapi umumnya hanya lima atau enam yang biasa ditemukan. Biasanya monosakarida digolongkan berdasarkan jumlah atom karbonnya, misalnya triosa (C3H6O3), tetrosa (C4H8O3), pentosa (C5H10O5) dan heksosa (C6H12O6). Dari golongan tersebut dapat dibagi lagi berdasarkan gugus fungsional yang ada, misalnya dari golongan heksosa ada aminoheksosa (C6H13O5N), deoksiheksosa (C6H12O5) dan asam heksuronat (C6H10O7). Contoh monosakarida adalah glukosa dan fruktosa.
2.1.3. Oligosakarida
Oligosakarida terdiri dari beberapa monosakarida (2-10) yang saling terikat oleh ikatan glikosidik. Tetapi ada juga yang mengklasifikasikan sendiri karbohidrat dengan dua gugus gula sebagai disakarida. Menurut Christian dan Vaclavik (2003) disakarida terdiri dari dua molekul monosakarida yang bergabung dengan ikatan glikosidik. Contoh disakarida di pangan adalah maltosa, selubiosa, dan sukrosa. Oligosakarida yang memiliki lebih dari tiga gugus gula contohnya adalah rafinosa dan stakiosa.
2.1.4. Polisakarida
Polisakarida merupakan polimer dari gula sederhana yang tersusun atas lebih dari sepuluh monomer gula sederhana. Contoh polisakarida di makanan adalah pati, pektin dan gum. Ketiganya adalah polimer karbohidrat kompleks dengan sifat yang berbeda, tergantung unit gula penyusunnya, tipe ikatan glikosidik dan derajat percabangan molekul.
2.2. Pentingnya Analisis Total Karbohidrat
Total karbohidrat yang ada dalam bahan pangan perlu diketahui dengan alasan: standards of
identity (pangan harus memiliki komposisi yang sesuai dengan regulasi pemerintah); nutritional labelling (menginformasi konsumen mengenai kadar nutrisi dalam bahan pangan); detection of adulteration (tiap tipe pangan memiliki 'fingerprint' karbohidrat); food quality (sifat fisikokimia
dari pangan seperti kemanisan, penampakan, stabilitas dan tekstur tergantung tipe dan stabilitas karbohidrat yang ada); ekonomi (agar lebih dapat menghemat biaya produksi bahan yang digunakan pada industri) dan food processing (efisiensi dari proses pangan banyak tergantung pada jenis dan kadar karbohidrat). Dalam berbagai studi mengenai bahan makanan penting untuk mengetahui persentasi kadar karbohidrat pada pangan yang diujikan sehingga nilai karbohidrat pada bahan lain dapat dikonversi menjadi nilai total pangan.
2.3. Total Karbohidrat dalam Bahan Pangan dan Metode Analisisnya
2.3.1. Definisi total karbohidrat
Total karbohidrat atau total karbohidrat menurut Badan Pengawasan Obat dan Makanan (2005) meliputi gula, pati, serat pangan dan komponen karbohidrat lain. Pernyataan jumlah total karbohidrat dalam gram penyajian yang dinyatakan dengan nilai gram terdekat, jika penyajian kurang dari 0,5 gram, jumlah kadarnya dapat dinyatakan sebagai nol dan jika penyajian lebih dari 0,5 gram dibulatkan ke kelipatan 1 gram terdekat. Total karbohidrat dapat dinyatakan dengan total karbohidrat by difference.
Total karbohidrat dalam pengukuran karbohidrat dengan metode langsung dinyatakan dalam bentuk persen yang setara dengan glukosa. Satuan glukosa (glucose equivalent) juga dapat diganti dengan larutan gula lain yang dijadikan sebagai larutan standar.
2.3.2. Metode analisis total karbohidrat
Sejumlah teknik analisis telah dikembangkan untuk mengukur jumlah dan tipe karbohidrat yang ada di bahan pangan. Kadar karbohidrat di bahan pangan dapat diketahui dengan menghitung persentase yang tersisa setelah semua komponen lain telah diukur (total carbohydrate by
difference), yaitu dengan persamaan (1.1) (SNI 01-2891-1992):
(1.1) Metode by difference ini masih digunakan oleh FDA, tetapi metode ini dapat menghasilkan nilai yang salah karena ada kemungkinan terjadi akumulasi kesalahan dari metode-metode yang digunakan untuk mengukur komponen lain, dan kemungkinan adanya komponen non karbohidrat yang terukur sebagai karbohidrat menyebabkan penyimpangan yang lebih besar. Pengukuran kadar karbohidrat secara langsung lebih baik karena didapat hasil lebih yang akurat.
2.3.2.1. Analisis karbohidrat langsung
Metode yang telah dikembangkan untuk analisis karbohidrat sangat banyak, dan tergantung juga oleh jenis analisis (kuantitatif atau kualitatif) dan tipe karbohidrat yang dianalisis. Sehingga metode pengukuran karbohidrat sangat beragam mulai dari metode kromatografi dan elektroforesis (Kromatografi Lapis Tipis, Kromatografi Likuid Kinerja Tinggi dan Kromatografi Gas); metode kimia (metode titrasi Lane Eynon, metode gravimetri Munson Walker, metode Luff Schoorl, metode kolorimetri seperti anthrone sulfat dan fenol sulfat); metode enzimatis; metode fisik (polarimetri, indeks refraktif, densitas dan infra merah) serta metode immunoassay.
Uji karbohidrat yang resmi ditetapkan oleh BSN dalam SNI 01-2891-1992 yaitu analisis total karbohidrat dengan menggunakan metode Luff Schoorl. Pada tahun 1936 International Commission for Uniform Methods of Sugar Analysis mempertimbangkan Metode Luff-Schoorl sebagai salah satu metode yang digunakan untuk menstandarkan analisis gula pereduksi karena metode Luff Schoorl saat itu menjadi metode yang resmi dipakai di pulau Jawa, di samping nominator lainnya yaitu metode Lane-Eynon. Tetapi pada saat itu metode kolorimetri belum banyak berkembang dan dalam catatan komisi itu terdapat agenda untuk melakukan penyeragaman analisis gula dengan metode kolorimetri.
Berikut ini adalah beberapa jenis analisis total karbohidrat langsung:
2.3.2.1.1. Analisis total karbohidrat dalam SNI 01-2891-1992
Seluruh senyawa karbohidrat yang ada dipecah menjadi gula-gula sederhana (monosakarida) dengan bantuan asam yaitu HCl dan panas. Monosakarida yang terbentuk kemudian dianalisis dengan Metode Luff-Schoorl. Prinsip analisis dengan Metode Luff-Schoorl yaitu reduksi Cu2+ menjadi Cu 1+ oleh monosakarida. Monosakarida bebas akan mereduksi larutan basa dari garam logam menjadi bentuk oksida atau bentuk bebasnya. Kelebihan Cu2+ yang tidak tereduksi kemudian dikuantifikasi dengan titrasi iodometri (SNI 01-2891-1992).
Reaksi yang terjadi (1.2):
Karbohidrat kompleks → gula sederhana (gula pereduksi) Gula pereduksi+ 2 Cu2+→ Cu 2O(s) 2 Cu2+(kelebihan) + 4 I-→ 2 CuI 2→ 2 CuI-+ I2 I2+ 2S2O32-→ 2 I-+ S4O6 2-(1.2) Osborne dan Voogt (1978) mengatakan bahwa Metode Luff-Schoorl dapat diaplikasikan untuk produk pangan yang mengandung gula dengan bobot molekuler yang rendah dan pati alami atau modifikasi.
Kemampuan mereduksi dari gugus aldehid dan keton digunakan sebagai landasan dalam mengkuantitasi gula sederhana yang terbentuk. Tetapi reaksi reduksi antara gula dan tembaga sulfat sepertinya tidak stoikiometris dan sangat tergantung pada kondisi reaksi. Faktor utama yang mempengaruhi reaksi adalah waktu pemanasan dan kekuatan reagen. Penggunaan luas dari metode ini dalam analisis gula adalah berkat kesabaran para ahli kimia yang memeriksa sifat empiris dari reaksi dan oleh karena itu dapat menghasilkan reaksi yang reprodusibel dan akurat (Southgate 1976).
2.3.2.1.2.
Analisis total karbohidrat dengan Metode Anthrone sulfat
Penggunaan Metode Anthrone untuk analisis total karbohidrat mulai berkembang sejak penggunaan pertama kali oleh Dreywood pada tahun 1946 untuk uji kualitatif. Dasar dari reaksi ini adalah kemampuan karbohidrat untuk membentuk turunan furfural dengan keberadaan asam dan panas, yang kemudian diikuti dengan reaksi dengan anthrone yang menghasilkan warna biru kehijauan (Sattler dan Zerban 1948) dalam Brooks et al (1986).
Anthrone, C6H4COC6H4CH2, adalah turunan dari anthraquinone. Senyawa ini diproduksi oleh reduksi katalitik dari anthraquinone oleh asam hidroklorat dengan keberadaan logam timah. Senyawa ini mungkin ada dalam bentuk keto atau enol, yang masing-masing dikenal dengan nama anthrone and anthranol. Reaksinya dapat dilihat pada persamaan (1.3):
(1.3)
Mekanisme pembentukan warna anthrone dengan gula telah diteliti. Hurd dan Isenhour (1932) dan Wolfrom et al (1948) mempostulasikan bahwa karbohidrat dan turunannya mengalami pembentukan cincin dalam keberadaan asam kuat dari mineral, seperti yang ditunjukkan untuk glukosa (1.4):
(1.4) Tiap tahap adalah pemecahan dari glukosa(I) menjadi 5-(hydroxymethyl)-2-furaldehyde(IV) menunjukkan dehidrasi baik pada double bond atau pembentukan cincin. Wolfrom et al. (1948) menunjukkan bukti spektroskopik untuk senyawa intermediate (II) dan (III) pada reaksi ini Sattler
and Zerban (1948) menyarankan bahwa pembentukan warna hijau pada reaksi anthrone tergantung oleh keberadaan 5-(hidroksimetil)-2-furaldehid, atau senyawa furfural yang mirip, yang dibentuk oleh reaksi asam sulfat pada karbohidrat.
Momose et al. (1957) melakukan kromatografi pada ekstrak benzene dari pewarna terhadap alumina dan menunjukkan bahwa bagian yang dapat larut dari benzene-terdiri dari beberapa pewarna yang memberikan pewarnaan yang berbeda dengan asam sulfat. Mereka menentukan berat molekul dari salah satu pewarna utama yaitu kurang lebih 530, dan mempostulasikan formula dari pewarna itu (C47H30O3). Mereka menyimpulkan bahwa 3 mol anthrone bereaksi dengan 1 mol glukosa, yang digambarkan dalam persamaan (1.5):
3C
14H
10O + C
6H
12O
6 C
47H
3O
30+ 5H
2O + CH
2O
(1.5)
Dari data analisis dan spektrum inframerah dari pewarna, dan mekanisme reaksinya dipertimbangkan, mereka menduga struktur yang mungkin adalah 1,2,5,- atau 1,3,5,-trianthronylidenepentane.
Ludwig dan Goldberg (1956) melaporkan adaptasi dari Metode Anthrone kolorimetri untuk analisis total karbohidrat secara kuantitatif pada pangan. Metode yang digunakan relatif cepat dan akurat serta lebih baik daripada metodologi analisis karbohidrat sebelumnya, yaitu metode Somogyi-Shaffer-Hartmann yang menggunakan teknik teknik iodometri dan prinsip gula pereduksi. Mereka menunjukkan bahwa persiapan hidrolisis dan deproteinisasi tidak perlu dilakukan ketika teknik anthrone digunakan.
Uji Anthrone ini memiliki kelebihan dalam hal sensitifitas dan kesederhanaan ujinya (Koehler 1952).Sejumlah kecil karbohidrat dapat memberikan warna yang terdeteksi dengan menggunakan spektrofotometer. Dreywood (1946) melakukan uji spesifisitas dari reaksi dan membuat daftar 18 jenis karbohidrat, termasuk beberapa turunan selulosa, yang memberikan hasil positif. Dia juga melaporkan hasil negatif terhadap kelompok besar nonkarbohidrat, termasuk sejumlah resin sintetik nonselulosa, asam organik, aldehid, fenol, lemak, terpena, alkaloid, dan protein. Nonkarbohidrat yang menunjukkan hasil positif hanya furfural, tetapi hasil positif ini cepat menghilang karena warna hijau dikaburkan oleh presipitat coklat. Morris (1948) juga menunjukkan spesifisitas anthrone untuk karbohidrat sangat tinggi, dan dia melaporkan reaksi positif untuk semua mono-, di-, dan polisakarida murni yang diujikan, juga sampel of dekstrin, dekstran, pati, polisakarida tumbuhan dan gum, polisakarida tipe II dan II dari pneumococcus, glukosida, dan senyawa asetat dari mono-, di-, dan polisakarida.
Kekurangan dari Metode Anthrone adalah ketidakstabilan dari reagen (anthrone yang dilarutkan dalam asam sulfat), sehingga perlu dilakukan persiapan reagen yang baru setiap hari. Dreywood (1946) memperhatikan bahwa panas yang dihasilkan oleh pelarutan asam sulfat merupakan bagian yang penting dalam uji. Morris (1948) melihat signifikansi dari panas pada reaksi anthrone dan menunjukkan bahwa pada sejumlah karbohidrat yang diberikan, intensitas warna bervariasi dengan jumlah panas yang dihasilkan. Oleh karena itu kurva standar juga perlu dibuat setiap hari.
Nilai total karbohidrat tidak dapat dinyatakan dalam persen karbohidrat, tetapi lebih baik dinyatakan dengan istilah glucose equivalents per cent, karena kepekatan warna yang dihasilkan dari reaksi anthrone bervariasi dengan tipe gula yang ada. Kepekatan warna yang sama contohnya, ditunjukkan oleh 100 µg. glukosa, 105 µg. maltosa, dan 111 µg glikogen. Gula murni lain selain glukosa dapat dikalkulasi dengan faktor konversi. Tetapi jika terdapat campuran karbohidrat yang tidak diketahui pada bahan pangan faktor konversi itu tidak dapat digunakan, dan hasilnya bukan persentase karbohidrat absolut, melainkan ekuivalen glukosa, yang dapat bervariasi dari nilai persentasi karbohidrat yang sebenarnya dengan jumlah yang tidak dapat ditentukan. Keganjilan ini tidak signifikan ketika nilai glucose equivalents per cent digunakan hanya sebagai basis untuk mengkonversi nilai total karbohidrat menjadi nilai total pangan (Beck dan Bibby 1961). Untuk tujuan ini glucose equivalents per cent hanya sebagai indeks dari persentasi absolute dari masing-masing karbohidrat dalam pangan.
2.4. Validasi dan Verifikasi Metode
Metode analisis memiliki beberapa atribut, seperti ketepatan, ketelitian, spesifisitas, sensitivitas, kemandirian, dan kepraktisan, yang harus dipertimbangkan ketika akan digunakan (Garfield et al. 2000). Informasi yang digunakan untuk mengambil keputusan harus seimbang dengan pertimbangan praktis seperti biaya, waktu, risiko, kesalahan, dan tingkat keahlian yang diperlukan. Selain itu suatu laboratorium yang akan menerapkan suatu metode perlu mempertimbangkan apakah data validasi yang ada mengenai metode tersebut cukup memadai atau apakah masih membutuhkan tindakan validasi ulang sebelum metode itu digunakan. Selanjutnya jika data validasi telah cukup memadai, laboratorium perlu mengetahui apakah level performa yang ditunjukkan oleh data validasi tersebut mampu dilaksanakan. Untuk mencapai level performa itu dibutuhkan analis yang kompeten serta peralatan dan fasilitas yang memadai (Jelita 2011).
Data validasi yang kurang memadai biasanya ada pada metode yang baru dikembangkan baik oleh laboratorium itu sendiri atau yang dikembangkan oleh pihak lain; metode yang digunakan
oleh laboratorium lain atau metode yang telah dipublikasi tetapi belum menjadi metode baku. Ketika data validasi yang ada telah memadai, yaitu seperti pada metode yang telah divalidasi oleh organisasi terstandarisasi seperti AOAC (Association of Official Analytical Chemists) Internasional, laboratorium umumnya hanya menjaga performa data dengan cara melakukan verifikasi metode.
Validasi metode analisis adalah suatu tindakan penilaian terhadap parameter tertentu, berdasarkan percobaan laboratorium, untuk membuktikan bahwa parameter tersebut memenuhi persyaratan untuk penggunaannya (Harmita, 2004). Berdasarkan Harvey (2000), validasi merupakan suatu proses evaluasi kecermatan dan keseksamaan yang dihasilkan oleh suatu prosedur dengan nilai yang dapat diterima. Sebagai tambahan, validasi memastikan bahwa suatu prosedur tertulis memiliki detail yang cukup jelas sehingga dapat dilaksanakan oleh analis atau laboratorium yang berbeda dengan hasil yang sebanding. Menurut AOAC (2002) validasi metode menunjukkan apakah suatu metode sesuai dengan tujuan yang diinginkan. Dalam praktiknya, memungkinkan untuk merancang percobaan yang akan dilakukan sehingga karakteristik validasi yang sesuai dapat diterapkan untuk mendapatkan hasil yang cukup dan menyeluruh mengenai kemampuan suatu prosedur analisis, seperti: spesifisitas, linearitas, rentang, akurasi (kecermatan), dan presisi (keseksamaan) (EMA, 1995).
Verifikasi metode adalah suatu tindakan validasi metode tetapi hanya pada beberapa beberapa karakteristik performa saja. Laboratorium harus menentukan karakteristik performa yang dibutuhkan. Spesifikasi analisis dapat menjadi acuan untuk merancang proses verifikasi. Rancangan yang baik akan menghasilkan informasi yang dibutuhkan serta meminimalisir tenaga, waktu, serta biaya. Pemilihan parameter validasi atau verifikasi tergantung pada beberapa faktor seperti aplikasi, sampel uji, tujuan metode, dan peraturan lokal atau internasional.
Adapun beberapa parameter analisis yang harus dipertimbangkan dalam validasi metode analisis :
2.4.1. Akurasi
Akurasi atau kecermatan adalah seberapa dekat suatu hasil pengukuran kepada nilai sebenarnya. Terkadang masalah dalam menentukan akurasi adalah ketidaktahuan terhadap nilai yang sebenarnya. Dalam beberapa tipe sampel kita dapat menggunakan sampel yang telah diketahui nilainya dan mengecek metode pengukuran yang kita gunakan untuk menganalisis sampel itu sehingga kita mengetahui akurasi dari prosedur yang diujikan, metode ini disebut dengan CRM (Certified Reference Method). Pendekatan lain adalah dengan membandingkan
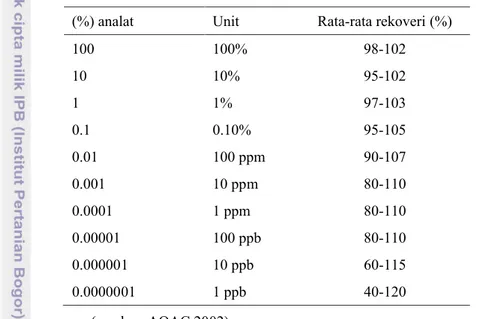
hasilnya dengan hasil yang dilakukan oleh lab lain (Smith, 2010) atau dengan menggunakan metode referen (Walton 2001). Akurasi juga dapat diketahui dengan melakukan uji rekoveri (Walton 2001). Hasil uji ini akurasi dapat dinyatakan sebagai persen perolehan kembali (recovery) analat yang ditambahkan pada sampel. Sampel ditambahkan (spiking) dengan standar yang telah diketahui jumlah dan kadarnya (EMA, 1995). Rentang nilai penerimaan kecermatan suatu metode akan bervariasi sesuai kebutuhannya (FAO, 1998). Adapun AOAC menetapkannya seperti dalam Tabel 1.
Tabel 1 Persentase rekoveri yang dapat diterima sesuai dengan konsentrasi analat (%) analat Unit Rata-rata rekoveri (%)
100 100% 98-102 10 10% 95-102 1 1% 97-103 0.1 0.10% 95-105 0.01 100 ppm 90-107 0.001 10 ppm 80-110 0.0001 1 ppm 80-110 0.00001 100 ppb 80-110 0.000001 10 ppb 60-115 0.0000001 1 ppb 40-120 (sumber: AOAC 2002)
2.4.2. Presisi
Presisi adalah ukuran yang menunjukkan derajat kesesuaian antara hasil uji individual, diukur melalui penyebaran hasil individual dari rata-rata jika prosedur diterapkan secara berulang pada sampel-sampel yang diambil dari campuran yang homogen (Harmita, 2004). Presisi dapat dibagi dalam dua kategori: keterulangan atau ripitabilitas (repeatability) dan ketertiruan (reproducibility). Ripitabilitas adalah nilai presisi yang diperoleh jika seluruh pengukuran dihasilkan oleh satu orang analis dalam satu periode tertentu, menggunakan pereaksi dan peralatan yang sama dalam laboratorium yang sama. Ketertiruan adalah nilai presisi yang dihasilkan pada kondisi yang berbeda, termasuk analis yang berbeda, atau periode dan laboratorium yang berbeda dengan analis yang sama. Karena ketertiruan dapat memperbanyak sumber variasi, ketertiruan dari analisis tidak akan lebih baik hasilnya dari nilai keterulangan (Harvey, 2000).
Presisi dalam hal ripitabilitas diukur dengan menghitung relative standard deviation atau simpangan baku relatif (RSD) dari beberapa ulangan dengan menggunakan rumus (1.6):
(1.6) Standar deviasi ripitabilitas bervariasi tergantung pada konsentrasi (AOAC 2002). Oleh karena itu hasil yang didapat dari perhitungan dibandingkan hasilnya dengan nilai yang ada di Tabel 2.
Tabel 2 Nilai presisi (RSD) sesuai dengan konsentrasi analat (%) analat Konsentrasi RSD (%) 100 100% 1 10 10% 1.5 1 1% 2 0.1 0.10% 3 0.01 100 ppm 4 0.001 10 ppm 6 0.0001 1 ppm 8 0.00001 10 ppb 15 (sumber: AOAC 2002)
Nilai yang didapat juga dapat dibandingkan atau dengan menggunakan rumus (1.7):
(1.7) dengan C adalah konsentrasi yang didapat dari rataan.
Nilai yang dapat diterima untuk ripitabilitas adalah antara 1/2 dan 2 kali dari nilai yang dijadikan sebagai pembanding. Ada juga yang menggunakan RSD Horwitz sebagai nilai pembanding, RSD Horwitz dihitung dengan rumus (1.8):
(1.8) Dengan menggunakan pembanding RSD Horwitz nilai yang dapat diterima untuk ripitabilitas adalah RSD yang terhitung dari ulangan yang ada harus kurang dari 2/3 dari nilai RSD Horwitz (Garfield 2000).
2.4.3. Spesifisitas
Spesifisitas dari metode analitik tertentu berarti metode itu hanya mendeteksi komponen yang diinginkan. Metode analitis dapat bersifat sangat spesifik untuk komponen tertentu atau pada beberapa kasus dapat menganalisis spektrum komponen yang luas (Smith, 2010).
Spesifisitas suatu metode diuji dengan membandingkan hasil dari sampel yang mengandung pengotor dengan hasil sampel yang tidak mengandung pengotor. Pada dasarnya, spesifisitas dapat diuji secara langsung atau tidak langsung. Pendekatan secara tidak langsung ditinjau dari penerimaan parameter akurasi. Pendekatan secara langsung ditinjau dari keberadaan komponen pengganggu (Ermer dalam Ermer dan Miller, 2005). Cara yang terakhir dilakukan dengan menambahkan sejumlah tertentu komponen pengganggu pada larutan standar murni. Jika diperkirakan tidak adanya komponen pengganggu pada sampel, spesifisitas dapat ditunjukkan dengan membandingkan hasil uji sampel dengan standar (EMA, 1995).
2.4.4. Limit Deteksi dan Limit Kuantitasi
Limit deteksi atau Limit of Detection (LOD) suatu metode analisis adalah jumlah terkecil dari analat yang dapat dideteksi namun jumlah ini belum tentu dapat dikuantisasi dengan presisi yang baik oleh metode tersebut. Limit kuantitasi atau Limit of Quantitation (LOQ) yang disebut juga limit determinasi adalah konsentrasi terendah dari analat yang dapat ditentukan secara kuantitatif dengan presisi dan akurasi yang dapat diterima (Ermer dalam Ermer dan Miller, 2005).
Giese (2004) menyatakan bahwa terdapat dua cara untuk menentukan LOD dan LOQ, yaitu dengan menentukan kurva kalibrasi menggunakan sepuluh level konsentrasi, atau melakukan analisis blanko berulang. Tetapi ada masalah dalam pendekatan menggunakan blanko karena seringkali sulit diukur dan variasinya sangat tinggi. Lebih lanjut, nilai yang didapat dengan pendekatan seperti ini tidak bergantung dari analat (AOAC 2002).
Limit deteksi hanya berguna untuk mengontrol ketidakmurnian yang tidak diinginkan yang konsentrasinya harus tidak lebih dari level tertentu dan mengontrol kontaminan dengan konsentrasi rendah, sedangkan materi yang bermanfaat harus ada pada konsentrasi yang cukup tinggi agar dapat menjadi fungsional. Limit deteksi dan determinasi seringkali bergantung pada kemampuan instrumen (AOAC 2002).
2.4.5. Linieritas
Linearitas metode analisis menunjukkan kemampuan suatu metode untuk memperoleh hasil uji, yang baik langsung maupun dengan definisi transformasi matematis yang baik, proporsional
dengan konsentrasi analat dalam sampel pada range tertentu (Leyva et al 2008). Linieritas dapat diuji secara informal dengan membuat plot residual yang dihasilkan oleh regresi linier pada respon konsentrasi dalam satu seri kalibrasi (Thompson et al. 2002).
Linieritas harus dievaluasi dengan pemeriksaan visual terhadap plot absorbansi yang merupakan fungsi dari konsentrasi analat. Jika hubungannya linier, hasil uji dievaluasi lebih lanjut secara statistik dengan perhitungan garis regresi. Dalam penentuan linieritas, sebaiknya menggunakan minimum lima konsentrasi (EMA, 1995). Rentang penerimaan linieritas tergantung dari tujuan pengujian. Pada kondisi yang umum, nilai koefisien regresi (r2) ≥ 0,99.
2.5. Matriks Sampel
Suatu metode harus dapat menunjukkan rekoveri dan ripitabilitas yang dapat diterima pada konsentrasi dan matriks yang mewakili kelompok sampel dimana metode itu hendak diterapkan (AOAC 2002). Suatu metode yang hendak diterapkan pada “pangan” secara umum, metode tersebut perlu diujikan pada jenis pangan yang dianggap mewakili kelompok pangan secara umum. Sampel yang yang dianggap mewakili dapat dipilih berdasarkan skema segitiga atau
triangle scheme yang disarankan AOAC Internasional (Gambar 1) (Sullivan dan Carpenter 1993).
Skema segitiga ini berdasarkan kadar karbohidrat, protein dan lemaknya yang mana dianggap memiliki pengaruh terbesar terhadap kemampuan metode analisis. Suatu kelompok pangan, yang diwakili oleh segitiga kecil, dikatakan memiliki kadar yang “tinggi”, “sedang” dan “rendah” berdasarkan kadar karbohidrat, protein dan lemaknya. Pangan kompleks diposisikan pada salah satu segitiga kecil—menurut kadar karbohidrat, lemak dan proteinnya (dengan persentase yang telah dinormalisasi menurut perbandingan dari ketiga komponen). Pemetaan ini dilakukan dengan meniadakan persentase kadar air dan kadar abu. Tiap sudut segitiga merupakan kelompok pangan yang terdiri dari 100% lemak, 100%protein, dan 100% karbohidrat.
Gambar 1. Matriks pangan berdasarkan kadar protein, lemak dan karbohidrat (Nielsen 2010). Nielsen (2010) mengatakan bahwa kemampuan suatu metode analisis dipengaruhi oleh matriks pangan (misalnya komponen dari pangan tersebut terutama lemak, protein dan karbohidrat). Matriks pangan merupakan tantangan terbesar bagi para analis pangan. Makanan dengan kadar lemak tinggi dan kadar gula tinggi dapat menghasilkan interferensi yang berbeda dengan makanan dengan kadar lemak rendah dan kadar gula rendah. Prosedur digesti dan tahap ekstraksi sangat penting bagi hasil analisis yang akurat. Hal ini tergantung pada matriks pangan. Kompleksitas dari berbagai sistem pangan seringkali membutuhkan lebih dari satu teknik dan prosedur untuk komponen spesifik tertentu, termasuk pengetahuan mengenai teknik mana yang sesuai untuk matriks pangan yang spesifik.
Metode analitik yang umum harus dapat menganalisis kesembilan kombinasi yang ada, menggantikan metode yang spesifik pada matriks tertentu (matrix dependent method). Misalnya dengan menggunakan metode yang dipengaruhi oleh matriks, kita mungkin dapat menggunakannya untuk menganalisis bahan yang rendah protein, dengan karbohidrat dan lemak sedang seperti coklat dan keripik kentang. Tetapi untuk bahan dengan protein tinggi, lemak rendah dan karbohidrat tinggi seperti susu rendah lemak, harus digunakan metode analisis yang lain. Hal ini cukup merepotkan dan kemungkinan nilai yang didapat dari hasil analisis kedua metode perlu dievaluasi (Nielsen 2010).
Validasi metode memerlukan pengetahuan mengenai identitas dari sampel yang akan dianalisis, karena jika tidak, meski banyak informasi berguna yang didapat, tetapi informasi itu akan terombang-ambing bagaikan kapal di lautan yang luas, tidak mengetahui dimana keberadaannya, tanpa penanda yang menunjukkan posisinya (AOAC 2002). Oleh karena itu selain melakukan studi literatur dilakukan uji proksimat terhadap sampel yang akan dianalisis untuk
mengonfirmasi komposisi dari sampel. Berikut data mengenai sampel yang akan digunakan dalam perbandingan metode:
2.5.1. Kecap manis
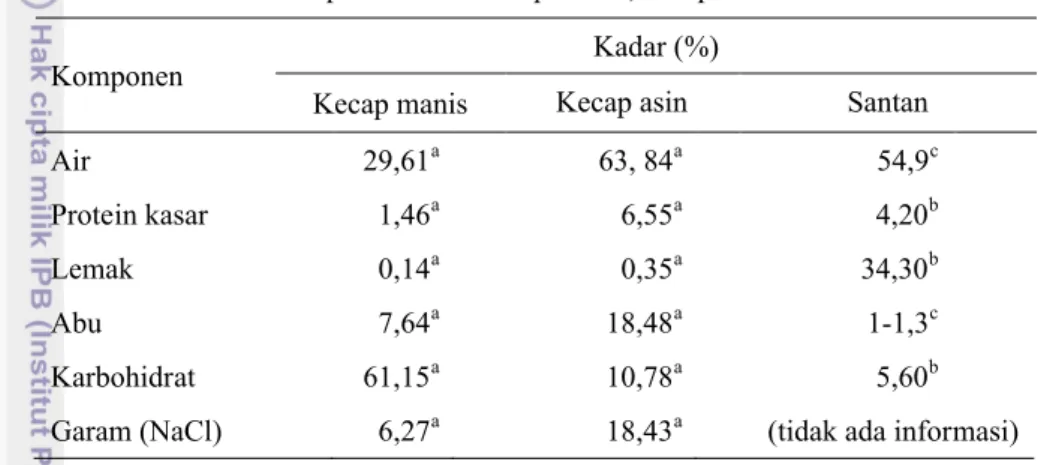
Kecap manis merupakan produk olahan kedelai, yang teksturnya kental dan berwarna coklat kehitaman (Suprapti 2005). Komposisi kimia kecap manis dapat dilihat pada Tabel 3.
Tabel 3 Komposisi kimia kecap manis, kecap asin dan santan
Komponen Kadar (%)
Kecap manis Kecap asin Santan
Air 29,61a 63, 84a 54,9c
Protein kasar 1,46a 6,55a 4,20b
Lemak 0,14a 0,35a 34,30b
Abu 7,64a 18,48a 1-1,3c
Karbohidrat 61,15a 10,78a 5,60b
Garam (NaCl) 6,27a 18,43a (tidak ada informasi) Sumber: aJudoamidjojo (1987) , bDirektorat Gizi (1967), c Woodroof (1979)
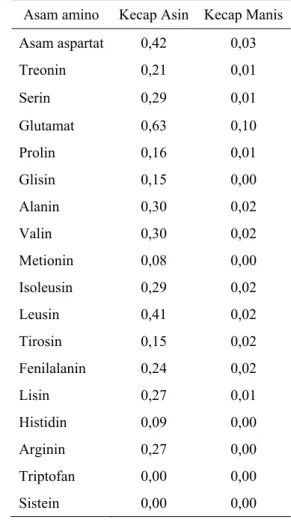
Kandungan gula dan viskositas yang tinggi dari produk ini disebabkan karena penambahan gula dalam proses pembuatannya. Komponen terbesar kecap manis adalah karbohidrat, terutama sukrosa, glukosa dan fruktosa (Kusumadewi, 2011). Kandungan gula kecap manis, yaitu 26-61%, lebih banyak dari kecap asin yang hanya 4-19% (Judoamidjojo 1987). Kandungan asam amino yang cukup tinggi dari kecap manis karena salah satu bahan yang digunakan untuk membuatnya adalah kedelai yang memiliki kandungan protein yang tinggi (Santoso 1994). Rincian jenis asam amino kecap manis dapat dilihat pada Tabel 4.
Dalam kecap manis, selain dari kedelai senyawa organik yang ada juga berasal dari gula merah. Senyawa organik dalam kecap manis adalah asam sitrat, tartarat, suksinat, laktat, format, piroglutamat, propionate dan butirat (Judoamidjojo et al 1985). Kecap yang bermutu tinggi berkadar garam 18%, gula minimal 40% dan pHnya berkisar antara 4,7-4,8 (Buckle et al 1988). Adapun persyaratan BSN untuk kecap manis (SNI 01-2543-1999) kadar garam minimal 3% dan total gula (dihitung sebagai sakarosa) minimal 40%.
Tabel 4. Kandungan asam amino kecap asin dan kecap manis (g/100g) Asam amino Kecap Asin Kecap Manis
Asam aspartat 0,42 0,03 Treonin 0,21 0,01 Serin 0,29 0,01 Glutamat 0,63 0,10 Prolin 0,16 0,01 Glisin 0,15 0,00 Alanin 0,30 0,02 Valin 0,30 0,02 Metionin 0,08 0,00 Isoleusin 0,29 0,02 Leusin 0,41 0,02 Tirosin 0,15 0,02 Fenilalanin 0,24 0,02 Lisin 0,27 0,01 Histidin 0,09 0,00 Arginin 0,27 0,00 Triptofan 0,00 0,00 Sistein 0,00 0,00 Sumber: Judoamidjojo et al (1985)
2.5.2. Kecap kedelai asin
Kecap kedelai asin atau yang biasa dikenal dengan nama kecap asin merupakan hasil fermentasi dari kedelai. Menurut definisi SNI 01-3543-994 kecap kedelai adalah produk cair yang diperoleh dari hasil fermentasi dan atau cara kimia (hidrolisis) kacang kedelai (Glycine max. L) dengan atau tanpa penambahan bahan makanan lain dan bahan tambahan makanan yang diizinkan. Warna dari kecap asin adalah coklat gelap. Tetapi warna ini bergantung pada proses penuaan atau
agingnya. Kecap asin mirip dengan kecap manis, hanya tanpa penambahan gula. Komposisi kimia
dari kecap kedelai dapat dilihat dari Tabel 3 dan kandungan asam aminonya dapat dilihat pada Tabel 4.
2.5.3. Santan
Berdasarkan SNI 01-3816-1995, santan adalah produk cair yang diperoleh dengan menyaring daging buah kelapa (Cocos nucifera) dengan atau tanpa penambahan bahan tambahan makanan yang diizinkan. Santan merupakan emulsi lemak dalam air (Kirk dan Otmer 1950) yang
distabilisasi secara alamiah oleh protein (globulin dan albumin) dan fosfolipida (Tangsuphoom dan Coupland, 2008). Senyawa δ-C8-laktone, δ-C10-laktone, dan n-oktanol merupakan komponen volatil utama dan memberikan karakteristik aroma pada santan kelapa (Lin dan Wilkens 2006),
Adapun komposisi kimia santan dapat dilihat di Tabel 3. Tetapi komposisi kimianya masih bervariasi tergantung pada varietas lokasi tumbuh, cara budi daya, kematangan buah, dan metode ekstraksi seperti jumlah penambahan air dan suhu ekstraksi. Menurut Seow dan Gwee (1997), komposisi kimia santan kelapa yang diekstraksi dengan tanpa penambahan air terdiri atas protein 2.6-4.4%; lemak 32-40%; air 50-54%; dan abu 1-1.5%.
2.5.4. Bahan Acuan
Semua metode instrumental membutuhkan bahan acuan, sekalipun untuk metode yang mengukur analat yang empiris. Analat yang empiris adalah analat yang nilainya tidak seperti senyawa kimia yang stoikiometris yang bersifat tetap. Analat empiris merupakan hasil dari penerapan prosedur yang biasa digunakan untuk mengukurnya, contohnya untuk kadar air, kadar abu, kadar lemak, kadar karbohidrat (by difference) dan kadar serat (AOAC 2002).
Bahan acuan memainkan peranan penting untuk mengetahui akurasi dalam melakukan validasi. Bahan acuan disini dapat diartikan sebagai bahan atau zat yang memiliki sifat-sifat tertentu yang cukup homogen dan stabil, yang telah ditetapkan untuk dapat digunakan dalam pengukuran atau dalam pengujian suatu contoh. Bahan acuan dapat digunakan untuk mengontrol presisi pengukuran walaupun bahan acuan tersebut tidak memiliki nilai acuan (assigned value), sedangkan untuk kalibrasi atau untuk mengontrol kebenaran pengukuran hanya bahan acuan yang memiliki nilai acuan yang dapat digunakan (Dara 2010). Kalibrasi dan pengontrolan analisis sangat penting, karena menyangkut kehandalan hasil pengujian. Untuk pengambilan keputusan yang krusial diperlukan hasil pengujian yang dapat dipercaya (Nuryatini 2010). Bahan acuan ini dapat diperoleh dari berbagai produsen bahan acuan seperti Puslit Kimia LIPI yang telah mengembangkan beberapa bahan acuan (in-house reference materials) khususnya untuk pengujian dalam bidang lingkungan dan pangan (Dara 2010).
Bahan acuan dapat dibagi menjadi dua yaitu Certified Reference Material (CRM) dan Standard Reference Material (SRM). CRM dapat ditelusur hingga standard internasional dengan ketidakpastian yang telah diketahui dan oleh karena itu dapat digunakan untuk mengukur semua aspek bias (bias metode, bias antarlab, and intralab) secara bersamaan, dengan asumsi bahwa tidak ada ketidaksesuaian matriks. Perlu dipastikan bahwa nilai ketidakpastian yang dimiliki cukup kecil sehingga dapat mendeteksi bias pada kisaran tertentu. Tetapi jika nilainya tidak cukup kecil,
penggunaan CRM masih dianjurkan, tetapi dengan disertai dengan pengujian tambahan. Jika diperlukan dan dapat dilakukan, sejumlah CRM yang sesuai dengan matriks dan konsentrasi analit sebaiknya diujikan (Thompson et al 2002).
SRM dapat digunakan jika tidak ada CRM. SRM adalah material yang telah dikarakterisasi dengan baik untuk tujuan validasi. Hal yang perlu diperhatikan adalah jika nilai bias tidak signifikan, hal ini bukan berarti merupakan bukti bahwa tidak adanya bias sama sekali. Akan tetap jika terdapat bias yang signifikan, hal ini menandakan perlunya investigasi lebih lanjut. SRM dapat berupa material yang telah dikarakterisasi oleh produsen CRM tetapi tidak dilengkapi dengan dokumen mengenai nilai ketidakpastiannya atau material yang telah terkualifikasi oleh sebuah manufakturer; materials yang dikarakterisasi dalam lab sebagai reference material; dan material yang didistribusikan dalam proficiency test. Meskipun ketertelusuran dari material tersebut dipertanyakan, jauh lebih baik untuk menggunakan material tersebut dibandingkan tidak melakukan pengukuran terhadap bias sama sekali. Material dapat digunakan dengan cara yang sama seperti CRM, sekalipun tidak ada nilai ketidakpastian yang tercantum, seluruh pengujian yang signifikan bergantung seluruhnya pada presisi yang dapat diamati dari hasil (Thompson et al 2002).