Vol 4, No 4, October - Decenûer 1995 Auloittttttune
response
2L5The
Mechanisms
of Autoimmune Response
:
Insights
into an Enigmatic
Repertoire
Wihaskoro
Sosrosenol'3, EndangHerminajeng2
Abstrak
Makalah ini mendiskusikan penelitian-penelitian tentang uekanisnre respon autoituunitas yang nenyebabkan terjadinya penyakit autoinun. Kegagalan toleransi
sel
B
danT
autoreaktrt kurang berfungsinyasel
T supresor, kerusakan nekanisne apoptosis, peningkattan ekspresi superantigen, efek saupittg sitokin, dan kenungkinan infeksiuikrobial
dianggap sebagai nrckanisne 7'angbertanggutrg jawab terhadap terjadinya respon autoiuunitas. Natnun, terlihat baluva sulit nenerapkan satu nekarisnte autoinwûîas untuk nenerangkan terjadinya satu nacan penl'akit tersebut; dengan kata lain, satu peq,akit autoitttutt nwtgkin akibat dari kombinasi ntekanisne tersebut. Perlu pula diteliti lebih lanjut utttuk uenggunakan lrcsil penelitian yang tnennkai nodel binatang percobaan pada uanusia
Abstract
Several possible nechanisns ofautoinutune response leading to tlrc developntent o;fautoitutnune diseases are discnssed briefly herein. Failure
of
both B and T cell tolerance, lack of suppressor T cell functiotts, an apoptosis defect, increased etpressiottof
suPerantigens, adverse effects of c1'tokines and possible uicrobial infecrions have all been proposed as tlrc tnechanisns of autoiuuune response. Yet, none of the proposed nechanisns seeis to e.rplain satisfactorill, and etclusively the developient of a single autoiutttwre disease, suggestirtg that the diseases nruy involve couple.r uechanistus ratlrcrtlnn
due toasingle
patlna1,. Whetherfindings based upon the aniual nrcdels can be eilrapolated in hwnans renruins also to be investigatedfurther, sinceit
v'ould provide a directionfor future bi ntodal tlrcrapy.Keywords : Inununoregulation, autoi ttuttuttity, autoi ttuttune diseases
The induction
of
the immune system
following
anantigen
recognition involves highly complex
interac-tion
of
all
immunocompetent cells
and nrolecules.It
isin this
respect
that
the
ability to
recognize both
self-andnon
self
antigens
is
a
key
feature
of
the immune
system. Thus,failure
to suppress theimmune
response againstself
antigens
would
result
in
theinducttion of
autoimmune
r"aponr";
Ihowever,
the exact mechanismby
which the autoimmune
response
occurs
remainsquestionable.
Distinct
mechanisms such
asfailure
of
deletion
of
autoreactive
T
and
B
cells
have
been reported.Yet,
whether these proposed mechanisms canexplain
the immunopathogenesisof
all
known
autoim-mune diseasesis
unclear,
since eachofthese
diseasesis
characteristically distinct
in
ternrs
of
the
autoan-tigen-activated
effector cells and/or
molecules. The
aim
of this paper
is to
discuss several
proposed
nrechanismsof
the
autoinrmune
responseand
its
as-sociation
with
thedevelopment of
certainautoinrmune
diseases.
Failure of self-T cell tolerance
Both intra and extrathymic
developnrent
of T
cells
which
result
in
the
ability
of the
cells to
discriminate
betweenself-
and nonself
antigens are perhaps oneof
the central
questions among
immunologist.
The
precise mechanism
by which
these phenomenaoccur
is still far from clear; however, two
debatable
path-ways, i.e.,clonal deletion
andclonal
anergy, have beenput forward
to
explain the T
cell
negative selection
during
its ontogeny
in
both
intra thynric
(central)
andextrathymic (peripheral) environment.2
Clonal delettion or
elinrination of
self-reactive
T
cells
is
considered
as the main
nrechanism
of
the
T
cell
Ith , Deparrttrcnt of Oral2L6
Sosroseno and Herninajeng Med J Indonesautoimmune
glomerulonephritis.la In
this study, I-Ecr
chain-derived peptides were presented
by
I-AD
molecules
of B
cells,
implying that
these
peptidescompeted
with
autoantigens
on
I-A
molecules
of
B
cells.
One
may assume
therefore
that B cells
indeedpresent autoantigens
to
autoreactive
T
cells, but
this
event
is
preventable
by
other unrelated
peptides.
It
should however
benoted that
clonal
deletion may,
tosome
extent,
occur
in
the peripheral
or
postthymic
sites, due to_anextremely
compelling response
to
im-mr,nog"nr.ll Not surpriiingty, peripheral self-T
cell
toleranceinvolves
amultistep
eventwhich
mayin turn
maintain both physical and functional depletion
of
autoreactive
T cells.
l5The evidence
implicating
therole
of T cells,particular-ly CD4
cells,
in
the development
of
autoimmune
dis-eases is
overwhelnring.
Thequestion
remainshowever
whether activated autoreactive
CD4
cells
areresulted
from the failure
of
central and/or peripheral
T
cell
tolerance.
In
nryastenia
gravis (MSG)
muscle
acetyl-choline receptor
(AChR)-specific CD4
cells helped
B
cells
to
produce autoantibodies
if
CD8
cells were
removed,l6 suggesting that this autoimmune
response is due to failure of CDScells to downregulate
autoan-tigen-activated CD4 cells
rather thanfailure
of central
T cell tolerance
per
se
(seealso
below). In
fact,
sup-pressed
AChR-specific antibody production
in
theMSG patient- derived
B andCD4 cell coculture
systemwas mediated
by
suppressor macrophages,
but not
CD8cells,"
supporting
the abovecontention.
Sinrilar-ly,
enhanced expressionof nryelin
basicprotein
(MBP)
in
MS
may be presented
by
local
APC such
asnricrogial cells
to naive CD4 cells which
are
in
turn
activated to
becomeautoantigen-reactive CD4
cells,lSsuggesting
that peripheral
T
cell
tolerance
fails
tooccur. Of
interest,
the
occurrence
of
this diseases is
strongly associated
with TCR VB-CDR3 expression.e
Thus, whelher
this disease
is due
to failure
of
central
and/or peripheral tolerance remains
to
be
elucidated.Failure
to
deleteclonaly
autoreactive
T
cells
can also be seenin the
development
of
autoimmune gastritis in
which immunisation of
gastricproton pump-derived
crand
Rsubunit
in the presence
of
adjuvant induced
theorgan specific
autoin-rmune
disease
in
an animal
model.le
Based upon thesefindings, it
seenrsplausible,
therefore, that failure
of
one
of
the clonal
selectien
theories
would only
mediate
specifically certain but
not
all types
of
autoimnrune
diseases.Should
both clonal
negative
T cell
selection theories
not besufficient to delete
thedevelopment of
autoreac-tive T
cells,it
needs otherexplanation
to deternrine the persistedautoinrnrune
responsein
this
cell
repertoire.
negative
selection intrathymically,
basedon the
fact
that
lack of
self-reactive
T cell
clones bearing
VB (T
cell
receptor
B locus)
o""r,.r.3
In
this
respectt,
thetransitional development
of
double negative to double
positive thymocytes requires the
presence
of
T
cell
receptor (TcR)
Rlocus,'
suggesting
that lack of
this
locuswould
resultin failure of deletion
ofself-reactive
T
cells. The
mechanisms
by
which clonal
deletion
occurs remains obscure.It
seems that nrajorhistocom-patibility (MHC)
molecule-bearing
thymic epithelial
and mesenchynrecells deternrine
suchselection.
Both
cells are
required
to
develop TCR-Cq4-CDS-
cells to
TCR*CD4*CD8*
cells
in
the thymus,)
whereasTCR-bearing thymocytes
which recognize
self
antigen-presenting
thymic dendritic cells would be negleted.o
How
autoreactive
T
cells
evade
the clonal
deletion-mediated negative selection remains to
beelucidated;
it seems
that,
increasedexpression
of endogenous
su-perantigens such asMls
in mice would
resultselective-ly
in
deletion
of
VB
locus-bearing
T
cells (see also
below;.7
However many investigators believe that
it is difficult
toreconcile
theclonal deletion
as a sole mechanismin
the T celldevelopment.
In
animal models
of
systemic
Iupuserythematosus
(SLE),
no clonal deletion
occurs andnormal TCR
VB expression can be detected.s It hasalso
beenargued
aVB
usage as themere explanation
of autoimmune
response due to thefact
thatin
autoim-nrune diseases such as
nrultiple
sclerosis(MS), sharing
TCR VB-CDR3 reeion
rather than
VB
alone
is
com-monly
,""n,e Clona'i anergy rather than
clonal deletion
can also be seen in a study
showing
thatpeptide-hyper-activated human
T
cell clones failed
to
proliferate,
following restimulation
with
optimal
doses
of
pep-tides.'"
Using
an in vitro system,it
appears thatfollow-ing
recognition
of
self
antigens,
T
cell
anergy
is
mediated
by lack
of lL-2
production
for self
growth,
perhapsdue
to
antigen presentation
function
of
non-profeisional antigen presenting cells (APC).ll'12
It
has,in this
respect, beenshown
thatin transgenic
nriceexpressing
MHC
class
II
on
pancreatic beta
cells, T
cellsrecognizing
transgenicproteins
presentedby islet
Vol 4, No 4, October - Decenùer j,995
One possible
mechanism
of T cell
tolerance
is
the"ignorance"
of
T
cells to
self
antigens.2o
In
this
proposed mechanism,
two possible concepts, i,e.,
af-finity binding
andaccessibility,
have been putforward.
Clonal
selection-evading autoreactive
T
cells would
normally have
low affinity
in
binding with self
an-tigen-bearing
APC.
Under certain not yet defined
cir-cumstances, increased expression
of
self
antigens
could occur and, in turn, attract the T cell-APC
bind-ing,
leading
to autoreactive
T cell
activation. This
elegant studies
using
three setsof
;i'i,'5,
il'
:"":':lï:::îf*"'in:
Kb
antibodies
andRIP-IL-
2mice
expressing RlP-controlledlL-2
g"n".2t,22
No cell
in-filtration
onpancreatic islets
"ould
b"
seenin RIP-Kb
X
Des-TcR Fr mice,
eventhough anti-Kb T cells
werenumerous.
However,
if
RIP-Kb
X
Des-TcR Fr
mice
were matched with RIP-IL-2 mice,
Bcell destruction
and diabetes
could occur
in
these
triply
transgenicmice.
Theseresults
suggesttherefore that high
levelsof IL-2 may
enhancethe expression
of self-antigens
which
are then capableof stimulating T cell-mediated
autoimmune
response.The accessibility-related
con-cept
is
presumably associated
with
the structure
of
antigens.
Sequesteredself antigen is commonly
unac-cessible
to be recognized
by T cells;
however,
somemolecules may appropriately
be presented andrecog-nized
by T cells which
arethen being activated.
The second concept can be seenfrom
astudy showing
thatwhole murine
cytochronre
c failed
to induce
T
cell
activation,
but peptide
8l-104
derived from
theseproteins
elicited
T cell
response,suggesting that
self
antigen-derived certain peptides which
areconrmonly
unexposed^may induce
autoimmune
responseof T cell
repertoire." However, the extrapolation
of
these
studies
in
the
development
of
autoimmune
diseases needsto
beelucidated.
Failure of self-B cell tolerance
Using
transgenic mice,
it
appears
that the fate
of
autoreactive B cells closely
resenrble thatof
Tcells. In
transgenic mice expressing
hen
egg lysosome,
inr-munisation
of
the lysosome has resultedin
suppressed'specific antibody
production without physical
lossof
B cells,
suggesting that autoreactive
B cells undergo
clonal anergy.'*
In sharp contrast, transgenic
nricecarrying
genesencoding anti-MHC antibodies
becometolerant
toappropriate self
antigensby deleting
MHC-specific
B cells,
suggesting
that
clonal deletion
oc-curs." Clonally
deletedself-
reactiveB
cells have also beenshown
by
using transgenic nrice
carrying
genesencoding anti-CD8.2 imnrunoglobuli
n nru chain.2oAuîoirtttttune
response
217Activated autoreactive B cells
which produce
autoan-tibodies are believed
to
play
a
crucial
role in
thedevelopment of certain autoimmune
diseases.2THow-ever, the mechanism
by which autoreactive
B cells can beactivated
toinduce
theautoimmune
response is notfully
understood.
In
lysosome transgenic mice,
self-reactive
B
cells were inactivated.zE
In this study,
if
continuous autoantigenic stimulation was
removed, theseputative cells would
then
beresponsive to LpS
or CD4 cells,
suggesting thatself-reactive B
cellsmay
be activated
polyclonally.
One possibility
is
rhattiJ,i:?i,"_.""1';:Tiilï,:1113ïTiïi{f
rrrien
Studies
using the lysosome transgenic mice
have alsoprovided
a
line of evidence that functionally silent
autoreactive
B cells can be activated
following
anintinrate
T-B
cell interaction. Thus,
autoantigen-ac-tivated CD4 cells help naive or autoreactive B cells to
produce specific autoantibodies.
A
support can
bedrawn
from
graft-versus-host- and chemical-induced
experimental systemic autoimmune
diseases.3lIn rhis
study,
Il-4-producing
CD4 cells were activated
by
alloantigens
andchemicals such
asmercury
andthey
in
turnprovided
signalsfor
Bcell activation
toproduce
autoantibodies such as anti-DNA
antibodies.
Like-wise,
in vivo depletion
of CD4 cells
with monoclonat
anti-CD4
cell antibodies
hasresulted
in inhibition
of
autoantibody
production
in an aninral model
of SLE
and
MS.32
'
'
During
the generation
of antibody diversity,
somatic
hypernrutation
of
rearranged immunoglobulin
V-region
glfes
is required
to produce
high affinity
an-tibodies."
It
is
in this respect
that anti-DNA
auto-antibodies from autoimnrune
nriceshow
extensivehy-permutation of
V-regiong.enes and appear to havehigh
affinity
to self-antigens.'* Indeed,
Ig-V
gene usageis
seen
ntore frequently
in
SLE patients than in normal
subjects.r) Thus,
it is possiblè
that following
secon-dary
autoantigen recognition, self-reactive
B
cells
which normally
producelow
affinity antibodies
and donot respond
to
self-antigens undergo somatic
hyper-ntutation. Despite
thefact
thatthis contention
remainsspeculative,
this pathrvay may reflect that
theseputa-tive B cells
evade theclonal
anergy.The role of strppressor
T (Ts) cells
2LB
Sosroseno and Henninajengcells can be divided into two types,
i.e.,IFN-1-
produc-ing Ts cells
type
I
andIl-4-producing
Tscells type2,
have sharpened the role
of
this T cell subpopulation in
the
immune.opons".36'3'I
'
The exact
role
of Ts cells in
the development
of
autoimmune
response isstill
debatable and appears todepend
upon the
nature
of
specific
autoimmune
dis-eases.In
the case
of
experimental autoimmune
inter-stitial
nephritis, tubular antigen-specific CD8
cells are theeffector
cells
capableof
developing cell-mediated
immune
response^anddamaging
renal tubular
base-ment membrane.'o
Surprisingly,
the
initial
activation
of
thesecells
are
inhibited
by CD4"
suppressor cetls(Tsl)
which
in
turn induce transforming growth factor
(TGF)-Rl-producing
CD8+
cells
(Is2)
acting
assup-pressot
cells
for
the
tubular-specific effector cell
ac-iiuity.3e'aoon
the otherhand,ôbservationsin
diseases such asuveitis,
MS and SLE suggested that Ts
(CD8)
cells
fail
to suppress the induction of autoreactiveCD4
effector
cells.al
In
experimental allergic
encephalo-myelitis (EAE),
ananimal
nrodel
of
human
MS,
lack
of
functionally active TS cells
occurs
in
genetically
susceptible animals, suggesting
that Ts
cells
areprotective
in
EAE,
perhaps
via
the
action
of
Ts
cell-àerived
TGF-
B.a2'4JThus,adoptive transfer of Ts cells
isolated
from
spontaneously recovery
EAE inhibited
the
induction
ofEAE in the
recipients and
proliferation
of myelin
basicprotein (MBP) specific T cell
lines in
vitro.s
Likewise, in vivo
depletion
of Ts (CD8)
cells
using a respective monoclonal antibody
increased thesusceptibility
to
develop
mercury
chloride-induced
autoinrmune
responsein-Brown Norway
rats.45Func-tionally impaired Ts cells in
these diseases have also been shown by using oral administration of therespec-tive
antigens.This
route
of
antigen administration
has led to induce.specific Ts cells and reduce the courseof
diseases such asMS, uveitis
and rheumatoid
arthritis
(RA) in both animal
modets and human studies.a6'a7
Moreover, depletion
of Ts
(CD8) cells
in vivo
by
injecting anti-CD8 cell
monoclonal antibodies
did
notprevent the induction
of
experimental
autoimnrune
uveitis (EAU)
in Lewis
rats,48
and
accumulated
evidences seem to suggest that Ts (CD8) cellsfunction
to
suppressS-antigen
or
interphotoreceptor
retinoid-binding protein-specific T
cetl
activity in
EAU.aeWhilst
the above discussion reveals that Ts suppressorcells
of
both CD4 and CD8
cell
phenotypes
play
acrucial role in
suppressingautoantigen-specific
T
cell
functions,
notall
modelsfollow
thispathway.
In MSG, suppressedAChR-specific
CD4 cell activation
was not associatedwith Ts (CD8) cell
functions, rather
it
wasmediated
by
suppressor macrophug"..
17As
yet,
noMed J Indones
clear explanation
can be forwardedfor
this
discrepan-cy.
Presumably, the precise
tinre at which CD8
cells
function
in this disorder
as seen in the murine nephritismodel
is
important
in
determining the role
of
these ce-llsin
viuo.38aoThe role of apoptosis (programmed cell
death)
Apoptosis
is a
phenomenon
of
the cell
death
driven
physiologically
by activated
genes
such as
Fasand shown
by
condensed
cellular
cytoplasm
andchromatin.)u
In
the immune
system,
along
with
thepossible
mechanisms as discussed above,clonal
dele-tion of
both thymocytes
and peripheral autoreactiveT
cetls has been demonstrated due to apoptosis.s I Yet,
it
has
been argued
that the negative selection
of
thymocytes may occur
due to simply_terminaldifferen-tiation
rather than due
to apoptosis.
/
Despite
thecon-troversial
evidences, apoptosis-induced
B
and
T
cell
tolerance are
relevant
to
the possibility that
autoim-mune response ispartly
due to impaired apoptosis.Since /pr gene
is
mapped
to the
same area
of
chromosonre
of
Fas gene,it
has been postulatedthere-fore
that spontaneously developed SLE
in
murine
homozygous for
lpr
is due
to
defect
of
Fas
g"n".52Indeed,
it
is now
recognized
that different
strains
of
nrice which
develop
spontaneousautoimmune
disor-ders have mutationin
apoptosis-associated genes.For
exanrple,
in MRL-lpr/pr
mice, defect
of
Fas
gene expression is due to insertion of a retrotransposoninto
the second intron
of
the Fas
gene.53l54Th"r"
phenomena may explainlynrphoproliferative
disorder,
lossof T cell
tolerance and
B cell
defect
occurring
in
these mice.The
apoptosis defect
seems alsoto
occur
in
SLE patients as seenin a study carried out
by Cheng
and colleaques.tt
In
this study,
increase
of
solubte
form of the
Fas nrolecules was accompanied
by
reduced apoptosis, suggesting
that
increased
expres-sion
of
these soluble nroleculeswould
result in altered
clonal
negative selection
of
autoreactive
cells
which
would in turn
leadto initiate
the
induction of
autoim-nrune disease.The apoptosis
defect may not
only
be associatedwith
the
failure
of
Fcs
geneexpression,
but also with
in-creased expression
of
Bcl-2
gene.Bcl-2 (B-cell
lym-phoma/leukemia-2)
geneis frequently
translocated in
imnrunoglobulin
locus
of follicular B-cell
lymphoma
andits expression is related
with cell survival;
he4cq
Vol 4, No 4, October - Decenber 1995
survival of B
cells
and aged transgenic 6icre^ carryingthis
gene developed
autoimmune
disease.ssIn
3t-È
patients, circulating CD4 and CD8 cells, but not B
cells,
expressedhigh level
of
Bcl-2 protein,
whereas increasedexpression
of Bcl-2 mRNA in
mononuclear
cells could
be seen,
suggesting that dysregulation
of
apoptosis^inlymphocyte population
may occur in thesepatients."
These
findings
demonstrate
that
altered
Bcl-2
expressionmay result in the
failure
of
autoreac-tive B
and/or T
cell
deletion
which in
turn accelerates
the autoimmune response. Theprecise mechanism by
which
alteredBcl-2
gene expression occursin
autoim-mune disease
is
however uncertain, since
several
evidences revealed thatBcl-2
genefailed
to induce thesurvival
of self-reactive B cells.) /Of interest,
insertion
of retroviral
geneinto
the
myeloid
leukemia
cell line
(HL-60 cell
line)
was able to over-expressthis g"n".@
By
analogous, one
may
assumethat
viral
infections
which
are suspectedto
associatewith
certain
autoim-mune diseases such as typeI
diabetesmellitus6l
may
increase the expressionof
this gene, leading to induce
longerlife-span
of both autoreactive B and T cells.The
APC
Autoinuune
response
219contention
remainsspeculative
and needs to beinves-tigated further.
The role of superantigens
Superanligens
such
terotoxins
B
(SEB),oz
group
A
st
3and mouse
mammary tumor
vir
been shownto
activate
T
cells specifically and more efficiently
than
in
the classicalMHC-peptide
interaetion.
Unlike
the
classical T cell recognition in which
antigens must be processed and presentedby
ApC in
small
peptides, superantigens can be presented as entire molecules.In
Food poisoning
andtoxic
shock syndrome
are among the diseases associatedwith
superantigens.The role
of
superantigens
in the
development
of
autoimmunity
has been a focusof
investigations, but
APC
As
As
Voc
vB/
vB/
TcR
B
A
M
HC
class
"
\
SA
V(
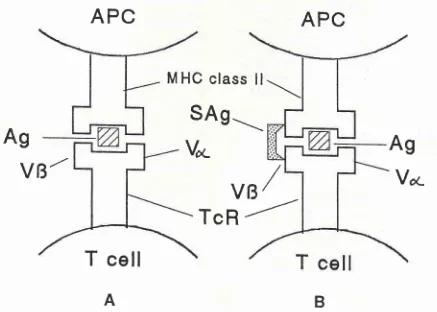
[image:5.595.68.505.361.673.2]T
cell
Figure
I'
Atttigen and superantigen recognition involvittg MHC and TcR ntolecule interactiott. cD4 and cDg ntoleculeantplifl, the
220
Sosroseno and Herninajengnot yet
precisely defined. Since such antigens
caninduce
polyclonally B cell
andspecifically T cell
ac-tivation,
oneor two cells in
theautoreactive cell
reper-toire
may beactivated; with
other words, superantigens may break the existenceof
self-tolerance. Rheumatoid
arthritis
(RA)
andMS
are amongautoimmune
diseasesin which
superantigens may
involve.o) In
the
caseof
MS, retroviruses
have been
believed
to
play a
role,
suggesting
microbial infections may be
involved.06Two
separatelines
of
evidencesmay support
therole
of
superantigensin MS. First, T cells
infiltration
in
thecentral nervous
systemin EAE
werepredominated by
Thl
cells.o'
Secondly,
superantigensinduce
preferen-tially
ThI
cell activation,
at least,via
theproduction
of
TGÈ-8.68-70
Furthermore,
increasednunlb"t of
VR17-positive T cells in
patientswith
RA,
conrpared to thosein
the controls
could
beobserved, suggesting that
thedevelopment of
RA
may
be associatedwith
superan-tigen
infections. "
Oneshould however
becautious
in
extrapolating
this
study
into a
general
conclusion,
since this study
did
not show whether
increasednum-ber
of
this
cell
population was
accon'rpaniedby
in-creased
expression
of superantigen-derived
peptides.A
direct
evidence showing
a
superantigen-RA
relationship
conresobviously from
animal
nrodels.In-jection
of
SEB resulted
in
high
nunrber
of
synovial
cells
in
VB8
TcR
transgenic
mice
(MRL
-
+
I
+),
compared
to
those
in
non
transgenic
MRL-lpr/pr
mice.
''
In
this study, higher
degreeof synovial
hyper-plasia was
seenin
transgenic mice than
in
non
trans-genic mice, suggesting
that superantigensmay induce
chronic
arthritis.
It
seemsplausible
thatsuperantigen-induced
autoreac-tive B or T
celI
activation
still
needsto encounter
therespective autoantigens,
in
order
to
develop
the
specific autoimnrune ,"rponr".65
In
EAE,
superan-tigen-activated
T
cells would then be
reactivated by
MBP
presentedby nricroglia
cells or astrocytes
in
thebrain,l6'73 ,ugg"riing
thai the
secondary
autoantigenchallenge
is
required
in
superantigen-nrediated
autoimmune
response.
Using SEB and
toxic shock
syndrome
toxin
(TSST)-
l,
a superantigen producedby
Staphylococcus aureus B,it
appears that bothsupelgl:
tigens bound
to
the
same
region
of
HLA-DR1./a'l)
These
studies also revealed that
whilst
the
binding
of
these superantigens
onto
theregion
of
DRI
molecules
was
dependent
upon the classical antigen
peptides,they
did not block
eachother completeiy,
suggestingthat the classical antigen peptides
direct
the
superan-tigen binding and that
superantigen-induced
T
cell
activation
may
still
be
dependent
upon
the
antigenpeptides. These
findings
iniply
therefore
that
the secondarychallenge
by
the respective autoantigens
isMed J Indones
indeed a
prerequisite
in
determining
therole
of
super-antigen-associated
autoimmune
response; with other
words,
superantigens
may
be
a predisposition
in
theinduction of
autoimmune
response. Careshould
how-ever betaken
in
extrapolating
the data basedupon
VBexpression
into the
possible
role
of superantigens
in
these diseases, since the ideaof
VB-associatedautoim-mune
diseasesstill
needsto
be investigated further.e
Indeed, Vcr
expression
would also be important in
determining
tight
antigen interaction
if
theMHC-super
is
weak.76The role of cytokines
During
antigen-driven imnrune response, signals
provided
by
cytokines
to
activate the
immunocom-petent cells and molecules are prerequisite.
Indeed,cytokines
are notonly
important
inprotection,
but alsoin maintaining autoreactive B
and Tcell activation
andinducing
tissue destruction.TTCytokines such
asIL-6
may
in
fact induce
secretion
of
proteinases such
asgranzymes
which
in
turn damage
target
cells
andrelease autoantigens as seen
in
Àultiple
sclerosis.TsTGF-B can
beprotective in this
disease as seenin
ananinal
model,
perhapsby inhibiting IL-1
production,
whereas
this
cytokine
has been
inrplicated
in
thedevelopnrent
of
BA,
by recruiting
leukocytes
in
thesynovial
tissues.'o
However, injection
of
monoclonal
anti-
TGF-B antibodies reduced the
severity of
strep-tococcal
cell wall-
induced
arthritis
in
a
rat
model.boIncreased
levels
of
IL-4,
IFN-1 and
IL-1,
but-sup-pressedTNF-a in
SLE
have also been reported.30The
exact role
of
theseimpaired
cytokine
levels
in
SLE
isstill
obscure;likely,
high
levelsof
IL-5
mayplay
arole
in
detern-riningthe
activation
of
CD5*
B
cells
which
may
in
turn produce
autoantibodies
in
SLE.8l
This
cytokine
profile of
SLE patients
isin fact parallel
with
studies
wing
the animal
nrodels.
For
example,
con-tinuous injection
of
anti-IL-10
antibodies
in
NZB/W
Fl
nrice resulted
in
reducing
the severity
of
spon-taneously developed
lupus-like autoimmunity
andin-creased serum levels
of
TNF-cr.82
These findings
suggest
therefore that
TNF-a
is protective
in
SLE.
In
insulin-dependent
diabetes
nrellitus,
monocyte-derived
IL-1
may be destructive
to
islet
beta
cells
which
woulcl
then
release
their autoantigens.6l
In-creased levels
of
IFN-1
in
Behçet syndronre have beensuggested to upregulate
NK
cell
activities.o'Likewise,
the induction
of
cellulal immune
responsein
blister
formation
as
well
as increased autoantigen gene
ex-pressionin
cultured keratinocytes
has been associatedwith excessive
production
of
IFN-1
in
bullous
penr-.
, 8J.85-Vol 4, No 4, October - Decenber 1995
The regulatory roles
of
cytokines such
as
IL-l
andTNF-a in
thedevelopment
of RA
areof
interest.
High
levels
of
IL-l
in
the
synovial
fluid
suggestthat
this
cytokine
plays acrucial
rolein
this disease. In responseto IL-1, synovial
fibroblasts
of RA
patients
producedMCAF
(monocyte chemotactic and
activating
factor)
and PGE2, suggesting that
IL-l
mayinvolve in
macro-phage accumulation and both pain and
edema
as-sociated
with
rheumatoid
synovitir.s6'87If
treotmentof
RA
patients
with
methotrexate
(MTX)
hasresulted in
reduced
IL-1
levels
of synovial
fluid,88 this
treatmentregiment
might
decrease theseverity
of RA.
Further-more,
one
of
the
TNF-a
activities
in
RA
may
beassociated
with hyaluronan
(HA),
a marker ofsynovial
proliferation, since
increased
HA
production
is
sig-nificantly
correlated
with
increased levels
of
this
cytokine.8e The most
important
roteof
thiscytokine
in
RA
is
perhapsrelated
with its
ability
to
induce
IL-l
production, but
suppress
GM-CSF production,
sug-gesting that
do_wnregulatedTNF-a production would
be
beneficial."
Indeed,
injection
of
anti-TNF-q,
an-tibodies before
or
during the arthritis
process
sig-n ificant
ly
supprers^sed theseverity
of
co I la gen-i nd ucedarthritis in
mice."
The role of microorganisms
Certain
microbial
antigen-derived peptides
which
are presentedby
MHC
classII
nrolecules sharehomology
with
self
peptidesderived
from HLA-DRa,
suggesting thatmicrobial
peptide-activated T cells
may be able torecognize self-antigen
presentedby
MHC
classII
andinitiate autoimmune
response.
This
phenomenon is
termed
as the antigenic
mimicry. For
example, in
ankylosing
spondylitis,
Klebsiella organisms
can
modify
HLA-827
negative
to
-827 positive like
per-sons,suggesting that
infection
dueto
theseorganisms
may
enhancethe individg.al
susceptibility to
develop
this
autoimmune
disease.v' Othermicroenvironmental
antigens such
asa
75 kD
heatshock
protein
(Hsp?O)of
Trypanosona brucei, RfbA protein
of
Sahnonella
typhinturiunr,
and
periplasmic protein
of
Treponenn
pallidum
haveshown
to have anrinoacid homology
atcertain position
with
HLA-DRcr
sequence,implying
that
these antigens
may trigger the
developnrent
of
certain autoinrmune
diseases.92The
antigenic
mimicry
hypothesis seems torely
on theexistence
of
HLA-DR
expression,
but
it nray
not
al-ways
be the
case
for
the development
of
certain
autoinrmune diseases such as
RA.
Someof
thepopula-tion carrying
HLA-DRl
or
DR4B do not develop this
disease,
suggesting
that adefinite
conclusion to show
Autoitanune
response
221a
correlation
of HLA-DR
expression
andRA
is
scan-ty.93
In fact,
nrycobacterium-induced
infection
showssinrilar
features seen in thisautoimmune
disease. Thus,elevated levels
of
rheumatoid factors, agalactosyl
im-nrunoglobulin
G,
and antibodies
to
mycobacterial
Hsp65 can be detected
in this infection,
suggesting thatrheumatoid
arthritis is
aslow
bacterial
infection.ea'esThe mechanism(s) underlying this
phenonrenon isun-known
and needsto
beelucidated
further. It
has beenpostulated
that
n'ricrobial antigens originated from
extra-articular
sitessuch
asthe gut
or lung would
be processed and presentedby synovial APC to
activate
T
cells
which in
turn
triggersynovial
macrophages torelease
tissue-danraging mediators.e) Perhaps,
the
slow
bacterial
infection
nray also be a good candidatein explaining
the pathogenesisof
Behçet's syndrome,
since
high
levels
of IgA
and
IgG
antibodies from
patients
with
this
diseasereact
with
65
to 70 kD
heatshock-protein (Hsp)
derived from
Streptococcus
san-guis.'o
It
is
unclear
how
thesenricrobial
antigens
in-duce
this
disease.Yet, it
may
be that the pathogenesisof
Streptococcal HSP65-induced Behçet syndrome
issimilar to
thatof
Mycobacterial-induced
RA,
since .S.sangais-derived
Hsp65 shareshomology
toMycobac-terial
Hsp65 and
high
levels
of
antibodies
specific
toboth microbial
antigens can
be
observed
in
both
autoimmune
disorders.96Of
interest,
Hsp6O-derivedamino
acids 336-35I
and
136- 150which
areT epitops
of
this syndronre
wereable to induce
uveitis in Lewis
rats,eT
suggesting
that nrycobacterial infections
nrayinduce both autoinrmune
diseases.CONCLUSION
Distinct
mechanisnrs
implicated
in
the induction
of
autoinrnrune
responsehave
beenproposed; however,
it
seenrsthat
noneof
them is able to explain
satisfac-torily
and exclusively
the
occurrence
of
a
single
autoinrnrune disease.
It
issafely
to saythat
theinduc-tion of
autoinrmune
responseis
aconrplex interaction
involving
nrany possible pathways.
Of inrportant,
other
effector
molecules
such as
conrplements,
idiotypic
antibodies
and adhesionntolecules
ntay alsoplay
acrucial role
in
the
induction
of
these diseases.Fresh ideas
with
substantial evidences, at
the presenttime, are
desperately
needed
to
delineate
this
phenomenon.
A slow
bacterial
infection
as a possibteetiology of
the autoinrnrune diseases is perhaps oneof
the
exanrples
to
open the dead lock, since
it
mayprovide
adilection for future
therapeutic
bimodal.
It
should
alsobe kept
in
nrind that
previously
describedworks in elucidating
the mechanisms
of
autoinrmune
222
Sosroseno and Henninajengextrapolation
of
these findings
in
humans
remainsspeculative
and needsto
beinvestigated further.
REFERENCES
l.
Sinha AA, Lopez MT, McDevitt HO. Autoirnmune diseases:the failure of self tolerance. Science 1990;248:1380-8. 2. Ramsdell F, Fowlkes BI. Clonal deletion versus clonal
aner-gy: the role ofthe thymus in inducing selftolerance. Science 199O;248:1342-8.
3. Blackman M, Kappler J, Marack P. The role of the T cell receptor in positive and negative selection of developing
T
cells. Science 199O;248:1334-40.
4. Palmer DB, Hayday A, Owen MJ. Is TCR R expression an
essential event in early thynrocyte developrnent. Inrnunol
Tcday 1993;14:460-2.
5. Anderson G, Jenkinson
EI,
Moore NC, OwenIIT.
MHC classlI-
positive epithelium and mesenchyrne cells are both requiredfor
T-cell
developmentin
the thymus. Nature1993;362:70-3.
6. Ardavin C, Wu
L,
Li
C-L, Shortrnan K. Tlryrnic dendritic cells and T cells develop simultaneously in the lhyrnus frorna common precursor population. Nature 1993;362:761-3. 7. MacDonald HR. Deletion !'ers&s anergy: the superantigen
paradi grn. Res Imnrunol 1992;1 43:307 -lO.
8. Baccalà R, Gonâles-Quintial R, Theofilopoulos AN. Lack ofevidence for central T-cell tolerance defects in lupus nrice and for VBdeleting endogenous superantigens in rats and humans. Res Immunol 1992;143:288-90.
9. Wilson
DB,
SteinmanL,
Gold DP. The V-region disease hypothesis: new evidence suggestsit
is
probably wrong. Immunol Today 1993; l4:376-80.10. Hewitt CRA, Lamb JR. Peptide-mediated anergy in hurnan
CD4'T
cells. Res hnmunol 1992;143:294-6.ll.
JenkinsMK.
Tlre role of cell divisionin
the inductionof
clonal anergy. Inrmunol Today 1992;13:69-73.
12. Miller IFAP, Morahan G. Peripheral T cell tolerance. Annu Rev Immunol 1992;10:5 l-69.
13. Lo D. Tolerance to peripheral antigens must involve non-deletion al mechanisms. Res Imm unol 1992;143 :29 6-303. 14. Merino
R,
IwamotoM,
FossatiL,
Muniesa P, Araki K,Takahashi
S,
et al.
Prevention
of
systenric
lupuserythematosus
in
autoimmune BXSB rniceby
trânsgene encoding I-Ec, chain. J Exp Med 1993;178:l 189-97. 15. Arnold B, Schônrich G, Hâmmerling GI. Multiple levelsof
peri pheral tole rance. Imm unol T oday 1993;1 4 :12
4.
16. Protti MP, Manfredi
AA,
Horton RM, BelloneM,
Conti-TronconiBM.
Myasthenia gravis:rccognitionof
a humanautoantigen
at
the molecular
level.
Immunol
Today 1993;14:363-8.17. Ofosu-Appiah
W,
MokhtarianF,
ShirazianD,
Grob D. Production of anti-acetyl choline receptor-alpha antibody in vitro by peripheral blood lymphocytes of patients withrnyas-thenia gravis: role
of
immunoregulatoryT
cells
and monocytes.I
Lab Clin Med 1994;124:231-41.18. Wucheçfennig KW, Weiner HL, Hafler DA. T-cell recog-nition of myelin basic prolein. Inrnrunol Today
l99l;12:217-82.
Med J Indones
19. Toh
BH,
Gleeson PA, VanDriel IR.
Autoimmunity: theparadigm
of
autoimmunegastritis. Today's
Life
Sci t993;5(2):18-27.20. Nossal GJV. Negative selection
of
lymphocytes. Cell 1994;76:229- 39.21.
Heath
WR,
Allison
I,
Hoffrnan
MW,
Schônrich
G, Hâmnrerling G, Arnold B, et al. Autoimmune diabetes as aconsecuence
of
locally
produced
IL-2.
Nature
1992;359:547-9.
22. Heath WR, Miller IFAP. Expression of two cr chains on the surface
of T
cellsin
TCR transgenic mice.J
Exp Med1993;178:1807-l
l.
23. Mamula
MI.
The inability to process a self-peptide allowsautoreactive
T
cells
to
escape tolerance.J
Exp
Med 1993:.1772567- 71.24. Goodnow CC, Adelstein S, Basten A. The need for central and peripheral tolerance
in
theB
cell repertoire. Science 199O;248:1372-9.25. Nemazee
D.
Mechanisms and meaningof
B-lymphocyte tolerance. Reslnrnunol
1992;143 :27 2-5.26.
Eibel
H,
BrombacherF, Kôhler G.
Analysisof
B-cell tolerancein
nrice expressing transgenic anti-CD8.2 im-mtrnoglobulin M molecules. Reslnmunol
19921,143:.276-8.27. Naparstek Y, Plotz PH. The role of autoantibodies in autoim-mune disease. Annu Rev Immunol 1993;l l:79-104. 28. Goodnow
CC,
Brink R,
AdamE.
Breakdownof
self-tolerance in anergy B lyurphocytes. Nature l99l;352:532-6.
29. Sosroseno
W,
HerminajengE.
Interleukin6
(IL-6):
the biochenristry and its role in B and T cell developrnènt. MedI
Univ Indon 1994;3:78-84.30. Linker-Israeli
M.
Cytokine abnormalitiesin
human lupus. Clin lrnrn unol hnrnunopathol 1992;63 :lO-2.31. Goldnran M, Druet P, Gleichrnann E. TH2 cells in systemic autoimrnunity: insights fronr allogeneic diseases and chemi-cally-induced autoimmunity. Imrnunol
Today
l99l;12:
223-7.
32. Waldman
H.
Manipulation
of
T-cell
responseswilh
mon oclonal an tibodi es. Ann u Rev Inrmunol 19 89 ;'l :407 -4 4.33. French
DL,
LaskovR,
SchadfMD.
The roleof
somatic hypern.rutationin
lhe
generationof
antibody diversity,.
Science 1989;244:1152-1.34. Shlonrchik M, Mascelli M, Shan H, Radic MZ, Pisetsky D, Marshak-Rothsteirr
A,
WeigertM.
Anti-DNA
antibodiesfrom
autoirnnrune mice ariseby
clonal expansion and somatic mutation. J Exp Med l99O;171:265-97 .35. Singh AK. Abnornralities in the regulation of variable region genes that encode for antibodies to DNA may be a central factor in the pathogenesis of systen-ric lupus erythematosus. Ann Rlreurn Dis 1993;52:378-83.
36. Bloorn BR, Salganre P, Diamond B. Revisting and revising suppressor T cells. lnrnunol Today 1992;13:l3l-5.
37. Kenreny DM, Noble
A,
Hohnes BJ, Diaz-Sanchez D.ln-mune regulation: â new role for the CDSt T cells. Immunol
Today 1994;15:107-10.
38. Meyers
CM,
Kelly
CJ. Effector nrechanismsin
organ-specific autoirnrnunity. L Clraracterization of a CD8t T cell line that rnecliates murine interstitial nephritis.I
Clin InvestVol 4, No 4, October - Decenber 1995
39. Neilson EG, McCafferty E, Mann R, Michaud L, Clayman
MD.
Tubular antigenderivatized cells inducea
disease-protective, antigen-specific, and idiotype-specific suppres-sor T cell network restricted by I-J nad IgH-V in mice rvith experimental interstitial nephritis. I Exp Med
1985;162:215-30.
40. Meyers CM,
Kelly
CI. Inhibition of nrurine nephritogenic effector T cells by a cione-specific suppressor factor.I
CIin Invest 1994;94 t2093-1O4.41, Tomer
Y,
ShoenfeldY.
The significance ofT
suppressorcells
in
the developmentof
autoimrnunity, J Autoimrnun 1989;2:739-58.42. Arnon R, Teitelbaum D. On the existence of suppressor cells. Int Arch Allergy Immunol 1993;lOO:2-7.
43.
Miller
A,
Lider O, RobertsAB,
SpornMB,
Weiner HL.Suppressor T cells generated by oral tolerization to myelin basic protein suppress both
in
vitro
andin
vivo irnrnuneresponses by the release of transforrning growth lactor B after
anti gen-specific triggering. Proc Natl Acad Sci 1992;89l.
42t-5.
44. Varriale S, Béraud E, Barbaria J, Galibert R, Bernard D. Regulation of experimental autoimmune encephalotnyelitis:
Inhibition
of
adoptive experimental autoinrrnune en-cephalomyelitis by'recovery-associated suppressor cells'. JNeuroimmunol 199 4;53t123 -3
l.
45. Mathieson PW, Stapleton
KI,
Oliviera DBG, LockwoodCM.
Immunoregulationof
mercuric chloride-inducedautoimmunity in Brown Norway rats: a role for CD8+ T cells revealed
by
in
vivo
depletion studies.Eur
J
Lnrnunol l99L;21:2105-9.46. Sosroseno W. The mechanisnrs
of
oral tolerance and inr-munotherapy. J Royal Soc Med 1995; Qn press).47. Weiner HL, Friedman A, Miller A, Khoury SI, Al-Sabbagh A, Santos L, et al. Oral tolerance: Immunologic meclranisms
and treatment of animal and human organ-specific autoirn-mune disease by oral adrninistration of autoantigens. Annu Rev Immunol 1994;L2:8O9-37 .
48. Calder
YL,
Zhao ZS, WangY,
BartonK,
Lightrnan SL.Effects of CD8 depletion on retinal soluble antigen induced
experimental autoimmune
uveoretinitis, lunrunology
1993;79:255-62.
49. Nussenblatt
RB.
Experirnental
autoinrntune uveitis:Mechanisnrs of disease and clinical therapeutic indications. Invest Optlrahnol Vis Sci l99l;32:3131-41.
50. Cohen JJ. Apoptosis. Immunol Today 1993; 14 :1264A.
51. Kabelitz
D,
PohlT,
PechholdK.
Activarion-induced celt death (apoptosis) of mature peripheral T lyrnphocytes. Iur-munol Today 1993; 14:338-9.52. Cohen PI-, Eisenberg
RA.
Thelpr
and g/r/in
systerrnic autoinrmunity:life
and deathin
the Fas lane. Inrruunol Today 1992;13:427-8.53, Wu
I,
Zhou T, He J, Mountz JD. Autoinrmune disease in mice due to integration of an endogenous retrovirus in anapoptosis gene. J Exp Med 1993;178:461-8.
54. Chu JL, Drappa J, Pannassa A, Elkon KB. The defect of Fas
mRNA expression in MRL-lpr nrice is associated with inser-tion of the retrotransposon, ETn.
I
Exp Med1993;178:723-30.
Autoitil,ttune
response
22355. Cheng I, Zhou T, Liu C, Shapiro JP, Brauer MJ, Kieler MC,
et al. Identification of a soluble forrn of the fas molecule that protect cells from fas-mediated apoptosis. Science 1994;
263;1759-6t.
56. Korsmeyer SL BcI-2: a repressor of lyrnphocyte death. Im-munol Today 1992; l3:385-6.
57. Nrinez G, Merino R, Grillot D, Gonzilez-Garcia M. Bcl-2 and Bcl-x:regulatoiy switches for lyrnphoid death and sur-vival. Irnrnunol Today L994;582-8.
58. Strasser A, Whittingharn S, Vaux DI, Bath ML, Adarns
IM,
Cory S, Harris AW. Enforced BCL2 expression in B-lym-phoid cells prolongs antibody responses and elicits
autoim-mune disease. Proc Natl Acad Sci USA 1991;88:8661-5. 59. Aringer Nd Wintersberger W, Steiner CW, Kiener H, Presterl
E, Jaeger U, et al. High levels of Bcl-2 protein in circulating
T
lynphocytes, but notB
lyrnphocytes,of
patients with systernic lupus erythernatosus. Arthr Rheun'r L994;37:1423-30.
60. Naurnovski L, Cleary ML. Bcl2 inhibits apoptosis associated
with terrninal differentiation
of
HL-60 myeloid leukemiacells. Blood 1994;83:2261-1 .
61. Bach JP. Insulin-dependent diabetes rnellitus as an
autoirn-nrune disease. Endocrine Rev 1994;15:516-42.
62. Marrack P, Kappler J, The staphylococcal enterotoxins and
their relatives. Science 1990;248:705-l
l.
63. Taylor fE, Ross DA, Goodacre
IA.
Group A streptococcal antigens and superantigens in tlre pathogenesis ofautoim-nrune artluitis. Eur J Clin Invest 1994;24:5l l-21.
64. Held W, Acha-Orbea H, MacDonald HR, Waanders GA.
Superantigens and retroviral infection: insiglrts frorn mouse
nrarnmaly turnor virus. lnnrunol Today 1994;15:184-90. 65. Acha-Orbea H. Bacterial and viral superantigens: roles on
autoirnmunity. Ann Rheurn Dis 1993;52:S6-516.
66. Christensen T, MQller-Larsen A, Haahr S.
A
retroviral im-plication in multiple sclerosis?TIM
1994;2:332-6. 67. Renno T,7æine R, Girard JIr{, Gillnni S, Dodelet V, OwensT. Selective enriclunenr of Trrl CD45RB|.*CD4* T cells in
autoimrnune
infiltrates
in
experirnental
allergic
en-cephalonryel i tis. Int Irnrnunol 199 4 ;6:3 47 -5 4.
68. Nagelkerken L, Gollob KI, Tielenrans M, Coffinan RL. Role of transfornring growth factor-R in the pr.eferential induction of T helper cells of type
I
by staphylococcal enterotoxin B.Eur
f
hrrnunol 1993;23:2306-lO.69. Schnritz J, Assenrnacher lr{, Radbruch
A.
Regulation of T helper cell cytokine expr.ession: functional dichotornyof
antigen-presenting cells. Eur J lnnrunol 1993;23:l9l-9. 70.
Gollob
KI,
NagelkerkenL,
ColfnranRL.
Endogenousretroviral superantigen preseutation by B cells induces the
developtnent of type
I
CD4'
T helper lyrnphocytes. EurI
Irn rnunol 1993 ;23 :25 65 -7
l.
7L.7agon G, Tuurang
IR, Li Y,
Friedntan SM, Grow MK.Increased frequency of VRlT-positive T cells in patients with rheurnatoid arthlitis. Afthr Rheunr l9g4;37
:l43L40.
72. Mountz JD, Zhout T, Long RE, Bluethmann H, Koopman
WI,
Edward CKIII. T
cell influence onsuperantigen-in-duced
althritis
in MRI--/pr/pr
nice. Arthr
Rheum 1994;37:ll3-24.73. Brocke S, Verorna T, Weissrnnn II-, Gijbels K, Steiman L. Infection and nrultiple sclerosis: a possible role for
224
Sosroseno and Hernûnajeng74. Kim J, Urban RG, Strominger JL, Wiley DC. Toxic shock
syndrome toxin-l complexed with a class II major
histocom-pa tibility molecule HLA-DR
l.
Science 199 4 ;266;187 0- 4.75. Thbodeau
I,
CloutierI,
l.avoie PM, Labrecque N. MouradW, Jardetzky T, et al. Subset of HLA-DRl molecules defined
by SEB and TSST-I binding. Science 1994;216:1874-8.
76. Woodland
DL,
BlackmanMA.
How do T-cell receptors,MHC molecules and superantigens get together? Inrmunol
Today 1993;14:208-12.
77. Brennan FM, Feldman M. Cytokines in autoimmunity.
Cur
Opin Immunol 1992;4 :7 5 4 -9.
78. Opdenakker G, Van Damme I. Cytokine-regulated proteases
in autoimmune diseases. lnmunol Today 1994;15:103-6.
79. Sosroseno W, Herminajeng E. The immunoregulatory role
of transforming gowth factor beta (TGF-B): a review. Br J
Biomed Sci 1995; (In press).
80. Wahl SM, Allen JB, Costa GL, Wong HL, Dasch JR.
Rever-sal of acute and chronic synovial inflammation by
anti-trans-forming growth factor R. J Exp Med 1993;177:225-30.
81. Herron LR, Coffrnan RL, Bond MW, Kotzin BL. Increased
autoantibody production by NZB/NZW B cells in response
to IL-5. J Imrnunol 1988;141:842-8.
82. Ishida H, Muchamuel T, Sakaguchi S, Adrade S, Menon S,
Howard M. Continuous administration of anti-interleukin
l0
antibodies delays onset of autoimmunity in NZB/W F1 nrice.
J Exp Med 1994;179:305-10.
83. Hamzaoui
K,
AyedK,
Slim
A,
HamzaM,
TouraineI.
Natural killer cell activity, interferon-gamma and antibodies
to herpes viruses in patients with Behçet's disease. Clin Exp
Immunol l99O;1 9 :28 -34.
84. Kaneko F, Minagawa T, Takiguchi
Y,
SuzukiM,
Itoh N.Role of cell-mediated inrrnune reaction in blister fornration
of bullous pemphigoid. Dermatology 1992;18 4 :34 -9.
85. Sugita Y, Nagatani T, ikezawa Z, Nornura K, Watanabe Y,
Uitto
I,
NakajirnaH.
Modulationof
bullous pernphigoidantigen
gene expressionby
1-interferonin
culiuredkeratinocytes. Br
I
Dermatol 1992;126:468 -7 3.86. Akahoshi T, Wada C, Eno H, Hirota K, Hosaka S, Takagishi,
et al. Expression
of
monocyte chemotatic and activatingfactor in rheumatoid arthritis. Arthr Rlleurn 1993;36:762-71.
Med J Indones
87. Hulkower KI, Wertheinrer SI, Levin W, Coffey fW,
Ander-son
CM,
ChenT,
etal.
Interleukin-lB induces cytosolicphospholipase
Az
and prostaglandin
H
synthasein
rheumatoid synovial fibroblasts. Arthr Rheum L994;37
:653-61.
88. Thomas R, Carroll GJ. Reduction
of
leukocyte andinter-leukin-lB concentration in the synovial fluid ofrheumatoid
anhritis patients tleated
with
rnethotrexate.Artlu
Rheum 1993;36:1244-52.89. Manicourt DH, Triki R, Fularda K, Devogelaer IP, de
Deux-chaisnes CN, Thonar EJMA. Levels
of
circulating tumornecrosis
factor
cr
and interleukin-6
in
patientswith
rheumatoid arthritis. Arthr Rheum 1993;36:490-9.
90. William RO, Feldmann M, Maini RN. Anti-TNF arneliorates
joint disease in rnurine collagen-induced arthritis. Proc Natl
Acad Sci USA 1992;89:9784-8.
91. Greczy
AF,
EdwardsCM,
McGuiganLE,
Sullivan JS.Cross-reacting determinants
in
ankylosing spondylitis. ISIAtlas Sci Immunol 1988;l:7-10.
92. Baurn H, Butler P, Davies H, Sternberg MIE, Burroughs AK.
Autoitnrnune disease and nrolecular minricry: an hypothesis.
TIBS 1993;18:,140-4.
93. Wordsworth P. Rheun'ratoid arthritis. Curr Opin lnmunol
1992;4:166- 9.
94. Rook GAW, Lydyald PM, Stanford JL. A reappraisal of the
evidence
that
rheurnatoidarthritis
and several otheridiopathic diseases are slow bactelial infections. Ann Rheum
Dis 1993;52:S30-8.
95.
McCulloch
J,
Lydyard PM, Rook GAW.
Rheurnatoidarthritis: how well do the theolies fit the evidence? Clin Exp
Imnrunol 1993;92:l-6.
96. Lehner T, Lavery E, Snrith R, van der Zæe R, Mizushirna Y,
Shinnick
T.
Association between the 65-kilodalton heat slrock protein, Streptococcu:; sangris, and the correspondingantibodies
in
Behçet's syndrome.
Infect
Immun
199 l;59:.1434-41 .
97. Stanford
MR,
Kasp E, WhistonR,
HasanA,
Todryk S,Shinnick T, et al. Heat shock plotein peptides reactive in
pâtients with Behcet's disease are uveitogenic in Lewis rats.