Harry is always available and is happy to help you, both inside and outside the lab. I would also like to thank Jay Winkler, who in many ways is the science engine of the laboratory.
Dodecaborate Clusters
Their work confirmed Longuet-Higgins' general qualitative description of chloro-dodecaborate and provided additional quantitative information, including a. Specifically, Wiersema and Middaugh demonstrated that the one-electron electrochemical oxidation of closo-dodecaborate results in the irreversible formation of [B24H23]3-.
![Figure 1.1. Crystallographic data reported by Lipscomb demonstrating the near-icosahedral symmetry of closo-[B 12 H 12 ] 2-](https://thumb-ap.123doks.com/thumbv2/123dok/10406532.0/22.918.175.778.124.756/figure-crystallographic-reported-lipscomb-demonstrating-icosahedral-symmetry-closo.webp)
Functionalization of closo -Dodecaborate
Synthetic method for perfunctionalized ether and ester derivatives of closo-[B12(OH)12]2- developed by Hawthorne and co-workers. An acetonitrile solution of closo-[B12(OH)12]2- and the relevant reactant is refluxed in the presence of a base, yielding the desired closo-[B12(OR)12]2-cluster.
![Figure 1.3. Synthetic method for perfunctionalized ether and ester derivatives of closo-[B 12 (OH) 12 ] 2- developed by Hawthorne and co-workers](https://thumb-ap.123doks.com/thumbv2/123dok/10406532.0/28.918.325.650.129.249/figure-synthetic-method-perfunctionalized-derivatives-developed-hawthorne-workers.webp)
Hypoelectronic Dodecaborates
Most of the reported hypercloso-B12(OR)12 clusters are indefinitely stable under ambient conditions, which is remarkable considering that even one-electron oxidation results in immediate decomposition of closo-[B12H12]2-. An important consequence of a1u depopulation is the appearance of a strong visible absorption band (~ 455 nm e > 2*104M-1cm-1).
Notably, hypercloso-B12(OCH2C6F5)12 is also able to photoinitiate polymerization of isobutene, which was previously not achieved in metal-free systems. Part of the work presented in this thesis aims to rationalize the photooxidative properties of hypercloso-B12(OR)12 clusters.28,37.
![Figure 1.4 Relationship between [B 12 (OR) 12 ] redox potentials and substituent Hammett constants](https://thumb-ap.123doks.com/thumbv2/123dok/10406532.0/32.918.161.813.181.849/figure-relationship-b-redox-potentials-substituent-hammett-constants.webp)
Concluding Remarks
Unpaired electron confined in icosahedral borane: synthesis and crystal structure of the blue, air-stable {[Closo-B12(CH3)12]·}– radical. Most of the results presented here have not yet been published and the discussion is sometimes speculative.
Background
Time-dependent density functional theory (TD-DFT) was used to explain the reactivity by comparing the LUMO energy of hypercloso-B12(OBz)12 and F600 with the HOMO energy of the styrene monomers. Label A indicates protons from the end of the olefinic chain; B/C, allylic protons of the chain end; D, methine protons.
Preliminary Photophysical Studies
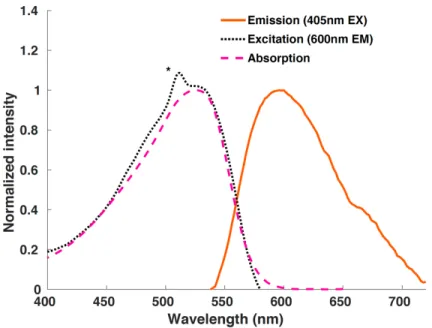
Notably, comparison of the three clusters indicated that increased fluorine content was correlated to both stronger preference for the purple state and more intense fluorescence. We wonder what the nature of the purple species was and whether it could be responsible for the observed fluorescence spectra.
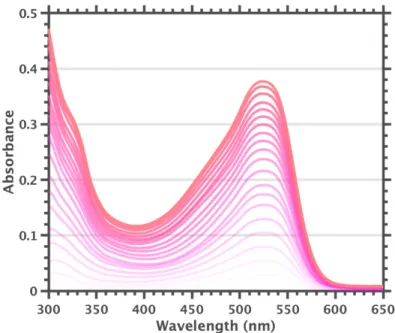
Comparison of the UV-visible absorption spectrum of the purple F60 samples showed an uncanny similarity to the published spectrum of [F60]1-. After electrochemical characterization, we used spectroelectrochemistry to show that the published extinction coefficient of the [F60]1-525 nm band was significantly underestimated, and that the actual spectrum matched well with the purple species we had during our preliminary work perceived.

Interactions with Haloarenes
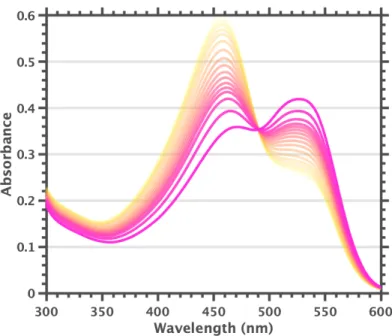
F840 samples show a prominent 525 nm band even in neat 1,2-difluorobenzene and addition of a speck of 1,2-diiodo-tetrafluorobenzene leads to complete conversion to [F84]1-. However, the strong 525 nm band and almost complete 455 nm bleach are the result of slow increase of the F600.

Solutions of F600 in 2-MeTHF, which appear to undergo complete conversion to [F60]1-, provide further support for assigning the 600 nm fluorescence band to [F60]1-. Consistent with room temperature data, F600 in toluene glass showed fluorescence centered around ~600 nm, likely originating from trace [F60]1-. Dimerization mechanisms similar to what we suggested for F600 in 2-MeTHF (section 2.5) may also play a role, but seem less likely because the 77 K emission spectrum of F600 in 2-MeTHF is straightforward.
Conclusions
The room temperature emission studies presented in this chapter show that the B120 excited state rapidly returns to the ground state through non-radiative decay. The group-solvent interaction studies presented above, along with computational work by Král and co-workers (Appendix A), support the pre-association of clusters and monomers. Although many questions remain, the work presented here provided sufficient insight to guide us toward the computational and mechanistic studies presented in Appendix A.
Experimental Details
A 1 vol% solution of trifluoroacetic acid was added to the sample in increments of 25 µl until a total of 500 µl was added. The sample solution was degassed by three 15-min freeze-pump-thaw cycles, after which the solution was transferred to the cuvette compartment. All steps after compound addition to the sample preparation vessel were performed under argon/vacuum using standard air-free techniques.
Introduction
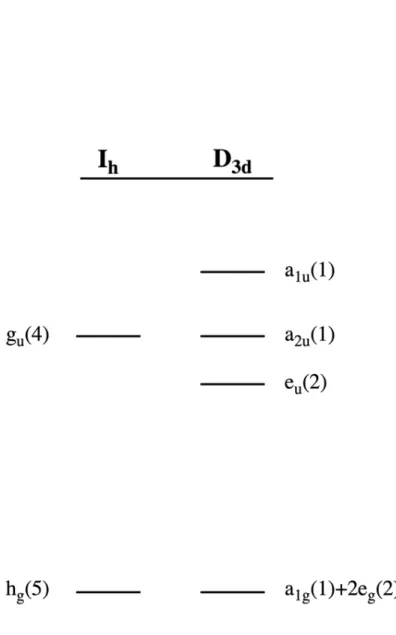
Breaking the gu degeneracy yields a non-degenerate a1u singly occupied molecular orbital (SOMO) in the case of hypocloso-[B12]1- or lowest unoccupied molecular orbital (LUMO) in the case of hypercloso-B120. Hypoelectronic states are stabilized by a structural distortion, which lowers the overall energy of the group. Increasing the partially filled or unfilled a1u and decreasing the filled eu is energetically favorable in hypoelectronic clusters.
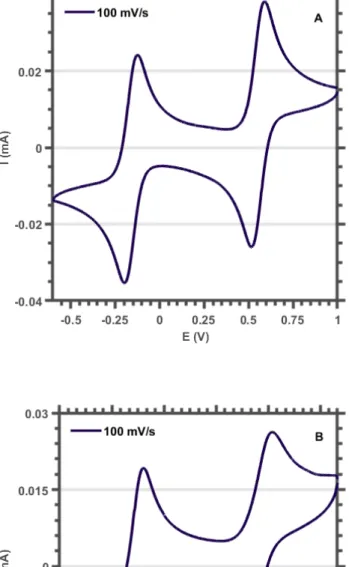
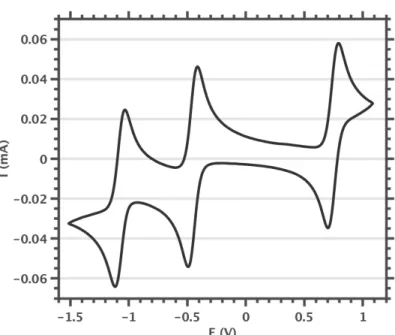
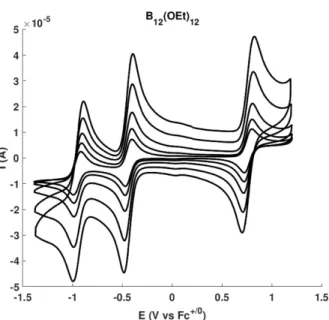
The reversibility of the waves was not negatively affected by the presence of water (see section 3.5). Linear relationship between peak currents and scan speed. ipox and ipred are the oxidative and reductive peak currents. E1/2 is the average of the reductive and oxidative peak potentials. ipox and ipred are the peak currents observed at the absolute maximum of the oxidative and reductive waves, respectively.
The absence of the strong absorption bands characteristic of Magic Blue further confirmed that the oxidant had been consumed. Solutions of Magic Blue in THF without 40 last for hours without appreciable consumption of the oxidant, implying that [4]1+ plays a key role. In the case of Magic Blue, in addition to solvent oxidation, there is evidence that chemical transformations involving Magic Blue or the tris(4-bromopheyl)amine product can occur after formation of [4]1+.
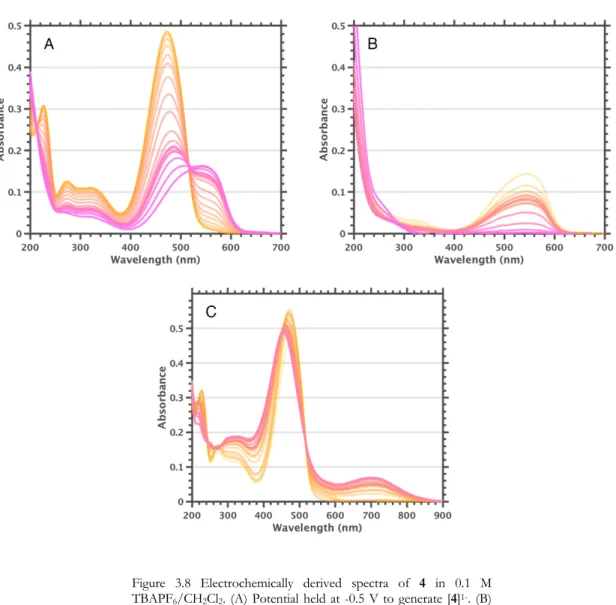
Conclusions
Experimental Details Compounds Compounds
A portable Pine WaveNow potentiostat was used to provide applied bias, while a Varian Cary 50 Bio UV-visible spectrometer was used to collect spectra. Reactivity was monitored by visual observations and UV-visible spectra collected on a Varian Cary 50 Bio UV-visible spectrometer. Samples were prepared in Schlenk cuvettes to facilitate collection of UV-visible spectra without compromising the samples.
The structural significance of the number of skeletal electron pairs in carboranes, the higher boranes and borane ions, and various transition metal carbonyl cluster compounds.
ELECTRONIC STRUCTURES OF PERFUNCTIONALIZED DODECABORATE CLUSTERS
Background
Oxidation yields an unstable open-shell species and leads to dissolution of the cluster.3 However, some perfunctionalized dodecaborate clusters bearing electron-donating groups are able to accommodate hypoelectronic states. Increasing the unpopulated SOMO(a1u) (singly occupied molecular orbital; in [B12]1-) or LUMO(a1u) (lowest unoccupied molecular orbital; in B120 and [B12]1+) and decreasing the fully populated HOMO -1(eu) set (HOMO-1 refers to the second lowest energy occupied molecular orbital) reduces the overall energy of the cluster. We decided to investigate paramagnetic hypocloso-[B12(OR)12]1- and recently discovered [B12(OR)12]1+ clusters using pulsed EPR techniques, hoping to verify and verify the delocalized description of the frontier orbitals to show that the spin density is limited to the core of the cluster.
This description has gained support from DFT calculations, although the details of the computational results vary considerably between different authors. The very weak 19F hyperfine coupling, which likely derives from through-space interactions, further substantiates a constrained description of the cluster core. In this chapter, we have restricted our quantitative calculations to the s-orbital spin density, which can be easily obtained from the isotropic component of the hyperfine tensor.
![Figure 4.1. Q-band ESE-EPR spectra of [1-3] 1- collected at 5 K and 34.11 GHz microwave frequency](https://thumb-ap.123doks.com/thumbv2/123dok/10406532.0/119.918.314.681.397.739/figure-band-ese-epr-spectra-collected-microwave-frequency.webp)
The ENDOR spectra at the center of [4-5]1+ are similar to spectra collected for the [1-3]1 anions, suggesting that the distribution of the SOMO(a2u) is similar to the SOMO(a1u) of the anions. . This is further supported by the ENDOR field dependency and the additional functions in the HYSCORE of [4]1+. This could be due to inherent differences in the orbital geometry between aryl-functionalized and alkyl-functionalized clusters, but a more likely cause is increased distortion in the cluster core of the more oxidized species.
![Figure 4.5. (Top) ESE-EPR spectra and (Bottom) pseudomodulated ESE-EPR spectra for [4-5] 1+ collected 32 K and 34.11 GHz ([5] 1+ ) or 34.13 GHz ([4] 1+ ) microwave frequency](https://thumb-ap.123doks.com/thumbv2/123dok/10406532.0/125.918.307.681.140.484/figure-ese-spectra-pseudomodulated-spectra-collected-microwave-frequency.webp)
Analogous considerations can be used to show that the ∼525 nm ES3 in open-shell hypocloso- [B12(OR)12]1- clusters correspond to a 2Eg¬2A1u transition. It is interesting to compare this value with the ES1 energy, as both provide indications of the energy splitting between the LUMO(a1u), SOMO(a2u), and HOMO-1(eu) orbitals. ES1 is the lowest energy excited state, ES2 the second lowest energy excited state, and ES3 the third lowest energy excited state (regardless of triplets).
![Figure 4.12. UV–vis–NIR spectra of TBA[1] and TBA[2] collected in dichloromethane-d 2](https://thumb-ap.123doks.com/thumbv2/123dok/10406532.0/133.918.272.671.139.474/figure-uv-nir-spectra-tba-tba-collected-dichloromethane.webp)
Conclusions
Experimental Details Compounds Compounds
Descriptions of the ESE-EPR, Davies ENDOR, Mims ENDOR and HYSCORE experiments can be found in the literature.20–27. The s-orbital spin density can be easily calculated by dividing aiso by the calculated isotropic hyperfine interaction for unit spin density (aiso0).28 The anisotropic part of the hyperfine tensor represents contributions from non-spherical orbitals (p, d, f). Orientationally selected two-dimensional ESEEM spectroscopy of the Rieske-type iron-sulfur cluster in 2,4,5-trichlorophenoxyacetate monooxygenase from Burkholderia Cepacia AC1100.
![Figure 4.15. Field dependent Davies ENDOR spectra of [1] 1- 1-collected at 5 K and 34.11 GHz microwave frequency](https://thumb-ap.123doks.com/thumbv2/123dok/10406532.0/142.918.322.667.126.473/figure-field-dependent-davies-spectra-collected-microwave-frequency.webp)
FINISHING REMARKS
Current State of Affairs
Interest in hypoelectronic dodecaborate clusters has grown markedly over the past decade. Although icosahedral dodecaborate clusters were first isolated about 70 years ago, 1 no hypoelectronic analogues were reported until the late 1990s, when the Hawthorne laboratory first isolated hypochlose-[B12Me12]1-(Me = CH3),2hypoxlose-[B12(OBz )12] 1- and hypercloso-B12(OBz)12. Bz = CH2C6H5).3 Development in the years following these initial reports was limited to the chemical isolation and electrochemical observation of additional hypoelectronic analogues, including a series of [B12(OR)12] clusters4 and perhalogenated hypochlorous-[B12X12]1- ( X = F , Cl, Br, I) analogues.5,6 A major breakthrough occurred when the Spokoyny laboratory published a more convenient route for the microwave-assisted synthesis of hypercloso-B12(OR)12,7 which was quickly followed by publications describing a number of interesting applications . It took us about two years to get a good handle on clusters in the specific settings we were interested in.
Electronic Structure and Super-oxidized Clusters
Given that the [B12]1+ ENDOR spectrum and magnitude of the 10/11B strong hyperfine tensor closely resemble [B12]1-, we are confident that we also see the symmetric a2u orbital as highly delocalized. 3,12,13 Considering that the redox potential of the clusters strongly depends on the identity of the R-group, this seems reasonable.4,7 Interestingly, however, the B-B bonding frontier orbitals have very little density per R-group. Our interpretation is that electron donation to the B12 core involves mainly oxygen lone pairs and that the role of the R-groups is primarily to modulate how tightly the oxygen holds its electrons.
Future Directions
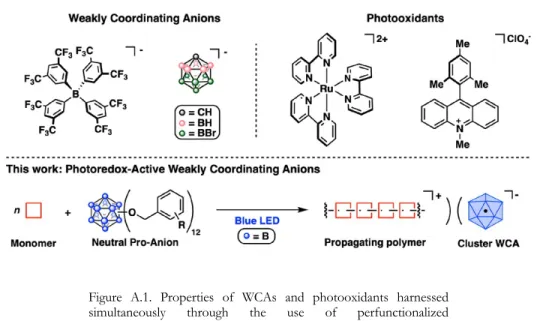
It seems unlikely that further oxidation to [B12]3+ (and beyond) can be achieved without compromising the integrity of the B12 pseudoicosahedron. I hope that our observations will provide guidance to future researchers who wish to conduct more in-depth research into the areas discussed here. This also suggests that 1−3 behave as weakly coordinating anions (WCA) upon single-electron reduction, as no incorporation of the cluster-based photoinitiators is observed in the analyzed polymeric products.
Introduction
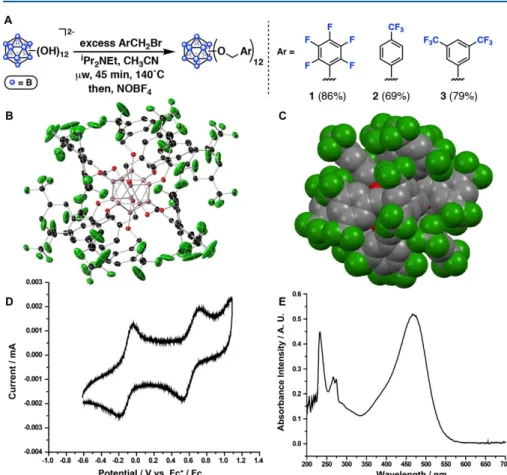
Perfunctionalized dodecaborane clusters of the type B12(OCH2Ar)12 (1; Ar = Ph, C6F5) initiate the polymerization of a number of styrenes as well as of isobutene.11 We therefore wondered whether species of this type could break new chemical space in the context of WCA chemistry.12 In general, the use of icosahedral boron clusters, typically the monoanionic carba-closo-undecaborate, [CB11H11]-, and its functionalized derivatives as WCAs is well known,13 and they continue to be widely used. effect, as for example shown in recent revelations by Nelson and co-workers.14 While B12(OCH2Ar)12 species can be easily isolated as charge neutral species,. Properties of WCAs and photooxidants exploited simultaneously through the use of perfunctionalized photoredox active boron cluster proanions. Here we provide evidence for the WCA behavior of [B12(OCH2Ar)12]-, photooxidatively generated by the reaction of 1-3 with a series of styrenes under blue light-.
Synthesis and Self-exchange Studies
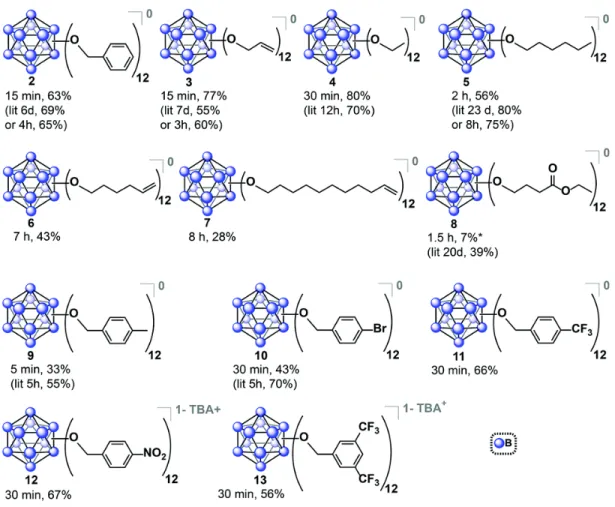
This study opens the door to previously unexplored photoredox-active WCAs. A) Synthetic protocol to generate neutral B12(OCH2Ar)12. Our ability to cleanly isolate [3]− and 3 by treating [3]2−/− with FeCl3 and NOBF4, respectively, prompted a solution-phase electron self-exchange study using 19F NMR spectroscopy to probe the kinetics of ET. At temperatures varying from 20 to 60 °C in 1,2-dichloroethane, we have recently shown that electron self-exchange between [NBu4][3] and 3 is slow on the NMR time scale, with kex < M−1 s −1.16 Self-exchange of electrons is known to occur between the 0/1− redox couple in Ru3O(OAc)6(CO)(L)2 groups [where L = 4-cyanopyridine, pyridine or 4-(dimethylamino)pyridine] strongly depends on the degree of charge density located on the auxiliary pyridyl ligands.17 Considering the burdensome steric profile of the pseudosphere 1-3 and taking into account that the highest occupied molecular orbital (HOMO) [3]- and the lowest unoccupied molecular orbital (LUMO) are of the 3 ground state are based exclusively on the boron cage,16 we attribute this low ET to poor electronic coupling between 3 and [3]− in the ground state.
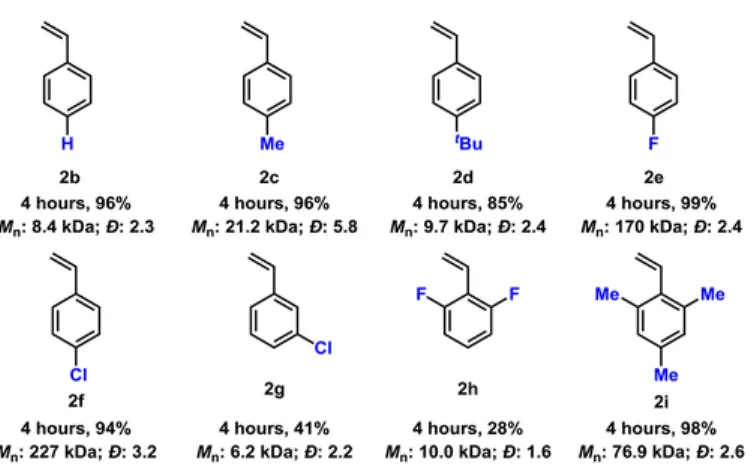
Styrene Polymerization
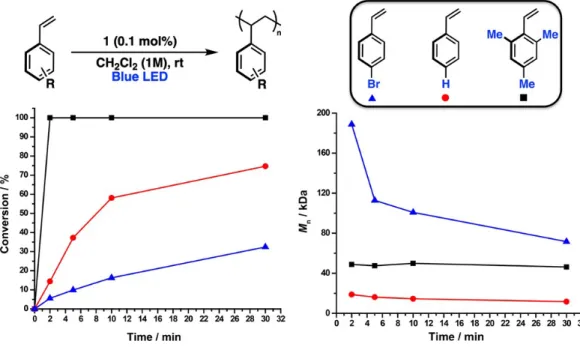
The bar graph shows the Mn values obtained for all polymerizations and reveals the unusually high Mn values obtained for S2−S4 in CH2Cl2 with 1. In general, we find that moderate Mn values are obtained in almost all cases , consistent with an uncontrolled cationic polymerization involving a countermovement that does not. We suggest that the unique distribution of molecular weights of polymers based on the solvent, initiator and monomer indicates both the weakly interacting behavior of the dodecaborate anions with the cationic chain end and an easily influenced equilibrium (vide infra) of these interactions by contact ion pairs to more efficiently solvated ion pairs; the location of this equilibrium ultimately determines the resulting Mn values.

Computational Analysis of Photoexcitation
In addition, the HOMO of the system is localized on styrene, again suggesting that ET from styrene after photoexcitation of the cluster is mechanistically reasonable. Interestingly, when modeling in CH3CN, both the HOMO and LUMO of the system are mostly located in the core of the cluster. Based on the solid crystal structure of 3 , the closest B−C or O−C intermolecular distances (analogous to initiator–substrate distances) are well within 15 Å.

Discussion of Photoinitiated Polymerization Studies
This suggests that the relationship between propagation rate and chain transfer rate for S4 is significantly different from that for S1 and S5 and that this relationship reaches a steady state much later in the polymerization. In the carbocation polymerization of styrene with SnCl4, Overman and Newton observed that the addition of durene or hexamethylbenzene caused an increase in the Mn of the resulting polystyrene. We found that the addition of durene (1 equivalent relative to 1) to the polymerization of S1 by 1 under standard conditions only slightly increased the molecular weight of the resulting polymer (14 vs. 8 kDa on average), but did not significantly change the yield of the resulting polystyrene compared to polymerization of styrene alone with 1.
Conclusion
Taming the cationic beast: new developments in the synthesis and application of weakly coordinating anions. Silver-free activation of bound gold(I) chlorides: use of [Me3NB12Cl11] as a weakly coordinated anion in homogeneous gold catalysis. Structure Effects of Lewis acids on the living cationic polymerization of p-methoxystyrene: a marked difference in polymerization behavior from vinyl ethers.
![Figure 1.2. Electronic structure of closo-[B 12 H 12 ] 2- calculated by Bambakidis and Wagner](https://thumb-ap.123doks.com/thumbv2/123dok/10406532.0/23.918.181.813.131.905/figure-electronic-structure-closo-b-calculated-bambakidis-wagner.webp)