저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게
l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다:
l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다.
l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다.
저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다.
Disclaimer
저작자표시. 귀하는 원저작자를 표시하여야 합니다.
비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다.
변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
이학석사 학위논문
KOt-Bu-mediated
transition-metal-free indole synthesis
전이금속 촉매를 배제하고
포타슘 터트-부톡사이드를 사용한 인돌 합성법
2020년 2월
서울대학교 대학원 화학부 유기화학 전공
김정윤
KOt-Bu-mediated
transition-metal-free indole synthesis
Supervisor: Prof. Hong Geun Lee
By
Jeongyun Kim
A Thesis for M.S. Degree in Organic Chemistry
2020
Department of Chemistry
The Graduate School
Seoul National University
Contents
Abstract
Introduction 1
Results and Discussion 7
Conclusion 22
Experimental Section 23
References 44
Abstract
KOt-Bu-mediated transition-metal-free indole synthesis
Jeongyun Kim Organic Chemistry Department of Chemistry The Graduate School Seoul National University
A single-electron transfer (SET)-mediated modular indole formation reaction from a 2-iodoaniline and a ketone has been developed. This transition metal- free reaction shows a broad substrate scope and unconventional regioselectivity trends. Moreover, important functional groups for further transformation are tolerated under the reaction conditions. Density functional theory studies reveal that the facile product formation and regiocontrol are governed by the internal metal coordination and torsional strain of the radical intermediates.
Keywords: Indole, radical, transition-metal-free, regioselectivity, DFT, electrocatalysis
Student Number: 2018-25699
1
1. INTRODUCTION
Since the structure of proposal by Adolf von Baeyer in 18691, indole has become a vital structure in diverse research areas, such as drug discovery and material science.2-3 For instance, indole and its derivatives exhibit various interesting biological properties and serve as a privileged structure of motif in medicinal chemistry.4-6 On the material science side, indole-based bipolar host materials were also designed and synthesized for organic light-emitting diode (OLED).7
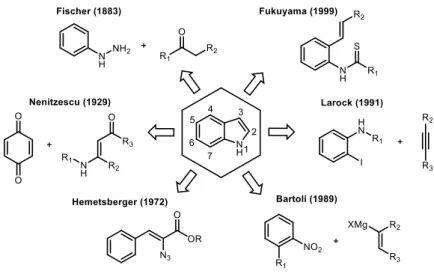
Due to these interesting functions, numerous synthetic protocols have been developed aiming at efficient and practical methods for the preparation of indole scaffolds over the past few decades.8-10 Representative methods include Fischer11-12, Nenitzescu13, Hemetsberger14, Bartoli15, Larock16, and Fukuyama17 indole syntheses, which are still widely used for the preparation of the core structure of diverse complex molecules (Figure 1).
Figure 1. Classic methods for the preparation of the indole
2
Nevertheless, research on the development of a new methodology is still underway to overcome certain limitations of existing methods, such as the issue of regioselectivity and functional group tolerance. Most methods reported in recent years are using transition-metal catalysts.18 However, these reactions have suffered from problems originating from (a) the toxicity and the expense of transition-metal catalysts, and (b) complications associated with handling moisture and air-sensitive materials, and (c) most importantly not tolerated C(sp2) –halogen bond which can be used as a functional handles for further functionalization via cross-coupling reaction.
19 Therefore, the demand for developing transition-metal-free reaction is continuously growing.
Among various reactivities not containing transition-metal catalysts, a radical- based reactivity has drawn our attention due to the wide range of functional group compatibility and green chemistry. In 2008, Itami and co-workers reported a KOt- Bu-promoted biaryl coupling between heterocycles and iodoarene. It was proposed reaction through aryl radical species (Scheme 1.1, a).20 In 2010, it was proved experimentally that the aryl radical species were generated from aryl halides.21 In this paper, the coupling of aryl halides with benzene derivatives mediated by KOt- Bu with the aid of a catalytic amount of 1,10-phenanthroline (Scheme 1.1, b). Of importance, the authors have utilized the 1,10-phenanthroline based “promoters” for the first time. Highly conjugated phenanthroline derivatives act as a single electron transfer (SET) mediator that provides an electron to the aryl halide to generate a radical anion, which subsequently undergoes the formation of a reactive aryl radical species together with a halide anion. After the establishment of mechanism report
3
associated with electron transfer from the complex A, intermolecular C–C bond formation reactions have been reported by Shirakawa and Hayashi22, Kwong and Lei23 (Scheme 1.1, c). Subsequently, the Shi group reported an intramolecular reaction that produces benzofuran derivatives (Scheme 1.1, d). The initially formed aryl radical undergoes 5-exo-trig cyclization. In this case, the phenyl group on the end of the alkyl chain is essential for the stabilization of the generated radical. The reactivity could be extended to intermolecular C-C bond formation. In 2015, Ollevier reported an -arylation of enolizable aryl ketone using KOt-Bu in DMF and mechanistic evidence for a radical process (Scheme 1.1, e).24 It was postulated that the solvent acts as the initiator of the overall process. The tert-butoxide anion abstracts a proton from DMF, generating carbamoyl anion, which can transfer an electron to the aryl halide. From this paper, the author has demonstrated that the radical addition is possible to alkene but also to enolate derivatives.
4
Scheme 1.1. KOt-Bu-mediated radical addition reactions
5
Compare to other strategies to generate aryl radical from aryl halide, the described approach has advantages of using mild reagent, affording various functional group tolerance. To validate the possibility of applying the reactivity to the preparation of indoles, we designed the radical addition reaction that was attempted with N-allyl-2- iodoaniline (Scheme 1.2). The substrate, which was prepared from the simple allylation of 2-iodoanilne in 90% yield underwent smooth 5-endo-trig radical cyclization in the presence of KOt-Bu and a catalytic amount of 1,10-phenanthroline promoter.25 Albeit the isolated yield was modest (15%), the experiment has demonstrated that the KOt-Bu promoted radical formation would apply to the synthesis of indole derivatives.
Scheme 1.2. Preliminary result
Based on these observations, we have postulated that an iodoaniline derivative can undergo a radical based C–C bond formation with enolates derived from ketone. In combination with a condensation event, the protocol ultimately provides the indole scaffolds (Scheme 1.3). Depending on the substituents, the modular approach should provide the indole structures in a highly convergent and programmable way.26
6
Scheme 1.3. Synthetic strategy
7
2. RESULTS AND DISCUSSION
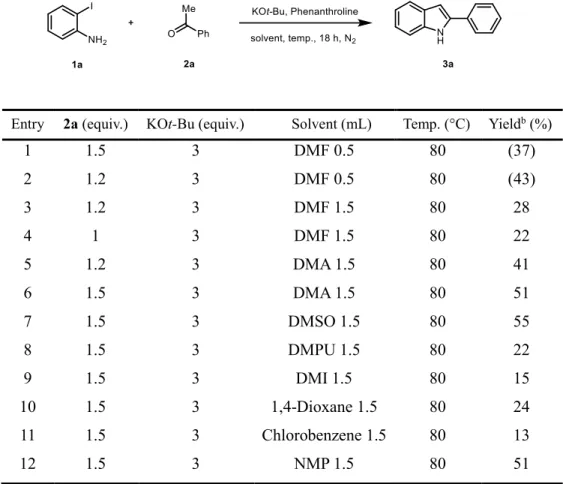
Initial optimization was carried out with 2-iodoaniline and acetophenone in the presence of KOt-Bu and 1,10-phenanthroline (Table 2.1). The desired 2- pheynlindole was isolated in 37% yield when the reaction was set up in 0.5 mL of DMF at 80 °C in 18 h (entry 1). Initially, the effect of ketone counterpart stoichiometry was evaluated (entries 1-2). It was hypothesized that the lower yield of entry 1 is originating from an insufficient amount of solvents for stirring.
Therefore, the reaction was performed with a sufficient amount of solvents and decrease the amount of ketone results lowering the yield (entries 3-4). The reaction was also performed under dimethylacetamide (DMA) solvent conditions with both 1.2 and 1.5 equivalent of ketones (entries 5-6). As a result, 1.5 equivalent of ketones were chosen as initially optimized stoichiometry. Next, a series of solvents, including dimethyl sulfoxide (DMSO), 1,3-dimethyl-3,4,5,6-tetrahydro-2-pyrimidinone (DMPU), 1,3-dimethyl-2-imidazolidinone (DMI), 1,4-dioxane, chlorobenzene, and N-methyl-2-pyrrolidone (NMP) were screened to improve the yield. In general, polar solvents such as DMSO and NMP were more effective than others (entries 6-12).
DMSO gave the best yield (55%), and further screening was conducted using DMSO as a solvent (entry 7).
8
Table 2.1. Optimization of the Reaction Conditions: Solvent Evaluation
aEntry 2a (equiv.) KOt-Bu (equiv.) Solvent (mL) Temp. (°C) Yieldb (%)
1 1.5 3 DMF 0.5 80 (37)
2 1.2 3 DMF 0.5 80 (43)
3 1.2 3 DMF 1.5 80 28
4 1 3 DMF 1.5 80 22
5 1.2 3 DMA 1.5 80 41
6 1.5 3 DMA 1.5 80 51
7 1.5 3 DMSO 1.5 80 55
8 1.5 3 DMPU 1.5 80 22
9 1.5 3 DMI 1.5 80 15
10 1.5 3 1,4-Dioxane 1.5 80 24
11 1.5 3 Chlorobenzene 1.5 80 13
12 1.5 3 NMP 1.5 80 51
aReaction was performed on a 0.5 mmol scale of 2-iodoaniline in the presence of phenanthroline 40 mol% and 3 equiv. KOt-Bu under N2 gas at 80 °C for 18 h. bThe yields of product was determined by gas chromatography with n-dodecane as an internal standard. Yields in parenthesis are isolated yield.
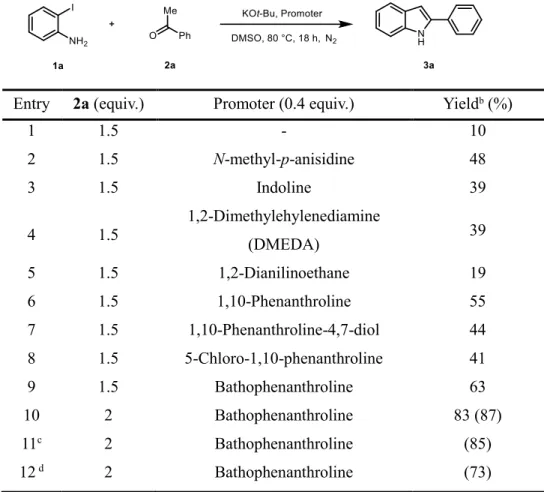
Next, the effect of promoters has been assessed (Table 2.2). Without a promoter, the reaction exhibited a significantly lower yield of efficiency (entry 1). Several of the reported promoters were proven to be inefficient for the desired transformation (entries 2-5). On the other hand, the use of 1,10-phenanthroline as a promoter provides 55% yield (entry 6). Further investigation associated with using 1,10-
9
phenanthroline derivatives has proven that the bathophenanthroline exhibited the highest reactivity (entries 7-9).
Table 2.2. Optimization of the Reaction Conditions: Promoter screening
aEntry 2a (equiv.) Promoter (0.4 equiv.) Yieldb (%)
1 1.5 - 10
2 1.5 N-methyl-p-anisidine 48
3 1.5 Indoline 39
4 1.5
1,2-Dimethylehylenediamine
(DMEDA) 39
5 1.5 1,2-Dianilinoethane 19
6 1.5 1,10-Phenanthroline 55
7 1.5 1,10-Phenanthroline-4,7-diol 44
8 1.5 5-Chloro-1,10-phenanthroline 41
9 1.5 Bathophenanthroline 63
10 2 Bathophenanthroline 83 (87)
11c 2 Bathophenanthroline (85)
12 d 2 Bathophenanthroline (73)
aReaction was performed on a 0.5 mmol scale of 2-iodoaniline in the presence of 3 equiv. KOt-Bu in 1.5 mL DMSO at 80 °C for 18 h under N2 gas. bThe yields of product was determined by gas chromatography with n-dodecane as an internal standard. Yields in parenthesis are isolated yield.
c20 mol% of bathophen was used. d10 mol% of bathophen was used.
10
The reaction could be further improved by utilizing 2 equivalent of ketone counterpart (entry 10). Finally, the effect of promoter loading was evaluated. There was no significant difference in yield when the amount of promoter was reduced from 40 mol% to 20 mol%, while distinctively lower yield was observed with 10 mol% of the promoter (entries 10-12). Therefore, further screening was carried out with 20 mol% of the promoter.
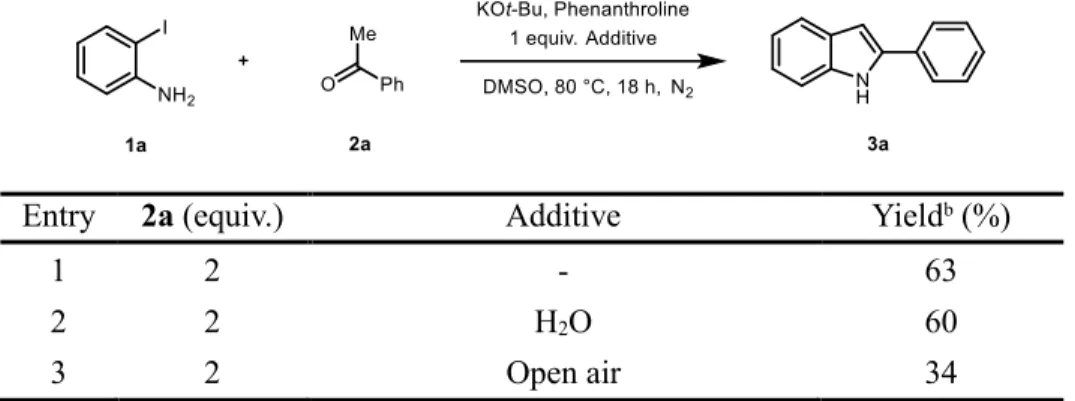
Additional experiments were conducted to determine the robustness of this reaction (Table 2.3). The addition of 1 equivalent of water did not significantly affect the yield of the reaction (entry 2). The moderate yield indicates that this reaction is insensitive to moisture. On the other hand, when the reaction was carried out under open-air conditions, the significantly lower yield was observed (entry 3), suggesting that the molecular oxygen can be detrimental to the reaction.
Table 2.3. Optimization of the Reaction Conditions: Additive Effect
aEntry 2a (equiv.) Additive Yieldb (%)
1 2 - 63
2 2 H2O 60
3 2 Open air 34
aReaction was performed on a 0.5 mmol scale of 2-iodoaniline in the presence of phenanthroline 20 mol% and 3 equiv. KOt-Bu in 1.5 mL DMSO at 80 °C for 18 h under N2 gas. bThe yields of product was determined by gas chromatography with n-dodecane as an internal standard. Yields in parenthesis are isolated yield.
Finally, time and temperature dependence experiments were examined to find optimal reaction conditions (Table 2.4). To evaluate the reaction temperature, the
11
reaction was set up at varying temperature ranging from 40°C to 80 °C (entries 1-4).
The significantly low yields were observed below 50 °C. Next, checked for time dependence (entries 5-11). Over time, the gradual increasing yield was observed. It was confirmed that the maximum yield was reached within 8 h. Thus, the optimum conditions were established from these results as follows: 0.5 mmol 2-iodoaniline, 2 equivalents acetophenone, 3 equivalents KOt-Bu, 20 mol% bathophenanthroline, and 1.5 mL DMSO at 60 °C for 8 h.
Table 2.4. Optimization of the Reaction Conditions: Temperature and Time Dependence
aEntry Temp.(°C) Time Yieldb (%)
1 80 18 h 87
2 60 18 h 83
3 50 18 h 70
4 40 18 h -
5 60 1.5 h 58
6 60 3 h 70
7 60 4.5 h 74
8 60 6 h 80
9 60 7 h 81
10 60 8 h 83
11 60 18 h 83
aReaction was performed on a 0.5 mmol scale of 2-iodoaniline in the presence of bathophen 20 mol%
and 3 equiv. KOt-Bu in 1.5 mL DMSO under N2 gas. bThe yields of product was determined by gas chromatography with n-dodecane as an internal standard..
12
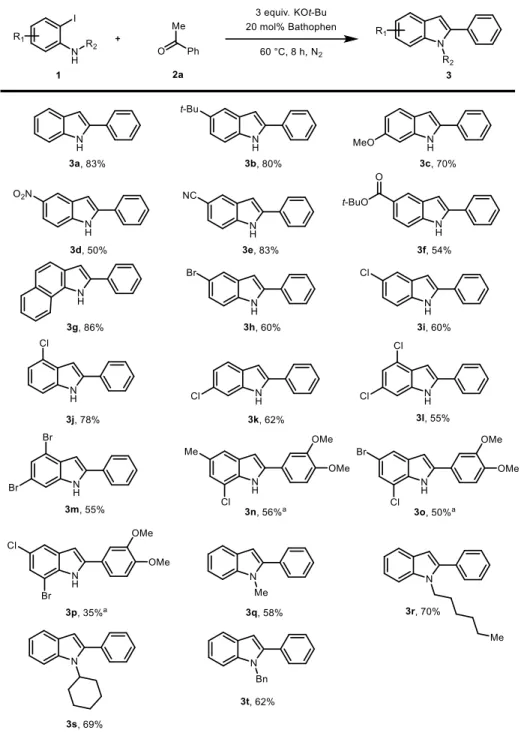
With the optimized reaction conditions in hand, we evaluated generality of the reaction with substituted 2-iodoanilines (Table 2.5). A wide range of electron- donating and electron-withdrawing substituents, including alkyl (3b), methoxy (3c), nitro (3d), cyano (3e), and alkoxy carbonyl (3f) groups afforded the corresponding indole products in moderate to good yield. Starting from 2-iodonaphthalen-1-amine, indole derivatives with an extended -system could also be accessed (3g).
Gratifyingly, the reaction also tolerated halogen substituent to give the desired indole product in synthetically useful yields (3h). Of importance, halogen atom could be appended all 4 positions of 6-membered ring of indole allowing for further modification through transition-metal-catalyzed cross-coupling (3i-k). It is noteworthy that the selective installation of a substituent at the 4-position of indole, which is difficult to realize in a single operation from readily accessible reaction partners, could be achieved with good yield (3j). In addition, substrates possessing two C–Cl or C–Br bonds could be successfully employed to provide the corresponding indole products with multiple handles for further elaboration (3l-m).
In the case of some substrates, the reaction was performed with 1-(3,4- dimethoxyphenyl)ethane-1-one due to the difficulty of purification (3n-p). However, relatively low yield of the reaction with 2-bromo-4-chloro-6-iodobenzene probably because of the steric hindrance of bromo substituent (3p). In addition, we investigated the reaction with N-substituted iodoanilines. Desired N-substituted indoles with aliphatic chain, secondary alkyl, and benzyl group were isolated in moderate to good yields (3q-3t).
13
Table 2.5. Scope with 2-iodoaniline derivatives
14
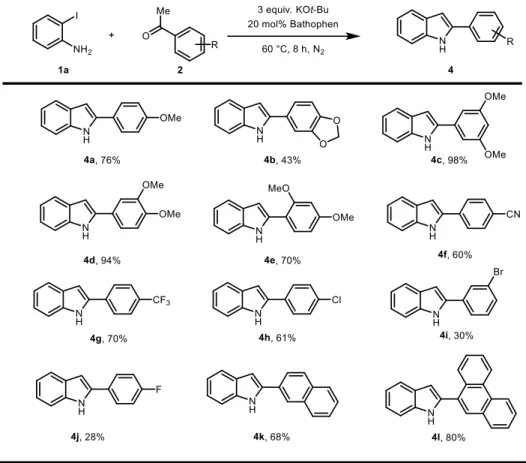
Next, the scope of the ketone counterpart was assessed by using various methyl ketones to afford 2-substituted indole derivatives (Table 2.6). The reaction using acetophenone with an electron-donating substituent on the para position proceeded smoothly (4a). The substrate with a dioxole acetal form was tolerated, while the yield was modest (4b). Dimethoxy substituted acetophenones were tested to confirm whether a steric or electronic effect was present (4c-e).
Table 2.6. Scope with acetophenone derivatives
The reaction with acetophenones that containing electron-withdrawing groups also gives desired products and again halogen atom could be retain (4f-j). Starting
15
from 1-(naphthalen-2-yl)ethan-1-one and 1-(phenanthren-9-yl)ethan-1-one, naphthyl, antracene indole derivatives could be also obtained as well (4k-l).
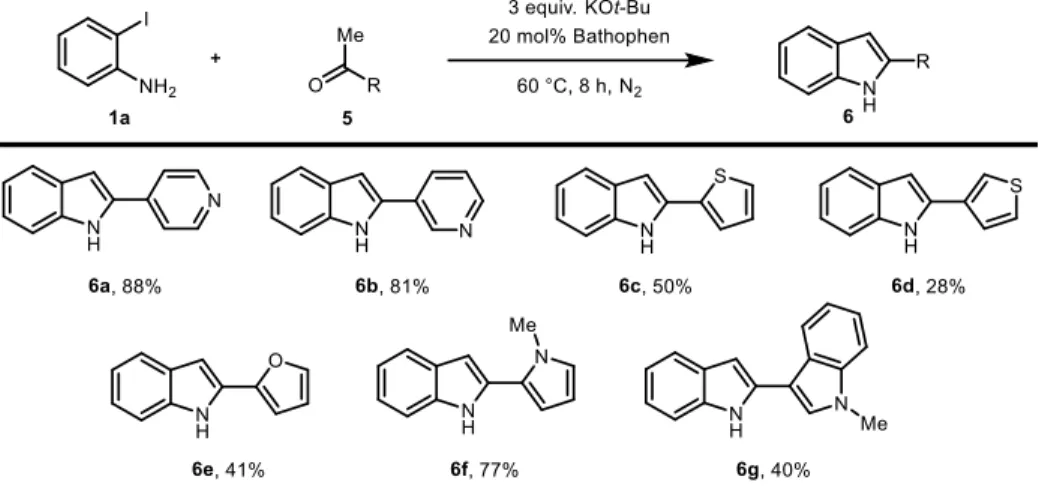
Other types of ketones were also investigated (Table 2.7). Pharmaceutically important heteroaryl groups such as pyridyl (6a-b), thiophene (6c-d), furyl (6e), and pyrrole (6f) group could be tolerated. Bisindole compound was isolated in 40% yield (6g).
Table 2.7. Scope with ketones containing a heterocycle
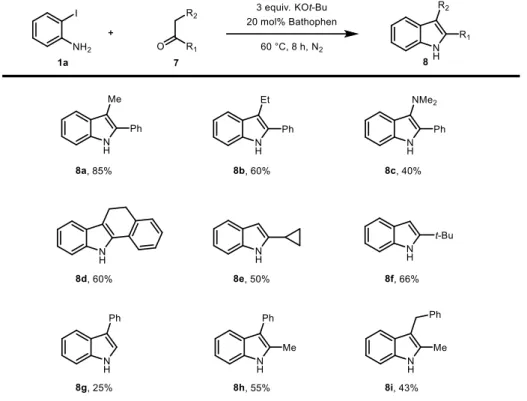
To extend the substrate scope, ketones with alkyl groups have been examined (Table 2.8). The reaction with substrates containing phenyl group proceeded through more favored enolate, which is stabilized by conjugation with phenyl group (8a-b).
Gratifyingly, heteroatom on the 3-position of indole compound also obtained in moderate yield (8c). Moreover, the reaction with -tetralone afforded corresponding indole product in 60% yield, which can be converted to 11H-benzo[a]carbazole
16
through oxidation reaction (8d). Aliphatic ketones such as cyclopropyl methyl ketone and 3,3-dimethylbutan-2-one also tolerated in this reaction (8e and 8f). The use of an aldehyde as a reaction partner, however, led to a significantly diminished yield of the desired 3-alkyl indole due to the competing aldol reaction (8g).
Interestingly, in the case of phenylacetone and benzylacetone, which have two possible positions for enolization, the product originating from thermodynamically more stable enolate was obtained (8h-8i).
Table 2.8. Scope with ketones with alkyl group
17
To gain better insight into the regioselectivity of the method, the indole formation reaction was attempted with ketone derivatives that can potentially provide an indolenine, a frequently observed product in existing protocols (e.g., Fischer indole synthesis) (Table 2.9).
Table 2.9. Regioselectivity trend of the reaction.
Although a preference arising from the formation of thermodynamic enolate has been observed when both of the possible products are indoles, a propensity to counteract that of conventionally approaches has been identified. Cyclization on the methine side of the ketone, which furnishes an indolenine product, was generally disfavored. Instead, the ring-formation event occurred preferentially on the methyl
18
or methylene side to provide the corresponding 2,3-disubstituted indole products (Table 2.9, 9aa, 9ab, 9ac, 9ad, 9ae, and 9af). To our knowledge, these are the first examples of single-step indolization with such a regiochemical trend under transition metal-free conditions.
Scheme 2.1. Possible reaction pathways.
0%
20%
40%
60%
80%
100%
0 h 2 h 4 h 6 h 8 h
Two possible pathways have been postulated for the mechanism of this reaction (Scheme 2.1, a). The first route involves SET of substrate metalloenamine condensate (path I, A) to provide an aryl radical intermediate B by the KOt- Bu/bathophen (P1) complex. The 5-endo-trig intramolecular cyclization and
19
subsequent oxidation forms the indole product (3a). Alternatively, aniline radical D can add to the potassium enolate in an intermolecular fashion to provide a ketyl radical intermediate (path II, E). Subsequent SET and condensation forms the product.
To further elucidate which pathway is perferable, DFT calculation studies were conducted in collaboration with Cheong group. DFT computations reveal a 4.9 kcal/cmol preference for radical D over B, in agreement with the second route.
Interestingly, the coordination of a potassium enolate to the nitrogen atom of D appear to play a key role in the C–C bond formation event. Control experiments using an iodobenzene and 2-iodo-N,N-dimethylaniline, which cannot participate in the coordination event, showed that the product formation is notably less efficient (Scheme 2.1, b). In addition, substantial quantities of the proto dehalogenation products were detected. These observations suggest the metal coordination play a key role in the success of the reaction.
We have examined the factors that govern the regiocontrol with DFT (Figure 2), specifically the preference for the formation of indole over the indolenine products in the case of methyl isopropyl ketone (9a, Table 2.9). The aniline radical/enolate adduct corrsponding to the indole is more stable than that of the indolenine by 15.5 kcal/mol, matching the observed experimental preference (denoted by * in Figure 2, a). The conformational rigidity arising from the potassium chelation between the oxygen and nitrogen in these adducts is key. These adducts can exist in two states (Figure 2, b) – a ketyl radical or a potassium radical. In the
20
absence of the steric influences from the methyl groups, the ketyl radical form is inherently favored – the corresponding acetaldehyde radical adduct which experiences none of the steric effects of the methyl isopropyl ketone prefers the ketyl radical form. In the indolenine pathway radical adduct, the potassium chelation results in an eclipsing interaction, preventing the formation of the preferred ketyl radical. This is in contrast to the indole pathway, where the corresponding radical exhibits no steric hindrance to achieve the stable ketyl radical.
Figure 2. Reaction coordinate diagram for the formation of two products.
Finally, the synthetic utility of the protocol was assessed (Scheme 2.2). The reaction could be conducted on a gram-scale to provide an indole product in practically effective yield (a). Moreover, the process was applied to the multistep synthesis of a nonsteroidal selective estrogen receptor modulator (SERM),
21
zindoxifene (b). The targeted drug molecule could be prepared in the most concise manner to date from readily available substrates.
Scheme 2.2. Synthetic utility of the reaction.
22
3. CONCLUSION
In conclusion, we have reported an efficient transition metal-free KOtBu-mediated protocol for indole synthesis. This method is advantageous in that no transition metal or light irradiation is required, and provides easy access to indoles bearing various functional groups, including C(sp2)–halogen bonds. Additionally, the developed strategy exhibits unconventional regioselectivity, that is rarely observed in other transition metal-free conditions. DFT studies suggest that intermolecular radical- enolate coupling is the preferred reaction pathway over intramolecular cyclization.
In addition, metal chelation and control of the torsional effects lead to the counter- Fischer regioselectivity.
23
4. EXPERIMENTAL SECTION
General Procedures
Reactions were carried out under an argon atmosphere with dry solvents under anhydrous conditions, unless otherwise noted. Anhydrous tetrahydrofuran (THF) and methylene chloride (CH2Cl2) were dried using a solvent purification system.
Anhydrous toluene and acetonitrile (CH3CN) were distilled according to the standard protocols. DMF, DMA, DMSO, DMPU, DMI, 1,4-Dioxane and NMP were purchased in anhydrous form and used without further purification. Reagents were purchased at the highest commercial quality and used without further purification, unless otherwise stated. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous materials, unless otherwise stated. All spectral data were acquired at 295 K. Chemical shifts (δ) are quoted in parts per million (ppm). The residual solvent peak, δH 7.26 and δC 77.0 for CDCl3, δH 2.50 and δC 39.5 for d6- DMSO was used as a reference. Coupling constants (J) are reported in Hertz (Hz) to the nearest 0.1 Hz. The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. NMR spectra were recorded on an Agilent 400-MR DD2 Magnetic Resonance System or Varian/Oxford As-500 instrument and calibrated using residue undeuterated solvent as internal reference. Reactions were monitored by thin-layers chromatography (TLC) carried out on 0.25 mm E. Merck silica gel plates (60F 254) using UV light as visualizing agent and an anisaldehyde or potassium permanganate, and heat as developing agents. E. Merck silica gel (60, particle size 0.040–0.063 mm) was used for flash column chromatography.
24 General procedure for reactions in Table 2.5
For compounds 3a-t.
To a schlenk tube equipped with a stirrer bar was added 1 (0.5 mmol, 1.0 equiv.), KOt-Bu (1.5 mmol, 3.0 equiv.), and bathophenanthroline (0.1 mmol, 0.2 equiv.). The tube was sealed and evacuated, back-filled with N2. Then 1.5 mL of DMSO was added to dissolved reagents followed by 2a (1 mmol, 2.0 equiv.). The reaction mixture was allowed to stir at 60 °C for 8 h. After completion, the reaction mixture was cooled to room temperature and diluted with EtOAc. The organic layers were combined and washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. This crude material was purified by column chromatography to give the desired indole product.
25 2-phenyl-1H-indole (3a)
Purified by silica gel chromatography (ethyl acetate/hexanes = 0/1 to 1/100) to provide 3a as a white solid (80 mg, 83%). 1H NMR (400 MHz, DMSO-) δ 11.52 (s, 1H), 7.86 (d, J = 7.5 Hz, 2H), 7.53 (d, J = 8.0 Hz, 1H), 7.46 (t, J = 7.5 Hz, 2H), 7.40 (d, J = 8.0 Hz, 1H), 7.31 (t, J = 7.5 Hz, 1H), 7.10 (t, J = 8.0 Hz, 1H), 7.00 (t, J = 7.5 Hz, 1H), 6.90 (s, 1H). 13C NMR (101 MHz, DMSO) δ 137.6, 137.1, 132.2, 128.9, 128.6, 127.4, 125.0, 121.5, 120.0, 119.3, 111.3, 98.6.
5-(tert-butyl)-2-phenyl-1H-indole (3b)
Purified by silica gel chromatography (ethyl acetate/hexanes = 0/1 to 1/100) to provide 3b as a white solid (99 mg, 80%). 1H NMR (400 MHz, DMSO) δ 11.40 (s, 1H), 7.85 (d, J = 7.4 Hz, 2H), 7.51 (d, J = 1.3 Hz, 1H), 7.44 (t, J = 7.7 Hz, 2H), 7.35 (d, J = 8.5 Hz, 1H), 7.29 (t, J = 7.4 Hz, 1H), 7.20 (dd, J = 8.6, 1.8 Hz, 1H), 6.85 (d, J = 1.6 Hz, 1H), 1.35 (s, 9H). 13C NMR (101 MHz, DMSO) δ 141.60, 137.60, 135.35, 132.43, 128.85, 128.49, 127.17, 124.84, 119.81, 115.68, 110.81, 98.81, 34.22, 31.81.
26 6-methoxy-2-phenyl-1H-indole (3c)
Purified by silica gel chromatography (ethyl acetate/hexanes = 0/1 to 1/100) to provide 3c as a white solid (78 mg, 70%). 1H NMR (400 MHz, DMSO) δ 11.38 (s, 1H), 7.81 (d, J = 7.6 Hz, 2H), 7.45 – 7.39 (m, 3H), 7.26 (t, J = 7.4 Hz, 1H), 6.92 (d, J = 1.9 Hz, 1H), 6.81 (d, J = 1.6 Hz, 1H), 6.69 (dd, J = 8.6, 2.3 Hz, 1H), 3.80 (s, 3H).
13C NMR (101 MHz, DMSO) δ 155.89, 138.00, 136.47, 132.49, 128.87, 126.88, 124.51, 122.97, 120.75, 109.63, 98.68, 94.36, 55.16.
5-nitro-2-phenyl-1H-indole (3d)
Purified by silica gel chromatography (ethyl acetate/hexanes = 0/1 to 1/100) to provide 3d as a white solid (60 mg, 50%). 1H NMR (400 MHz, DMSO) δ 12.29 (s, 1H), 8.52 (d, J = 2.2 Hz, 1H), 8.01 (dd, J = 8.9, 2.3 Hz, 1H), 7.89 (d, J = 7.2 Hz, 2H), 7.56 (d, J = 8.9 Hz, 1H), 7.50 (t, J = 7.7 Hz, 2H), 7.38 (t, J = 7.4 Hz, 1H), 7.16 (d, J
= 1.4 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 141.44, 141.03, 140.35, 131.04, 129.11, 128.48, 127.96, 125.46, 116.98, 111.65, 100.88.
27 2-phenyl-1H-indole-5-carbonitrile (3e)
Purified by silica gel chromatography (ethyl acetate/hexanes = 0/1 to 1/100) to provide 3f as a white solid (90 mg, 83%). 1H NMR (400 MHz, DMSO) δ 12.12 (br s, 1H), 8.06 (d, J = 1.5 Hz, 1H), 7.91− 7.88 (m, 2H), 7.56 (dt, J = 8.5, 1 Hz), 7.52−4.47 (m, 2H), 7.45 (dd, J = 8.5, 1.5 Hz, 1H), 7.38 (tt, J = 7.5, 1 Hz, 1H), 7.05 (d, J = 1.5 Hz, 1H); 13C NMR (101 MHz, DMSO) δ 140.2, 138.8, 131.1, 129.0, 128.4, 128.2, 125.5, 125.4, 124.2, 120.6, 112.4, 101.5, 99.3
tert-butyl 2-phenyl-1H-indole-5-carboxylate (3f)
Purified by silica gel chromatography (ethyl acetate/hexanes = 0/1 to 1/150) to provide 3f as a white solid (79 mg, 54%). 1H NMR (400 MHz, DMSO) δ 11.88 (s, 1H), 8.18 (s, 1H), 7.90 – 7.85 (m, 2H), 7.69 (dd, J = 8.5, 1.6 Hz, 1H), 7.47 (m, 3H), 7.35 (t, J = 7.4 Hz, 1H), 7.05 (d, J = 1.5 Hz, 1H), 1.57 (s, 8H). 13C NMR (101 MHz, DMSO) δ 165.99, 139.52, 139.31, 131.64, 128.97, 128.11, 127.86, 125.16, 122.68, 122.54, 122.32, 110.97, 99.83, 79.67, 27.99.
28 2-phenyl-1H-benzo[g]indole (3g)
Purified by silica gel chromatography (ethyl acetate/hexanes = 0/1 to 1/100) to provide 3g as a white solid (105 mg, 86%). 1H NMR (400 MHz, DMSO) δ 12.12 (s, 1H), 8.70 (d, J = 8.2 Hz, 1H), 8.00 (d, J = 7.7 Hz, 2H), 7.93 (d, J = 8.1 Hz, 1H), 7.68 (d, J = 8.6 Hz, 1H), 7.59 (t, J = 7.5 Hz, 1H), 7.48 (dd, J = 8.0, 5.8 Hz, 3H), 7.42 (t, J = 7.5 Hz, 1H), 7.32 (t, J = 7.3 Hz, 1H), 7.05 (d, J = 2.0 Hz, 1H).13C NMR (101 MHz, DMSO) δ 136.41, 132.44, 131.76, 130.04, 128.87, 128.46, 127.08, 125.33, 125.10, 124.70, 123.73, 122.09, 121.25, 120.56, 120.36, 100.92
5-bromo-2-phenyl-1H-indole (3h)
Purified by silica gel chromatography (ethyl acetate/hexanes = 0/1 to 1/100) to provide 3h as a white solid (82 mg, 60%). 1H NMR (400 MHz, DMSO) δ 11.76 (s, 1H), 7.85 (d, J = 7.3 Hz, 2H), 7.71 (d, J = 1.8 Hz, 1H), 7.47 (t, J = 7.7 Hz, 2H), 7.39 – 7.31 (m, 2H), 7.21 (dd, J = 8.6, 1.9 Hz, 1H), 6.88 (d, J = 1.5 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 139.18, 135.81, 131.66, 130.53, 128.99, 127.89, 125.20, 124.00, 122.15, 113.27, 111.90, 98.27.
29 5-chloro-2-phenyl-1H-indole (3i)
Purified by silica gel chromatography (ethyl acetate/hexanes = 0/1 to 1/100) to provide 3i as a white solid (68 mg, 60%). 1H NMR (400 MHz, DMSO) δ 11.75 (s, 1H), 7.85 (dd, J = 8.2, 1.0 Hz, 2H), 7.56 (d, J = 2.0 Hz, 1H), 7.53 – 7.19 (m, 5H), 7.09 (dd, J = 8.6, 2.1 Hz, 1H), 6.88 (d, J = 1.5 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 139.18, 135.81, 131.66, 130.53, 128.99, 127.89, 125.20, 124.00, 122.15, 113.27, 111.90, 98.27.
4-chloro-2-phenyl-1H-indole (3j)
Purified by silica gel chromatography (ethyl acetate/hexanes = 0/1 to 1/100) to provide 3j as a white solid (89 mg, 78%). 1H NMR (400 MHz, DMSO) δ 11.92 (s, 1H), 7.95 – 7.90 (m, 2H), 7.48 (t, J = 7.7 Hz, 2H), 7.43 – 7.39 (m, 1H), 7.35 (t, J = 7.4 Hz, 1H), 7.13 – 7.07 (m, 2H), 6.96 (d, J = 1.5 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 138.72, 137.86, 131.53, 128.98, 127.95, 127.21, 125.29, 124.16, 122.35, 118.96, 110.44, 96.72.
30 6-chloro-2-phenyl-1H-indole (3k)
Purified by silica gel chromatography (ethyl acetate/hexanes = 0/1 to 1/100) to provide 3k as a white solid (70 mg, 62%). 1H NMR (400 MHz, DMSO) δ 11.74 (s, 1H), 7.88 – 7.83 (m, 2H), 7.54 (d, J = 8.4 Hz, 1H), 7.50 – 7.43 (m, 3H), 7.33 (m, 4.2 Hz, 1H), 7.03 (dd, J = 8.4, 1.9 Hz, 1H), 6.91 (d, J = 1.4 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 138.80, 137.53, 131.78, 128.96, 127.73, 127.46, 126.11, 125.09, 121.39, 119.76, 110.84, 98.82.
4,6-dichloro-2-phenyl-1H-indole (3l)
Purified by silica gel chromatography (ethyl acetate/hexanes = 0/1 to 1/100) to provide 3l as a white solid (68 mg, 55%). 1H NMR (400 MHz, DMSO) δ 11.89 (s, 1H), 7.59 (d, J = 7.6 Hz, 2H), 7.42 (t, J = 7.6 Hz, 2H), 7.34 (t, J = 7.2 Hz, 1H), 7.21 (s, 1H), 7.11 (s, 1H), 6.83 (d, J = 1.2 Hz, 1H); 13C NMR (100 MHz, DMSO) δ 139.1, 137.1, 131.2, 129.2, 128.4, 127.7, 126.9, 126.2, 125.2, 120.5, 109.6, 98.4.
31 4,6-dibromo-2-phenyl-1H-indole (3m)
Purified by silica gel chromatography (ethyl acetate/hexanes = 0/1 to 1/100) to provide 3m as a white solid (97 mg, 55%). 1H NMR (400 MHz, DMSO) δ 12.08 (s, 1H), 7.94 – 7.88 (m, 2H), 7.58 (s, 1H), 7.48 (d, J = 7.9 Hz, 2H), 7.41 – 7.35 (m, 2H), 6.89 (d, J = 1.5 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 139.64, 137.71, 131.00, 129.02, 128.37, 128.30, 125.40, 124.14, 113.82, 113.57, 113.40, 98.57.
5-bromo-7-chloro-2-(3,4-dimethoxyphenyl)-1H-indole (3o)
Purified by silica gel chromatography (ethyl acetate/hexanes = 0/1 to 1/50) to provide 3o as a white solid (92 mg, 55%). 1H NMR (400 MHz, DMSO) δ 11.47 (s, 1H), 7.57 (d, J = 1.7 Hz, 2H), 7.54 – 7.50 (m, 1H), 7.33 (d, J = 1.8 Hz, 1H), 7.03 (d, J = 8.5 Hz, 1H), 6.91 (d, J = 1.9 Hz, 1H), 3.89 (s, 3H), 3.81 (s, 3H). 13C NMR (101 MHz, DMSO) δ 149.12, 148.86, 141.57, 134.22, 131.03, 124.15, 123.85, 123.07, 119.03, 118.28, 111.88, 110.00, 104.06, 99.37, 55.81, 55.58.
32 1-methyl-2-phenyl-1H-indole (3q)
Purified by silica gel chromatography (ethyl acetate/hexanes = 0/1 to 1/70) to provide 3q as a yellow solid (60 mg, 58%). 1H NMR (400 MHz, DMSO) δ 7.61–
7.56 (m, 3H), 7.54–7.49 (m, 3H), 7.45 (t, J = 7.5 Hz, 1H), 7.19 (t, J = 7.5 Hz, 1H), 7.08 (t, J = 7.5 Hz, 1H), 6.59 (s, 1H), 3.75 (s, 3H); 13C NMR (101 MHz, DMSO) δ 141.0, 138.1, 132.2, 129.0, 128.6, 127.9, 127.4, 121.4, 120.0, 119.5, 110.1, 101.1, 31.1.
1-benzyl-2-phenyl-1H-indole (3t)
Purified by silica gel chromatography (ethyl acetate/hexanes = 0/1 to 1/150) to provide 3t as a white solid (88 mg, 62%). 1H NMR (400 MHz, DMSO) δ 7.76-7.74 (m,1H), 7.50-7.43 (m, 5H), 7.34- 7.31 (m, 3H), 7.24-7.20 (m, 3H), 7.11-7.09 (m, 2H), 6.73 (s,1H), 5.43 (s, 2H). 13C NMR (101 MHz, DMSO): δ 141.8, 138.2, 138.0, 132.7, 129.2, 128.8, 128.6, 128.4, 128.0, 127.2, 126.0, 121.9, 120.6, 120.2, 110.6, 102.4, 47.7
33 General procedure for reactions in Table 2.6
For compounds 4a-l
To a schlenk tube equipped with a stirrer bar was added 1a (0.5 mmol, 1.0 equiv.), KOt-Bu (1.5 mmol, 3.0 equiv.), and bathophenanthroline (0.1 mmol, 0.2 equiv.). The tube was sealed and evacuated, back-filled with N2. Then 1.5 mL of DMSO was added to dissolved reagents followed by 2 (1 mmol, 2.0 equiv.). The reaction mixture was allowed to stir at 60 °C for 8 h. After completion, the reaction mixture was cooled to room temperature and diluted with EtOAc. The organic layers were combined and washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. This crude material was purified by column chromatography to give the desired indole product.
34 2-(benzo[d][1,3]dioxol-5-yl)-1H-indole (4b)
1H NMR (400 MHz, DMSO) δ 11.38 (s, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.44 (d, J = 1.6 Hz, 1H), 7.36 (d, J = 8.1 Hz, 2H), 7.07 (t, J = 7.0 Hz, 1H), 7.02 – 6.95 (m, 2H), 6.78 (d, J = 1.5 Hz, 1H), 6.07 (s, 2H). 13C NMR (101 MHz, DMSO) δ 147.88, 146.71, 137.64, 136.88, 128.68, 126.53, 121.23, 119.75, 119.29, 118.71, 111.08, 108.71, 105.56, 101.16, 97.97.
2-(3,4-dimethoxyphenyl)-1H-indole (4d)
1H NMR (400 MHz, DMSO) δ 11.41 (s, 1H), 7.54 – 7.34 (m, 4H), 7.13 – 6.92 (m, 3H), 6.81 (d, J = 1.4 Hz, 1H), 3.87 (s, 3H), 3.80 (s, 3H). 13C NMR (101 MHz, DMSO) δ 149.03, 148.44, 137.96, 136.89, 128.81, 125.13, 121.07, 119.66, 119.22, 117.51, 112.16, 111.01, 108.96, 97.63, 55.64.
35 2-(2,4-dimethoxyphenyl)-1H-indole (4e)
1H NMR (400 MHz, DMSO) δ 11.08 (s, 1H), 7.73 (d, J = 8.6 Hz, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.44 (d, J = 8.1 Hz, 1H), 7.09 – 7.03 (m, 1H), 7.00 – 6.95 (m, 1H), 6.82 (d, J = 1.5 Hz, 1H), 6.71 (d, J = 2.4 Hz, 1H), 6.66 (dd, J = 8.6, 2.4 Hz, 1H), 3.94 (s, 3H), 3.82 (s, 3H). 13C NMR (101 MHz, DMSO) δ 159.99, 157.28, 136.19, 134.88, 128.56, 128.37, 120.76, 119.50, 118.93, 113.70, 111.13, 105.57, 100.11, 99.01, 55.63, 55.60, 55.34, 55.32.
4-(1H-indol-2-yl)benzonitrile (4f)
1H NMR (400 MHz, DMSO) δ 11.76 (s, 1H), 8.03 (d, J = 8.4 Hz, 2H), 7.88 (d, J = 8.4 Hz, 2H), 7.58 (d, J = 7.9 Hz, 1H), 7.46 (d, J = 8.2 Hz, 1H), 7.23 – 7.13 (m, 1H), 7.11 – 7.01 (m, 2H). 13C NMR (101 MHz, DMSO) δ 137.72, 136.54, 135.60, 132.83, 128.41, 125.34, 122.79, 120.70, 119.84, 119.03, 111.65, 109.19, 101.56.
36 2-(4-(trifluoromethyl)phenyl)-1H-indole (4g)
1H NMR (400 MHz, DMSO) δ 11.75 (s, 1H), 8.06 (d, J = 8.2 Hz, 2H), 7.79 (d, J = 8.3 Hz, 2H), 7.57 (d, J = 7.9 Hz, 1H), 7.46 (d, J = 8.1 Hz, 1H), 7.16 (m, 1H), 7.04 (t, J = 7.2 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 137.55, 135.86, 128.44, 125.83, 125.79, 125.33, 122.43, 120.53, 119.70, 111.58, 100.73, 39.94, 39.73, 39.52, 39.31, 39.10.
2-(4-chlorophenyl)-1H-indole (4h)
1H NMR (400 MHz, DMSO) δ 11.58 (s, 1H), 7.88 (d, J = 8.6 Hz, 2H), 7.53 (t, J = 7.7 Hz, 3H), 7.40 (d, J = 8.2 Hz, 1H), 7.11 (t, J = 7.1 Hz, 1H), 7.00 (t, J = 7.1 Hz, 1H), 6.93 (d, J = 1.6 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 137.22, 136.35, 131.76, 131.12, 128.89, 128.53, 126.60, 121.87, 120.16, 119.50, 111.35, 99.32.
37 2-(3-bromophenyl)-1H-indole (4i)
1H NMR (400 MHz, DMSO) δ 11.62 (s, 1H), 8.09 (s, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 7.9 Hz, 1H), 7.48 (d, J = 8.0 Hz, 1H), 7.41 (m, 2H), 7.13 (t, J = 7.6 Hz, 1H), 7.05 – 6.96 (m, 2H). 13C NMR (101 MHz, DMSO) δ 137.29, 135.85, 134.59, 131.01, 129.89, 128.43, 127.24, 123.98, 122.46, 122.11, 120.33, 119.59, 111.44, 99.91.
2-(4-fluorophenyl)-1H-indole (4j)
1H NMR (400 MHz, DMSO) δ 11.53 (s, 1H), 7.94 – 7.86 (m, 2H), 7.53 (d, J = 7.8 Hz, 1H), 7.40 (d, J = 8.0 Hz, 1H), 7.30 (t, J = 8.9 Hz, 2H), 7.13 – 7.06 (m, 1H), 7.00 (t, J = 7.4 Hz, 1H), 6.86 (d, J = 1.4 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 162.76, 160.33, 137.11, 136.72, 128.90, 128.87, 128.64, 127.02, 126.94, 121.57, 120.02, 119.42, 115.92, 115.71, 111.28, 98.66.
38 2-(naphthalen-2-yl)-1H-indole (4k)
1H NMR (400 MHz, DMSO) δ 11.71 (s, 1H), 8.39 (s, 1H), 8.02 (dt, J = 20.5, 5.2 Hz, 2H), 7.93 (t, J = 7.4 Hz, 2H), 7.59 – 7.48 (m, 3H), 7.47 – 7.42 (m, 1H), 7.15 – 7.10 (m, 1H), 7.07 – 7.00 (m, 2H). 13C NMR (101 MHz, DMSO) δ 137.55, 137.38, 133.26, 132.28, 129.68, 128.69, 128.41, 127.86, 127.69, 126.70, 126.01, 123.82, 122.80, 121.81, 120.14, 119.45, 111.33, 99.56.
2-(phenanthren-9-yl)-1H-indole (4l)
1H NMR (400 MHz, DMSO) δ 11.71 (s, 1H), 8.94 (d, J = 8.0 Hz, 1H), 8.86 (d, J = 8.0 Hz, 1H), 8.43 – 8.37 (m, 1H), 8.12 – 8.02 (m, 2H), 7.82 – 7.62 (m, 5H), 7.52 (d, J = 8.0 Hz, 1H), 7.24 – 7.15 (m, 1H), 7.14 – 7.07 (m, 1H), 6.82 (d, J = 1.3 Hz, 1H).
13C NMR (101 MHz, DMSO) δ 136.70, 136.44, 130.94, 130.27, 129.95, 129.65, 129.51, 128.79, 128.36, 128.06, 127.31, 127.26, 127.19, 127.02, 126.34, 123.41, 122.88, 121.51, 120.11, 119.35, 111.40, 102.77.
39 General procedure for reactions in Table 2.7
For compounds 6a-g.
To a schlenk tube equipped with a stirrer bar was added 1a (0.5 mmol, 1.0 equiv.), KOt-Bu (1.5 mmol, 3.0 equiv.), and bathophenanthroline (0.1 mmol, 0.2 equiv.). The tube was sealed and evacuated, back-filled with N2. Then 1.5 mL of DMSO was added to dissolved reagents followed by 5 (1 mmol, 2.0 equiv.). The reaction mixture was allowed to stir at 60 °C for 8 h. After completion, the reaction mixture was cooled to room temperature and diluted with EtOAc. The organic layers were combined and washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. This crude material was purified by column chromatography to give the desired indole product.
40 2-(pyridin-4-yl)-1H-indole (6a)
1H NMR (400 MHz, DMSO) δ 11.82 (s, 1H), 8.62 (dd, J = 4.7, 1.5 Hz, 2H), 7.82 (dd, J = 4.6, 1.6 Hz, 2H), 7.59 (d, J = 7.9 Hz, 1H), 7.48 (d, J = 8.1 Hz, 1H), 7.19 (m, 2H), 7.09 – 7.02 (m, 1H). 13C NMR (101 MHz, DMSO) δ 150.23, 139.10, 137.65, 134.67, 128.25, 122.94, 120.80, 119.87, 119.06, 111.72, 101.72.
2-(pyridin-3-yl)-1H-indole (6b)
1H NMR (400 MHz, DMSO) δ 11.74 (s, 1H), 9.16 (d, J = 2.0 Hz, 1H), 8.51 (dd, J = 4.7, 1.3 Hz, 1H), 8.25 – 8.19 (m, 1H), 7.58 (d, J = 7.9 Hz, 1H), 7.50 – 7.43 (m, 2H), 7.15 (dd, J = 11.2, 3.9 Hz, 1H), 7.07 – 7.01 (m, 2H). 13C NMR (101 MHz, DMSO) δ 148.18, 146.31, 137.44, 134.60, 132.06, 128.54, 128.21, 123.93, 122.19, 120.38, 119.70, 111.51, 99.95.
41 2-(thiophen-2-yl)-1H-indole (6c)
1H NMR (400 MHz, DMSO) δ 11.54 (s, 1H), 7.51 (m, 3H), 7.36 (d, J = 8.1 Hz, 1H), 7.14 (dd, J = 4.9, 3.7 Hz, 1H), 7.12 – 7.07 (m, 1H), 6.99 (t, J = 7.4 Hz, 1H), 6.66 (d, J = 1.6 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 136.77, 135.48, 132.38, 128.46, 128.12, 125.14, 123.50, 121.72, 119.88, 119.52, 111.12, 98.71.
2-(furan-2-yl)-1H-indole (6e)
1H NMR (400 MHz, DMSO) δ 11.54 (s, 1H), 7.77 – 7.72 (m, 1H), 7.52 (d, J = 7.8 Hz, 1H), 7.38 (d, J = 8.1 Hz, 1H), 7.10 (dd, J = 7.9, 7.1 Hz, 1H), 7.00 (dd, J = 7.8, 7.1 Hz, 1H), 6.87 (d, J = 3.3 Hz, 1H), 6.72 – 6.68 (m, 1H), 6.62 (ddd, J = 3.3, 1.8, 0.7 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 147.75, 142.60, 136.63, 129.37, 128.24, 121.78, 120.13, 119.52, 111.94, 111.25, 105.84, 97.71.
42 2-(1-methyl-1H-pyrrol-2-yl)-1H-indole (6f)
1H NMR (400 MHz, DMSO) δ 11.18 (s, 1H), 7.53 (d, J = 7.8 Hz, 1H), 7.39 (d, J = 8.0 Hz, 1H), 7.12 – 7.07 (m, 1H), 7.03 – 6.98 (m, 1H), 6.90 – 6.86 (m, 1H), 6.56 (dd, J = 3.8, 1.5 Hz, 2H), 6.13 (dd, J = 3.5, 2.8 Hz, 1H), 3.82 (s, 3H). 13C NMR (101 MHz, DMSO) δ 136.18, 130.96, 128.83, 126.04, 124.85, 121.05, 119.64, 119.13, 110.80, 108.51, 107.42, 98.02, 35.63.
1'-methyl-1H,1'H-2,3'-biindole (6g)
1H NMR (400 MHz, DMSO) δ 11.23 (s, 1H), 8.03 (d, J = 7.8 Hz, 1H), 7.82 (s, 1H), 7.52 (dd, J = 7.9, 4.1 Hz, 2H), 7.37 (d, J = 8.0 Hz, 1H), 7.26 (m, 1H), 7.23 – 7.17 (m, 1H), 7.07 – 7.02 (m, 1H), 7.00 – 6.95 (m, 1H), 6.76 (d, J = 1.4 Hz, 1H), 3.87 (s, 3H). 13C NMR (101 MHz, DMSO) δ 137.11, 136.10, 133.72, 129.21, 127.26, 125.02, 121.89, 120.38, 119.97, 119.87, 119.12, 118.90, 110.54, 110.27, 107.73, 96.97, 32.77.
43 General procedure for iodination
Method A: For compounds 1d, 1e, 1g, 1l, and 1m.27
Aniline derivatives (1 equiv.), I2 (1 equiv.) and Ag2SO4 (1 equiv.) in EtOH (15 ml) were stirred for 1 hour at room temperature. After this time, the crude mixture was filtered to eliminate the precipitate of inorganic salt and then washed with a saturated Na2S2O3 solution. The organic phase was separated, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by chromatography
Method B: For compounds 1b, 1c, 1f, 1h, 1i28, 1j, 1k28, 1n, 1o, and 1p.29
Aniline derivatives (1 equiv.) and NIS (1 equiv.) were dissolved in glacial AcOH (3.0 mL) and stirred at r.t. for 48 h to give a brown solution. Analysis by GC-MS indicated clean conversion. The mixture was added to aqueous sodium bicarbonate (5 equiv.), stirred at r.t. for another 10 min, and extracted with EtOAc (3× 20 mL).
The combined organic layers were washed with saturated aqueous sodium thiosulfate (20 mL) then brine (20 mL) and dried over anhydrous Na2SO4. Flash chromatography afforded desired product.
44
5. REFERENCES
1. Baeyer, A.; Emmerling, A., Synthese des Indols. Ber. Deutsch. Chem. Ges.
1869, 2 (1), 679-682.
2. Van Order, R. B.; Lindwall, H. G., Indole. Chem. Rev. 1942, 30 (1), 69-96.
3. Taber, D. F.; Tirunahari, P. K., Indole synthesis: a review and proposed classification. Tetrahedron 2011, 67 (38), 7195-7210.
4. Chadha, N.; Silakari, O., Indoles as therapeutics of interest in medicinal chemistry: Bird's eye view. Eur. J. Med. Chem. 2017, 134, 159-184.
5. Kaushik, N. K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C. H.; Verma, A. K.;
Choi, E. H., Biomedical Importance of Indoles. Molecules 2013, 18 (6), 6620-6662.
6. Sravanthi, T. V.; Manju, S. L., Indoles — A promising scaffold for drug development. European Journal of Pharmaceutical Sciences 2016, 91, 1-10.
7. Chen, Y.; Wei, X.; Cao, J.; Huang, J.; Gao, L.; Zhang, J.; Su, J.; Tian, H., Novel Bipolar Indole-Based Solution-Processed Host Material for Efficient Green and Red Phosphorescent OLEDs. ACS Applied Materials & Interfaces 2017, 9 (16), 14112-14119.
8. Gribble, G. W., Recent developments in indole ring synthesis—
methodology and applications. J. Chem. Soc., Perkin Trans. 1 2000, (7), 1045- 1075.
9. Inman, M.; Moody, C. J., Indole synthesis – something old, something new. Chem. Sci. 2013, 4 (1), 29-41.
10. Humphrey, G. R.; Kuethe, J. T., Practical Methodologies for the Synthesis of Indoles. Chem. Rev. 2006, 106 (7), 2875-2911.
11. Fischer, E.; Jourdan, F., Ueber die Hydrazine der Brenztraubensäure. Ber.
Deutsch. Chem. Ges. 1883, 16 (2), 2241-2245.
12. Robinson, B., The Fischer Indole Synthesis. Chem. Rev. 1963, 63 (4), 373- 401.
13. Nenitzescu, C., Derivatives of 2-methyl-5-hydroxyindole. Bull. Soc. Chim.
45 Romania 1929, 11, 37-43.
14. Hemetsberger, H.; Knittel, D., Synthese und Thermolyse von α- Azidoacrylestern. Monatshefte für Chemie / Chemical Monthly 1972, 103 (1), 194- 204.
15. Bartoli, G.; Palmieri, G.; Bosco, M.; Dalpozzo, R., The reaction of vinyl grignard reagents with 2-substituted nitroarenes: A new approach to the synthesis of 7-substituted indoles. Tetrahedron Lett. 1989, 30 (16), 2129-2132.
16. Larock, R. C.; Yum, E. K., Synthesis of indoles via palladium-catalyzed heteroannulation of internal alkynes. J. Am. Chem. Soc. 1991, 113 (17), 6689-6690.
17. Tokuyama, H.; Yamashita, T.; Reding, M. T.; Kaburagi, Y.; Fukuyama, T., Radical Cyclization of 2-Alkenylthioanilides: A Novel Synthesis of 2,3- Disubstituted Indoles. J. Am. Chem. Soc. 1999, 121 (15), 3791-3792.
18. Mancuso, R.; Dalpozzo, R., Recent Progress in the Transition Metal Catalyzed Synthesis of Indoles. Catalysts 2018, 8 (10), 458.
19. Zhou, Q.-L., Transition-Metal Catalysis and Organocatalysis: Where Can Progress Be Expected? Angew. Chem. Int. Ed. 2016, 55 (18), 5352-5353.
20. Yanagisawa, S.; Ueda, K.; Taniguchi, T.; Itami, K., Potassium t-Butoxide Alone Can Promote the Biaryl Coupling of Electron-Deficient Nitrogen Heterocycles and Haloarenes. Org. Lett. 2008, 10 (20), 4673-4676.
21. Sun, C.-L.; Li, H.; Yu, D.-G.; Yu, M.; Zhou, X.; Lu, X.-Y.; Huang, K.; Zheng, S.-F.; Li, B.-J.; Shi, Z.-J., An efficient organocatalytic method for constructing biaryls through aromatic C–H activation. Nat. Chem. 2010, 2 (12), 1044-1049.
22. Shirakawa, E.; Itoh, K.-i.; Higashino, T.; Hayashi, T., tert-Butoxide-Mediated Arylation of Benzene with Aryl Halides in the Presence of a Catalytic 1,10- Phenanthroline Derivative. J. Am. Chem. Soc. 2010, 132 (44), 15537-15539.
23. Liu, W.; Cao, H.; Zhang, H.; Zhang, H.; Chung, K. H.; He, C.; Wang, H.;
Kwong, F. Y.; Lei, A., Organocatalysis in Cross-Coupling: DMEDA-Catalyzed Direct C−H Arylation of Unactivated Benzene. J. Am. Chem. Soc. 2010, 132 (47), 16737- 16740.
24. Pichette Drapeau, M.; Fabre, I.; Grimaud, L.; Ciofini, I.; Ollevier, T.; Taillefer, M., Transition-Metal-Free α-Arylation of Enolizable Aryl Ketones and Mechanistic
46
Evidence for a Radical Process. Angew. Chem. Int. Ed. 2015, 54 (36), 10587-10591.
25. Chatgilialoglu, C.; Ferreri, C.; Guerra, M.; Timokhin, V.; Froudakis, G.;
Gimisis, T., 5-Endo-trig Radical Cyclizations: Disfavored or Favored Processes? J.
Am. Chem. Soc. 2002, 124 (36), 10765-10772.
26. Barolo, S. M.; Lukach, A. E.; Rossi, R. A., Syntheses of 2-Substituted Indoles and Fused Indoles by Photostimulated Reactions of o-Iodoanilines with Carbanions by the SRN1 Mechanism. J. Org. Chem. 2003, 68 (7), 2807-2811.
27. Rambabu, D.; Raja, G.; Yogi Sreenivas, B.; Seerapu, G. P. K.; Lalith Kumar, K.; Deora, G. S.; Haldar, D.; Rao, M. V. B.; Pal, M., Spiro heterocycles as potential inhibitors of SIRT1: Pd/C-mediated synthesis of novel N-indolylmethyl spiroindoline-3,2′-quinazolines. Bioorg. Med. Chem. Lett. 2013, 23 (5), 1351-1357.
28. Bernini, R.; Cacchi, S.; Fabrizi, G.; Filisti, E., 2-Arylhydroxytyrosol Derivatives via Suzuki−Miyaura Cross-Coupling. Org. Lett. 2008, 10 (16), 3457-3460.
29. Sharp, P. P.; Banwell, M. G.; Renner, J.; Lohmann, K.; Willis, A. C., Consecutive Gold(I)-Catalyzed Cyclization Reactions of o-(Buta-1,3-diyn-1-yl-)- Substituted N-Aryl Ureas: A One-Pot Synthesis of Pyrimido[1,6-a]indol-1(2H)-ones and Related Systems. Org. Lett. 2013, 15 (11), 2616-2619.