PENGKONSTRUKSIAN
BIDIRECTED OVERLAP GRAPH
UNTUK DNA
SEQUENCE
ASSEMBLY
ALBERT ADRIANUS
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM
INSTITUT PERTANIAN BOGOR
PERNYATAAN MENGENAI SKRIPSI DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA
Dengan ini saya menyatakan bahwa skripsi berjudul Pengkonstruksian Bidirected Overlap Graph Untuk DNA Sequence Assembly adalah benar karya saya dengan arahan dari komisi pembimbing dan belum diajukan dalam bentuk apa pun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir skripsi ini.
Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
ABSTRAK
ALBERT ADRIANUS. Pengkonstruksian Bidirected Overlap Graph Untuk DNA SequenceAssembly. Dibimbing oleh WISNU ANANTA KUSUMA.
DNA sequencing technologies dapat digunakan untuk memecah gen bakteri menjadi jutaan potongan kecil yang disebut reads. Namun demikian, reads tersebut perlu disambung menjadi contigs agar dapat digunakan. Terdapat 2 metode untuk menyambungkan reads. Yang pertama dengan menggunakan overlap layout consensus (OLC) dan yang lainnya dengan menggunakan de Bruijn graph. Hal penting dalam metode OLC adalah bagian overlap dari masing-masing reads. Pada penelitian ini dikembangkan sebuah sistem untuk membuat bidirected overlap graph yang nantinya akan menghitung berapa jumlah reads yang saling overlap satu sama lain. Suffix array digunakan untuk menentukan fase/bagian overlap dari setiap reads dengan mengindeks setiap suffix dari reads. Waktu adalah parameter penting dalam DNA assembly karena DNA assembly membutuhkan banyak waktu. Untuk mengurangi waktu dilakukan perubahan dari masing-masing suffix dan prefix menjadi suatu nilai tertentu yang bersifat tunggal dan mencari overlap dengan membandingkan setiap reads. Cara ini memberikan dampak positif daripada perbandingan dengan menggunakan string. Perbandingan waktu yang diperlukan antara perbandingan angka dan perbandingan string cukup signifikan. Untuk 2000 dan 5000 reads, sistem dapat memberikan hasil 100% akurat untuk jumlah node dan edge.
Kata kunci: bidirected overlap graph, DNA assembly, overlap layout consensus
ABSTRACT
ALBERT ADRIANUS. Bidirected Overlap Graph Construction For DNA SequenceAssembly. Supervised by WISNU ANANTA KUSUMA.
High-throughput DNA sequencing technologies can be used to decipher a bacterial genome to millions of fragments called reads. However, reads must be assembled to contigs before it can be used. There are two methods to assemble reads. The first one is using overlap layout consensus (OLC) methods and the other one by using de Bruijn graph. The main thing in the OLC is the overlapping phase. In this project, we will develop a system to build bidirected overlap graph for counting how many reads overlapping each other. Suffix array is used to determine overlapping phase from each reads by indexing every reads’s suffix. Time is an important parameter for DNA assembly. To reduce time, we convert every suffix and prefix to a unique number and search for an overlap by comparing each reads. The method gives a positive result than the string method. The different of time consumed by the first method and the second method is fairly significant. For 2000 and 5000 reads, the system can give a 100% accuracy for the total numbers of node and edge.
Skripsi
sebagai salah satu syarat untuk memperoleh gelar Sarjana Ilmu Komputer
pada
Departemen Ilmu Komputer
PENGKONSTRUKSIAN
BIDIRECTED OVERLAP GRAPH
UNTUK DNA
SEQUENCE
ASSEMBLY
ALBERT ADRIANUS
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM
INSTITUT PERTANIAN BOGOR
Penguji:
Judul Skripsi : Pengkonstruksian Bidirected Overlap Graph Untuk Dna Sequence Assembly
Nama : Albert Adrianus NIM : G64090109
Disetujui oleh
Dr Wisnu Ananta Kusuma, MT ST Pembimbing
Diketahui oleh
Dr Ir Agus Buono, MSi MKom Ketua Departemen
PRAKATA
Pertama, penulis ingin mengucapkan syukur kepada Tuhan Yang Maha Esa atas selesainya penelitian dan tugas skripsi yang berjudul Pengkonstruksian Bidirected Overlap Graph Untuk DNA SequenceAssembly.
Penulis juga ingin berterima kasih kepada Bapak Wisnu Ananta Kusuma selaku pembimbing skripsi yang telah membimbing dengan sabar dan senantiasa memberi saran ketika penulis membutuhkan pertolongan. Selain itu, penulis juga ingin berterima kasih kepada orangtua yang selalu mendukung penulis dalam studi dan kepada saudari Nesya Nova Febriane yang membantu dalam proses pengumpulan data. Penulis berharap agar hasil karya ini dapat bermanfaat bagi orang lain.
DAFTAR ISI
DAFTAR TABEL vi
DAFTAR GAMBAR vi
DAFTAR LAMPIRAN vi
PENDAHULUAN 1
Latar Belakang 1
Perumusan Masalah 2
Tujuan Penelitian 2
Ruang Lingkup Penelitian 2
TINJAUAN PUSTAKA 2
DNA sequencing 2
Sequence assembly 3
OLC (Overlap Layout Consensus) 3
EDENA 4
Bidirected overlap graph 5
METODE 5
Pengumpulan data DNA sequence dan analisis kebutuhan 5
Penganalisisan Data 6
Pengevaluasian jumlah node dan edge serta waktu eksekusi 11
HASIL DAN PEMBAHASAN 13
Hasil 13
Pembahasan 15
SIMPULAN DAN SARAN 18
Simpulan 18
Saran 18
DAFTAR TABEL
1 Contoh-contoh assembler untuk setiap metode 3
2 Nilai masing-masing basa nitrogen 10
3 Proses pencarian overlap 11
4 Matriks untuk perhitungan jumlah node dan edge 13 5 Perbandingan jumlah node dan edge untuk 5000 reads (angka dan string) 13
DAFTAR GAMBAR
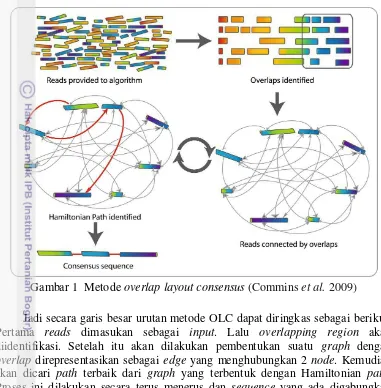
1 Metode overlap layout consensus 4
2 Contoh bidirected overlap graph 5
3 Contoh hasil simulasi metasim 6
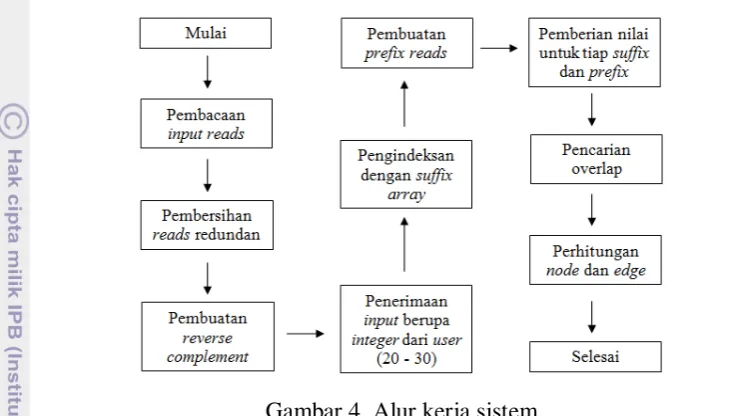
4 Alur kerja sistem 7
5 Pengindeksan suffix array 9
6 Suffix array setelah diurutkan 9
7 Kode pemberian nilai suffix/prefix 12
8 Kode pemberian nilai jumlah karakter suffix/prefix 12
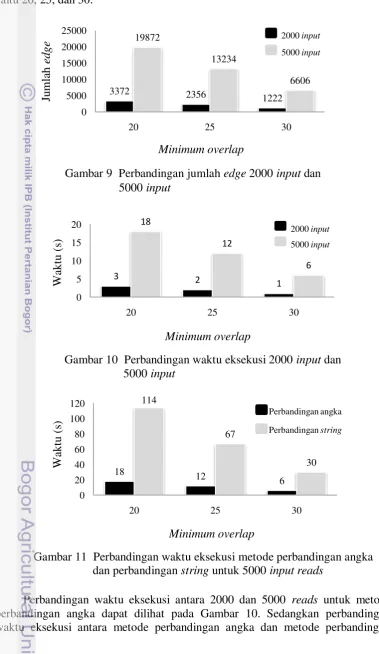
9 Perbandingan jumlah edge 2000 input dan 14
10 Perbandingan waktu eksekusi 2000 input dan 14
PENDAHULUAN
Latar Belakang
Teknologi DNA sequence assembly telah berkembang pesat dan mempunyai peran penting dalam pembelajaran mengenai genome. Teknik DNA sequence assembly muncul karena hingga saat ini belum ada teknologi yang dapat memecah/menguraikan DNA suatu organisme yang menghasilkan whole genome dalam satu kali percobaan. Sebelumnya teknologi yang digunakan untuk memecah DNA adalah teknologi Sanger. Dengan Sanger, dihasilkan potongan-potongan reads dengan panjang 400-800 base pairs (bp) namun membutuhkan waktu yang agak lama. Seiring berjalannya waktu, ditemukan suatu teknologi yaitu high throughput sequencing technologies. Dengan teknologi tersebut, gen bakteri dimungkinkan untuk diuraikan hanya dalam satu eksperimen dan dengan biaya yang tidak tinggi (Brenner et al. 2000). Hasil dari penguraian ini berupa jutaan potongan reads dengan panjang 35-50 bp. Namun demikian, potongan reads tersebut baru dapat digunakan dengan menyambung reads tersebut menjadi potongan-potongan yang lebih panjang (contigs). Karena itu diperlukan metode untuk menyambung (assemble) reads yang ada. Secara garis besar metode yang digunakan untuk menyambung reads ada dua yaitu metode overlap layout consensus (OLC) dan dengan menggunakan graf de Bruijn (Kusuma et al. 2011). Pada penelitian ini yang digunakan adalah metode OLC.
Sampai saat ini, pengembangan software untuk melakukan penyambungan reads terus dilakukan. Salah satu software yang menggunakan metode OLC untuk menyambung reads adalah Edena. Pada Edena, data input yang berupa potongan-potongan short reads akan diproses menjadi output yang berupa potongan-potongan contigs serta jumlah node dan edge yang terbentuk dari proses-proses yang digunakan di dalam metode OLC. Proses-proses tersebut terdiri atas proses penghilangan data reads yang redundan, pembuatan bidirected overlap graph, penghilangan transitive edges, serta pembersihan graph (Hernandez et al. 2008). Semua proses tersebut harus dilakukan secara berurutan.
Proses penghilangan data reads yang redundan dilakukan untuk membuat proses selanjutnya menjadi lebih simple. Hal ini dikarenakan apabila jumlah reads semakin banyak akan membuat overlap graph menjadi semakin kompleks. Hal ini diperparah apabila ada reads yang sama karena dapat menyebabkan cycle. Selanjutnya proses yang menjadi bahasan utama dalam penelitian ini yaitu pembuatan bidirected overlap graph. Proses ini digunakan untuk mendeteksi overlap dari masing-masing reads. Proses penghilangan transitive edges dan pembersihan graph secara garis besar digunakan untuk membuat graph yang sudah ada menjadi lebih simple.
2
overlap dari masing-masing reads. Adapun input yang digunakan adalah potongan-potongan reads dengan panjang yang sama dan output yang dihasilkan adalah jumlah node dan edge yang dihasilkan dari proses yang ada.
Perumusan Masalah
Perumusan masalah pada penelitian ini adalah untuk melihat apakah metode yang digunakan dapat membuat bidirected overlap graph dengan ketepatan tinggi dan memiliki waktu eksekusi yang cepat. Data keluaran dan waktu eksekusi akan dibandingkan dengan data keluaran dan waktu eksekusi yang dihasilkan dengan metode perbandingan string. Diharapkan dengan metode yang dipakai dapat menghasilkan keluaran dengan ketepatan tinggi dan waktu eksekusi yang lebih cepat.
Tujuan Penelitian
Penelitian ini bertujuan untuk mengkonstruksi bidirected overlap graph untuk DNA sequenceassembly.
Ruang Lingkup Penelitian
Ruang lingkup penelitian ini adalah:
1 Penelitian ini hanya sampai tahap pengkonstruksian bidirected overlap graph. 2 Data read yang dipakai adalah potongan Acidiphilium multivorum AIU301
plasmid pACMV4 DNA dengan panjang sama yaitu 35 bp. 3 Jumlah read yang dipakai adalah 2000 buah dan 5000 buah.
4 Data yang dipakai berekstensi txt dan data output dari software MetaSim. 5 Data read yang dipakai error free.
6 Jumlah minimum overlap yang digunakan adalah 20, 25, dan 30
TINJAUAN PUSTAKA
DNA Sequencing
Namun demikian, Sanger sequencer menghasilkan potongan/fragmen yang lebih sedikit daripada next generation sequencer (Kusuma et al. 2011) sehingga next generation sequencer lebih banyak digunakan dalam penelitian. Teknik DNA sequencing secara garis besar terbagi menjadi 2 yaitu shotgun sequencing dan map-based sequencing. Teknik shotgun sequencing lebih banyak digunakan dalam penelitian mengenai DNA sequence (Chaisson et al. 2004). Teknik shotgun sequencing terdiri atas Sanger sequencing, 454 sequencing, dan Illumina sequencing (Abegunde 2010).
Sequence Assembly
DNA sequence assembly merupakan proses penyatuan/perakitan potongan-potongan reads untuk menentukan whole DNA suatu organisme. Teknik penyatuan DNA sequence secara garis besar terbagi atas metode OLC, Eulerian path, dan align-layout consensus. Pada metode align layout consensus, sequence hanya dibandingkan dengan hasil sequence assembly yang sudah ada dan paling dekat dengan sequence yang dipakai. Saat ini terdapat banyak assembler yang dikembangkan dari metode-metode tersebut. Beberapa assembler dapat dilihat pada Tabel 1. Namun demikian, setiap assembler walaupun dikembangkan dari metode yang sama, tetap mempunyai pendekatan masing-masing yang berbeda satu dengan yang lain. Selain pendekatan yang berbeda, penggunaan dari masing-masing assembler pun juga berbeda. Sebagai contoh Phrap assembler digunakan untuk proses penyatuan dengan input berupa long reads. Berbeda halnya dengan Edena yang digunakan untuk proses penyatuan short reads.
Tabel 1 Contoh-contoh assembler untuk setiap metode
Jenis metode Assembler
OLC Phrap, TIGR, Edena
Eulerian Path Velvet, Euler
Align-layout consensus AMOS, Mosaik OLC
4
yang dapat digunakan adalah Edena. Edena digunakan untuk menyatukan reads-reads yang termasuk ke dalam short reads.
Jadi secara garis besar urutan metode OLC dapat diringkas sebagai berikut. Pertama reads dimasukan sebagai input. Lalu overlapping region akan diidentifikasi. Setelah itu akan dilakukan pembentukan suatu graph dengan overlap direpresentasikan sebagai edge yang menghubungkan 2 node. Kemudian akan dicari path terbaik dari graph yang terbentuk dengan Hamiltonian path. Proses ini dilakukan secara terus menerus dan sequence yang ada digabungkan untuk menghasilkan final consensus sequence yang merepresentasikan whole genome suatu organisme (Commins et al. 2009).
Edena
Edena merupakan suatu assembler yang digunakan untuk menyatukan potongan reads yang termasuk ke dalam short reads dengan pendekatan overlap layout consensus. Secara sederhana, tahapan penting dalam Edena dapat dibagi menjadi beberapa tahapan. Tahapan pertama adalah tahap penghilangan reads yang redundan. Tahapan kedua adalah tahapan pembentukan suatu bidirected overlap graph. Overlap graph tersebut terdiri dari node yang berupa potongan-potongan reads dan edge yang berupa overlap dari masing-masing reads yang ada. Namun demikian overlap yang didata hanya overlap yang melebihi panjang minimum overlap yang telah diberlakukan. Tahap ketiga adalah tahap pembersihan graph yang dilakukan dengan penghilangan transitive edge dan pembersihan bubbles. Tahap terakhir adalah proses penyatuan contigs sebagai output (Hernandez et al. 2008).
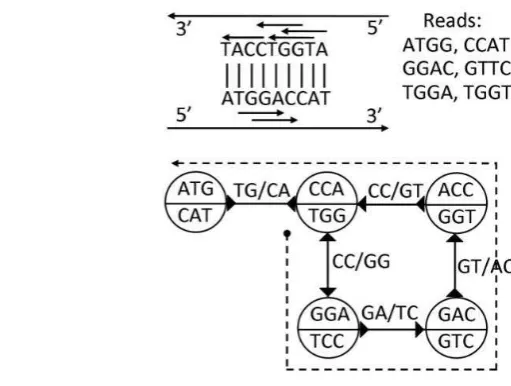
Bidirected Overlap Graph
Bidirected overlap graph merupakan suatu graph dengan node dan edge yang ada merepresentasikan reads yang saling overlap satu sama lain. Setiap edge dalam bidirected overlap graph mempunyai tanda panah di kedua ujung dari edge tersebut. Karena itu, terdapat 4 cara untuk menghubungkan dua node berdasarkan perpaduan dari tanda panah di kedua ujung edge (Hernandez et al. 2008). Contoh bidirected overlap graph dapat dilihat pada Gambar 2.
METODE
Pada penelitian ini akan dilakukan beberapa tahap yang akan dilakukan. Tahap-tahapan tersebut antara lain pengumpulan data DNA sequence dan analisis kebutuhan, penganalisisan data, dan pengevaluasian jumlah node dan edge pada graph yang dihasilkan serta waktu eksekusi yang diperlukan.
Pengumpulan Data DNA Sequence dan Analisis Kebutuhan
Data yang digunakan merupakan potongan-potongan reads dengan panjang 35 bp berjumlah 2000 dan 5000 reads yang diambil dari simulasi dengan software MetaSim. Setiap reads merupakan kombinasi dari basa-basa nitrogen yang terdiri dari Adenine, Cytosine, Guanine dan Thymine. Data yang diperlukan merupakan data yang bebas dari redundan. Organisme yang dipakai pada penelitian ini adalah Acidiphilium multivorum AIU301 plasmid pACMV4 DNA dengan panjang sequence asli 40 588 bp.
Dalam simulasi menggunakan MetaSim, digunakan error model dengan pilihan exact. Hal tersebut dilakukan agar data yang dihasilkan nantinya merupakan data yang bebas kesalahan (error free). Data yang error free itu sendiri merupakan data dimana setiap basa-basa nitrogen dalam fragmen-fragmen hasil tidak digantikan oleh basa nitrogen lainnya sehingga hasil akhirnya
6
merupakan potongan-potongan yang sama dengan organisme aslinya. Namun demikian hasil simulasi dari software MetaSim masih memungkinkan memiliki data yang redundan. Data redundan yang dimaksud adalah adanya reads yang sama persis satu dengan yang lain. Pada penelitian ini data masukan yang dibutuhkan merupakan data yang tidak redundan. Hal tersebut diperlukan agar graph yang akan dibentuk tidak kompleks. Contoh hasil dari simulasi dengan menggunakan software MetaSim dapat dilihat pada Gambar 3.
Gambar 3 Contoh hasil simulasi MetaSim
Data yang akan diambil hanyalah bagian reads saja. Bagian keterangan tidak akan dibaca oleh sistem. Contoh bagian data reads yang akan dibaca oleh sistem adalah “ATCCTCGACGAACAGCCAGTCGCGCAGGTTGGAGC”. Data yang digunakan nantinya bukan saja reads yang merupakan input, namun demikian juga reverse complement dari setiap reads yang ada. Reverse complement itu sendiri merupakan reade yang urutannya dibalik lalu ditukar dengan komplemennya. Dalam hal ini basa nitrogen A diganti dengan basa nitrogen T, basa nitrogen T diganti dengan basa nitrogen A, basa nitrogen C diganti basa nitrogen G, dan basa nitrogen G diganti dengan basa nitrogen C. Akhirnya data yang digunakan oleh sistem adalah reads yang tidak redundan dan reverse complement dari setiap reads yang tidak redundan itu.
Dari hasil analis yang telah dilakukan, diperoleh kebutuhan perangkat lunak yang digunakan dalam pengembangan sistem. Perangkat lunak yang digunakan untuk mengembangkan sistem ini adalah Dev-C++ 5.4.1. Sedangkan perangkat
keras yang digunakan untuk mengembangkan sistem ini adalah notebook bertipe Asus A42J series, CPU Intel Core i3-380M 2.53 GHz dengan memori 4 GB.
Penganalisisan Data
pembuatan reverse complement, pengindeksan dengan menggunakan suffix array, pencarian overlap antar reads dan reverse complement, serta perhitungan node dan edge sebagai hasil keluaran dari sistem. Alur kerja sistem secara ringkas dapat dilihat pada Gambar 4.
Pembacaan input reads
Pertama-tama sistem akan membaca data masukan yang berupa potongan-potongan reads hasil simulasi dari software MetaSim. Data yang akan dibaca oleh sistem hanyalah data input yang merupakan potogan dari DNA, sedangkan data yang berupa keterangan tidak dibaca oleh sistem. Potongan-potongan reads tersebut mempunyai panjang 35 bp. Data yang digunakan sebanyak 2000 dan 5000 buah.
Pembersihan inputreads redundan
Reads yang telah dibaca oleh sistem akan diperiksa satu persatu. Apabila ada reads identik yang berjumlah lebih dari satu maka sistem hanya akan mengambil salah satu reads. Hal ini agar data input yang akan digunakan untuk proses selanjutnya bebas dari data yang redundan. Data yang bebas redundan tersebut kemudian akan dimasukan ke dalam sebuah vector untuk digunakan pada proses selanjutnya.
Pembuatan reverse complement
Setelah itu sistem akan membuat reverse complement dari masing-masing reads yang menjadi masukan. Reverse complement itu sendiri merupakan reads yang isinya ditukar menurut pasangan basa nitrogennya (A menjadi T, C menjadi G, G menjadi C, dan T menjadi A) dan setelah itu urutan dari basa-basa nitrogen tersebut diurutkan terbalik. Hal ini dikarenakan high throughput sequencing technologies menguraikan DNA dari 2 sisi, dari sisi kiri dan sisi kanan. Karena itu, hasil potongan-potongan reads tidak diketahui apakah merupakan potongan dari rantai primer atau merupakan bagian dari rantai sekunder.
8
Penentuan jumlah minimum overlap
Poin penting dalam penelitian ini adalah jumlah minimum overlap, karena akan menentukan banyaknya jumlah reads overlap satu sama lain (node) dan jumlah overlap secara keseluruhan (edge). Hal ini juga dikarenakan apabila jumlah minimum overlap terlalu sedikit akan membuat graph yang dibentuk menjadi semakin kompleks. Graph yang terbentuk semakin kompleks karena semakin kecil jumlah minimum overlap maka jumlah reads yang saling overlap satu sama lain akan semakin banyak. Hal tersebut akan membuat jumlah node dan edge yang dihasilkan akan semakin banyak. Sedangkan apabila jumlah minimum overlap terlalu besar, maka akan menyebabkan semakin sedikit reads yang saling overlap. Hal tersebut akan menyebabkan terjadinya dead end path pada graph ketika proses penyambungan kumpulan reads yang saling overlap. Karena jumlah minimum overlap menjadi penting maka selanjutnya sistem akan meminta jumlah minimum overlap yang diharapkan oleh user. Jumlah minimum overlap yang digunakan dalam rentang 20 sampai 30.
Pengindeksan dengan suffix array
Tahap selanjutnya yaitu masing-masing reads akan diindeks dengan menggunakan suffix array. Suffix array merupakan perkembangan dari suffix tree. Suffix array adalah sebuah list terurut dari semua suffix suatu kata (Manberdan Myers 1993). Misalkan terdapat sebuah string “abracadabra”. Hal pertama yang akan dilakukan adalah proses pemasangan indeks ke masing-masing karakter dari string tersebut. Sebagai contoh pada string “abracadabra”, a akan diberi indeks 1, b diberi indeks 2, r diberi indeks 3, a diberi indeks 4, c diberi indeks 5, a diberi indeks 6, d diberi indeks 7, a diberi indeks 8, b diberi indeks 9, r diberi indeks 10, dan a diberi indeks 11. Setelah pemasangan indeks terhadap masing-masing karakter dari string tersebut kemudian dibuat suffix dari string tersebut sesuai dengan indeks yang diberikan. Dalam kasus dengan string “abracadabra” maka akan terdapat 11 suffix sesuai indeks yang ada yaitu “abracadabra”, “bracadabra”, “racadabra”, “acadabra”, “cadabra”, “adabra”, “dabra”, “abra”, “bra”, “ra”, dan “a”. Untuk lebih jelasnya dapat dilihat pada Gambar 5. Setelah didapatkan indeks seperti di atas maka kemudian akan diurutkan secara lexicographical atau berdasarkan abjad seperti pada Gambar 6.
Pembuatan prefix
Pada tahap ini akan dicari prefix dari setiap reads dan reverse complement yang ada. Prefix yang dicari juga mempunyai ketentuan tertentu. Prefix akan didata apabila memiliki jumlah karakter lebih besar dari jumlah minimum overlap yang dimasukan oleh user sebelumnya. Proses yang dilakukan sebenarnya sama dengan proses pencarian suffix, namun demikian bedanya pada tahap ini yang dicari adalah bagian prefix.
Pemberian nilai pada suffix dan prefix
Setelah masing-masing reads dan reverse complement telah memiliki prefix dan suffix, maka setiap prefix dan suffix diubah menjadi angka. Adapun pengubahan ini dilakukan untuk membuat waktu yang dibutuhkan pada proses
Gambar 5 Pengindeksan suffix array
10
selanjutnya menjadi lebih singkat. Proses pengubahan dari string menjadi double dilakukan dengan ketetapan tertentu. Aturan pertama adalah dengan cara membuat angka tertentu bagi masing-masing huruf dari basa nitrogen yang ada. Penentuan nilai bagi masing-masing basa nitrogen dapat dilihat pada Tabel 2.
Tabel 2 Nilai masing-masing basa nitrogen
Basa nitrogen Nilai
A 1.000000
T 0.110000
G 0.111100
C 0.111111
Setelah itu dari masing-masing nilai dari huruf basa nitrogen yang ada dikalikan dengan posisinya dan dengan suatu nilai tertentu. Lalu semua nilai tersebut dijumlahkan hingga menjadi sebuah nilai tunggal yang merepresentasikan suffix dan prefix dari masing-masing reads dan reverse complement yang ada (aturan pertama). Nilai hasil tersebut memungkinkan adanya suatu collision yang yaitu keadaan dimana ada dua suffix/prefix yang memiliki nilai yang sama namun bentuk fisik yang berbeda, karena itulah diperlukan nilai kedua yaitu nilai jumlah karakter suatu suffix/prefix (aturan kedua). Nilai suatu suffix/prefix (nilai suatu overlap) dapat dicari dengan formula seperti berikut:
Basa nitrogen “A”:
Nilai suatu suffix/prefix = maxi • a • 0.11 i=1
Basa nitrogen “T”:
Nilai suatu suffix/prefix = maxi=1 i • a • 0.1111 Basa nitrogen “G”:
Nilai suatu suffix/prefix = maxi=1 i • a • 0.111111 Basa nitrogen “C”:
Nilai suatu suffix/prefix = maxi=1 i • a • 0.11111111 Keterangan:
i : posisi suatu variabel a : nilai variabel pada posisi i
max : jumlah maksimal variabel suatu suffix atau prefix
Nilai jumlah karakter suffix/prefix = a max
i=1
Keterangan:
a : nilai variabel pada posisi i
max : jumlah maksimal variabel suatu suffix atau prefix
Cuplikan kode program untuk pemberian nilai suatu suffix/prefix dan nilai jumlah karakter suffix dan prefix dapat dilihat pada Gambar 7 dan Gambar 8. Pencarian bagian overlap
Setelah didapatkan nilai dari tiap prefix dan suffix menurut aturan pertama dan kedua, maka akan dilakukan proses pencarian overlap. Proses pencarian overlap dilakukan dengan membandingkan nilai dari masing-masing prefix dan suffix dari reads serta reverse complement satu sama lain.
Proses perbandingan dilakukan dengan mengurangkan nilai dari masing-masing suffix yang ada dengan nilai dari masing-masing prefix yang ada. Suffix yang digunakan bukan hanya berasal dari bagian reads saja tetapi juga dari bagian reverse complement. Demikian juga prefix yang digunakan merupakan prefix dari bagian reads dan bagian reverse complement. Hal ini menyebabkan adanya 4 proses perbandingan yang dilakukan oleh sistem untuk menemukan bagian overlap dari masing-masing reads. Dalam hal ini, apabila nilai aturan pertama dan aturan kedua bernilai 0 maka reads/reverse complement dengan suffix tersebut memiliki overlap dengan reads/reverse complement dengan prefix yang bersangkutan. Untuk lebih jelasnya dapat dilihat pada Tabel 3.
Tabel 3 Proses pencarian overlap Jenis Keterangan
S reads - P reads
Nilai suffix dari masing-masing read dikurangkan dengan nilai prefix dari masing-masing reads
S reads - P RC
Nilai suffix dari masing-masing read dikurangkan dengan nilai prefix dari masing-masing reverse complement
S RC - P reads
Nilai suffix dari masing-masing reverse complement dikurangkan dengan nilai prefix dari masing-masing reads
S RC - P RC
Nilai suffix dari masing-masing reverse complement dikurangkan dengan nilai prefix dari masing-masing reverse complement Pengevaluasian jumlah node dan edge serta waktu eksekusi
12
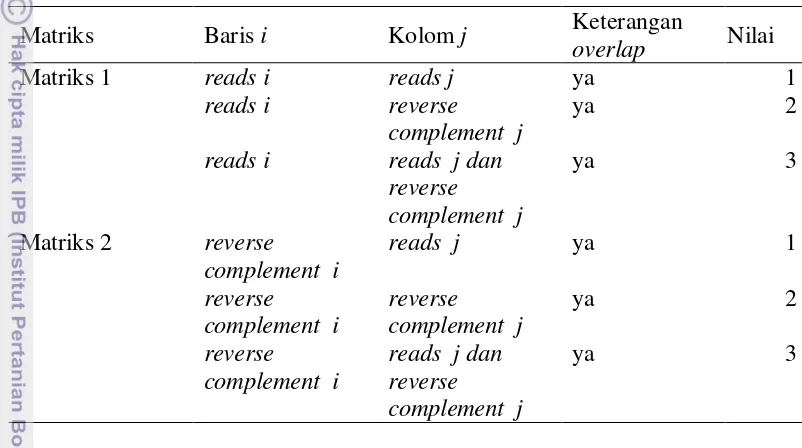
Setelah proses pencarian overlap dilakukan maka setiap reads yang saling overlap akan dimasukan ke dalam suatu matriks yang mempunyai ukuran n × n. Huruf n merupakan perwakilan dari jumlah reads keseluruhan. Pada sistem ini digunakan 2 matriks. Matriks pertama digunakan untuk menandai bagian overlap pada reads dan matriks kedua digunakan untuk menampung bagian overlap dari reverse complement. Pada matriks 1, apabila reads ioverlap dengan reads j maka matriks 1 baris i dan kolom j akan berisi nilai integer 1. Apabila reads i overlap dengan reverse complement j maka matriks 1 baris i dan kolom j akan berisi nilai integer 1. Sedangkan apabila reads i overlap dengan reads j dan reverse complement j maka nilainya akan menjadi 3. Hal yang sama juga diberlakukan pada matriks 2. Apabila reverse complement i overlap dengan reads j maka matriks 2 baris i dan kolom j akan berisi nilai integer 1. Apabila apabila reverse complement i overlap dengan reverse complement j maka matriks 2 baris i dan kolom j akan berisi nilai integer 1. Sedangkan apabila reverse complement i overlap dengan reads j dan reverse complement j maka nilainya akan menjadi 3. Setelah semua matriks terisi maka dicari jumlah node dan edge dari kedua matriks tersebut. Secara ringkas dapat dilihat pada Tabel 4.
Gambar 7 Kode pemberian nilai suffix/prefix
Selain jumlah node dan edge, salah satu hal yang diperhatikan dalam penelitian ini adalah waktu eksekusi yang diperlukan oleh metode perbandingan angka dan metode perbandingan string untuk jumlah input yang sama. Waktu eksekusi dihitung setelah user memasukan input berupa nilai panjang minimum overlap yang diinginkan. Waktu ekseskusi digunakan untuk membandingkan antara metode perbandingan angka dan metode perbandingan string. Satuan waktu yang digunakan adalah detik (s).
Tabel 4 Matriks untuk perhitungan jumlah node dan edge
Matriks Baris i Kolom j Keterangan
overlap Nilai
Matriks 1 reads i reads j ya 1
reads i reverse complement j
ya 2
reads i reads j dan reverse complement j
ya 3
Matriks 2 reverse complement i
reads j ya 1
reverse complement i
reverse complement j
ya 2
reverse complement i
reads j dan reverse complement j
ya 3
HASIL DAN PEMBAHASAN
Hasil
Pada penelitian ini jumlah minimum overlap yang digunakan adalah 20, 25, dan 30. Hasil penelitian yang dicari adalah jumlah node dan edge dan waktu eksekusi yang dibutuhkan. Perbandingan jumlah node dan edge antara penggunaan metode perbandingan angka dan metode perbandingan string dapat dilihat pada Tabel 5.
Tabel 5 Perbandingan jumlah node dan edge untuk 5000 reads (angka dan string) Perbandingan angka Perbandingan string
Minimum
overlap Jumlah node Jumlah edge
Minimum
overlap Jumlah node
Jumlah edge
20 4 704 19 872 20 4 704 19 872
25 4 436 13 234 25 4 436 13 234
30 3 469 6 606 30 3 469 6 606
14
hitam mewakili jumlah input 2000. Garis vertikal merupakan jumlah edge sedangkan garis horizontal merupakan jumlah minimum overlap yang digunakan yaitu 20, 25, dan 30.
Pembahasan
Perbandingan waktu eksekusi antara 2000 dan 5000 reads untuk metode perbandingan angka dapat dilihat pada Gambar 10. Sedangkan perbandingan waktu eksekusi antara metode perbandingan angka dan metode perbandingan
Gambar 9 Perbandingan jumlah edge 2000 input dan 5000 input
3372 2356
1222 19872
13234
6606
0 5000 10000 15000 20000 25000
20 25 30
Series1 Series2
2000input
5000 input
Gambar 10 Perbandingan waktu eksekusi 2000 input dan 5000 input
3
2 1
18
12
6
0 5 10 15 20
20 25 30
Series1 Series2
2000input
5000 input
Gambar 11 Perbandingan waktu eksekusi metode perbandingan angka dan perbandingan string untuk 5000 input reads
18 12
6 114
67
30
0 20 40 60 80 100 120
20 25 30
Series1 Series2
Perbandingan angka
Perbandingan string
Minimum overlap
Minimum overlap
Minimum overlap
Juml
ah
edge
W
aktu
(s
)
W
aktu
(s
string untuk 5000 reads dapat dilihat pada Gambar 11. Perhitungan waktu dimulai ketika user telah memasukan nilai jumlah minimum overlap sebagai input yang dibutuhkan oleh sistem.
Pembahasan
Beberapa hal yang akan dibahas adalah mengenai jumlah node, edge, dan waktu. Pada pembahasan ini akan dicoba untuk menjawab apakah metode yang dipakai mempunyai ketepatan yang baik dan waktu eksekusi yang lebih cepat dari metode perbandingan string.
Ketepatan hasil node dan edge
Hasil node dan edge yang dihasilkan oleh sistem akan dibandingkan dengan hasil node dan edge yang dihasilkan dengan metode perbandingan string. Perbandingan dilakukan dengan jumlah reads 5000 buah. Dapat dilihat pada Tabel 5, perbandingan antara jumlah node dan edge dari metode perbandingan angka (metode sistem) dan metode perbandingan string. Dari hasil yang didapat, hasil sistem mempunyai ketepatan 100% dibandingkan dengan metode perbandingan string untuk jumlah reads 5000 buah. Hal ini menyatakan bahwa metode yang digunakan pada sistem (perbandingan angka) berhasil memberikan hasil yang maksimal. Ketepatan dari sistem ini tak lepas dari nilai suatu suffix/prefix dan nilai jumlah karakter overlap yang digunakan. Nilai dari suatu suffix/prefix diharapkan mempunyai nilai yang unik. Unik yang dimaksud adalah bahwa nilai suatu suffix/prefix dari suatu suffix atau prefix sama satu sama lain apabila suffix dan prefix tersebut mempunyai karakter yang terdiri dari basa-basa nitrogen dengan jumlah yang sama dan dengan urutan yang sama. Namun demikian karena hasil suffix dan prefix yang dihasilkan dari reads dan reverse complement sangat banyak maka dapat menyebabkan ada beberapa nilai yang tidak unik. Maksud tidak unik disini adalah bahwa ada suffix atau prefix yang mempunyai nilai suatu suffix/prefix yang sama tetapi sebenarnya mempunyai karakter penyusun yang berbeda baik urutan maupun jumlahnya. Karena itulah nilai kedua digunakan. Nilai kedua (nilai jumlah karakter overlap) digunakan ketika sistem menemukan dua nilai suatu suffix/prefix yang sama. Jadi setiap suffix dan prefix dibandingkan dengan menggunakan 2 nilai dengan harapan bahwa hasil yang didapat maksimal. Untuk jumlah reads input 5000 buah, sistem dapat memberikan ketepatan 100%.
Jumlah edge antara input 2000 dan 5000 reads
16
lain. Karena edge merupakan perwakilan dari jumlah overlap maka dengan semakin banyaknya input maka akan membuat jumlah edge semakin banyak. Karena itulah semakin banyak jumlah input yang dimasukan maka akan membuat jumlah edge dan node semakin banyak yang akhirnya akan berdampak pada kekompleksan dari graph yang terbentuk dan waktu eksekusi yang diperlukan lebih lama.
Namun demikian terdapat sebuah hal penting yang perlu diperhatikan dalam penggunaan data dari MetaSim yaitu pemilihan organisme, jumlah reads, serta panjang reads. Misalnya dipilih suatu organisme dengan panjang 2000 bp maka apabila dipilih panjang potongan reads 35 dengan jumlah 2000 buah, akan membuat graph yang dihasilkan menjadi sangat kompleks. Hal ini dikarenakan setiap reads akan saling overlap satu sama lain, tidak ada satupun reads yang tidak overlap. Sehingga diperlukan pemilihan yang benar untuk panjang reads dan jumlah hasil agar tidak membuat graph menjadi kompleks.
Evaluasi waktu
Waktu merupakan esensi penting dalam penelitian ini. Hal ini dikarenakan metode perbandingan angka diharapkan selain memiliki ketepatan tinggi tetapi juga memberikan waktu eksekusi yang lebih cepat daripada perbandingan string. Waktu eksekusi mulai dihitung tepat saat user telah memasukan nilai jumlah minimum overlap yang diharapkan. Sebelumnya akan dibahas perbedaan waktu eksekusi antara pemprosesan input 2000 dan 5000 buah. Dari Gambar 10 dapat dilihat perbedaan waktu eksekusi dengan jumlah input berbeda yaitu 2000 dan 5000 buah. Waktu eksekusi untuk input dengan jumlah data yang lebih kecil (2000 buah) lebih cepat daripada waktu eksekusi untuk jumlah data yang lebih besar (5000 buah). Hal ini dikarenakan semakin banyaknya jumlah input data maka proses pengindeksan dan pencarian overlap akan semakin lama. Jumlah suffix dan prefix yang dibuat semakin banyak sehingga akan menambah waktu untuk perbandingan suffix dan prefix. Selain itu nilai jumlah minimum overlap yang dipilih juga menentukan waktu eksekusi yang diperlukan. Semakin besar nilai jumlah minimum overlap maka jumlah suffix dan prefix yang dihasilkan dalam pengindeksan akan semakin sedikit dan juga sebaliknya apabila nilai jumlah minimum overlap semakin kecil maka jumlah suffix dan prefix yang dihasilkan akan semakin banyak. Hal ini dikarenakan suffix akan diindeks mulai dari panjang reads (35 bp) – 1 sampai dengan nilai jumlah minimum overlap yang dimasukan oleh user. Sebagai contoh antara nilai jumlah minimum overlap 25 dan 30. Ketika user memasukan nilai jumlah minimum overlap 25 maka suffix dan prefix yang terbentuk akan memiliki panjang dari 34, 33, 32, … , 27, 26, 25 sehingga setiap 1 reads akan menghasilkan 10 suffix dan 10 prefix. Beda halnya dengan ketika user memasukan nilai minimum overlap 30. Ketika user memasukan nilai minimum overlap 30 maka panjang suffix dan prefix yang dihasilkan akan memiliki panjang mulai dari 34 sampai 30. Dengan demikian 1 reads hanya akan menghasilkan 5 suffix dan 5 prefix.Semakin banyaknya jumlah suffix dan prefix akan membuat waktu eksekusi semakin lama sehingga jumlah minimum overlap 30 akan memiliki waktu eksekusi yang lebih cepat daripada nilai minimum overlap 25.
perbandingan waktu eksekusi antara kedua metode (metode perbandingan angka dan metode perbandingan string). Untuk perbedaan waktu eksekusi akan dipakai jumlah input 5000 buah yang diproses oleh kedua metode yang ada. Dari Gambar 11 dapat dilihat perbedaan yang signifikan antara waktu eksekusi yang diperlukan antara kedua metode. Waktu eksekusi yang digunakan untuk metode perbandingan angka secara keseluruhan jauh lebih cepat daripada waktu eksekusi metode perbandingan string. Bahkan dapat dilihat secara keseluruhan waktu yang digunakan metode perbandingan string 5 kali lipat daripada waktu eksekusi dengan metode perbandingan angka. Hal ini dapat terjadi karena metode proses perbandingan angka menggunakan proses pengurangan sedangkan metode perbandingan string menggunakan proses penyamaan karakter dari masing-masing prefix dan suffix. Proses pengurangan membutuhkan waktu yang lebih cepat daripada proses penyamaan masing-masing string suffix-prefix dan hal itu telah dibuktikan dari hasil yang telah didapat dari penelitian ini. Hal tersebut kemudian memberikan hasil positif pada penelitian ini karena dapat membentuk bidirected overlap graph dengan waktu yang lebih cepat dan ketepatan 100%. Metode perbandingan angka menghasilkan output lebih cepat daripada perbandingan string dikarenakan untuk membandingkan 2 buah string, komputer akan merubahnya dahulu menjadi angka yang kemudian akan dibandingkan satu sama lain. Dengan metode perbandingan angka, waktu yang diperlukan untuk mengubah string menjadi angka dapat dihemat. Sistem ini mempunyai kompleksitas O(n2). Hal tersebut dikarenakan sistem menggunakan nested loop sebanyak 2.
Pembuktian ketepatan metode perbandingan angka
18
360 buah. Untuk jumlah masukan tersebut, metode perbandingan angka menghasilkan jumlah edge yang sama persis dengan metode perbandingan string yaitu berjumlah 336 414 buah. Sehingga, sampai dengan jumlah input 80 000, metode perbandingan angka yang digunakan mempunyai nilai ketepatan 100% dan nilai error collision rate sampai dengan jumlah input 80 000 adalah 0%.
SIMPULAN DAN SARAN
Simpulan
Penelitian ini telah berhasil menghasilkan suatu sistem yang dapat membentuk bidirected overlap graph. Dari hasil dan pembahasan dapat disimpulkan bahwa semakin banyak jumlah input yang dimasukan maka waktu eksekusi akan semakin banyak. Demikian pula dengan semakin kecilnya nilai jumlah overlap yang dimasukan maka akan membuat waktu eksekusi semakin lama. Namun demikian dari penelitian ini didapatkan suatu sistem yang dapat menghemat waktu eksekusi yang diperlukan dengan ketepatan hasil 100% pada jumlah input 5000 buah. Hal ini dikarenakan metode yang digunakan dalam sistem adalah metode perbandingan angka yang ternyata memerlukan waktu yang lebih cepat daripada perbandingan string dalam pencarian overlap.
Saran
Namun demikian setiap sistem mempunyai kekurangan. Karena itu untuk ke depannya, sistem ini dapat dikembangkan lagi. Beberapa saran yang dapat diberikan oleh penulis:
1 Menambah ragam organisme yang dipakai sebagai input dari sistem
2 Menambah jumlah input reads yang digunakan sehingga dapat melihat sejauh mana metode perbandingan angka dapat digunakan.
DAFTAR PUSTAKA
Abegunde T. 2010. Comparison of DNA sequence assembly algorithms using mixed data sources[tesis]. Saskatoon (CA): University of Saskatchewan. Brenner S, Johnson M, Bridgham J, Golda G, Lloyd DH, Johnson D, Luo SJ,
McCurdy S, Foy M, Ewan M, et al. 2000. Gene expression analysis by massively parallel signature sequencing (MPPS) on microbead arrays. Nat Biotechnol. 18:630-634.
Chaisson M, Pevzner P, Tang H. 2004. Fragment assembly with short reads. Bioinformatics. 20(13):2067-2074.
Commins J, Toft C, Fares MA. 2009. Computational biology methods and their application to the comparative genomics of endocellular symbiotic bacteria of insects. Biol Proced Online. 11:52-78. doi: 10.1007/s12575-009-9004-1. Hernandez D, François P, Farinelli, Østerås M, Schrenzel J. 2008. De novo
bacterial genome sequencing: millions of very short reads assembled on a desktop computer. Genome Res. 18(5):802-9. doi: 10.1101/gr.072033.107. Kundeti VK, Rajasekaran S, Dinh H, Vaughn M, Thapar V. 2010. Efficient
parallel and out of core algorithms for constructing large bi-directed de Bruijn graphs. BMC Bioinformatics. 11:560. doi: 10.1186/1471-2105-11-560.
Kusuma WA, Ishida T, Akiyama Y. 2011. A combined approach for de novo DNA sequence assembly of very short reads. IPSJ Transaction on Bioinformatics. 3(10):21-33. doi: 10.2197/ipsjtbio.4.21.
20
Lampiran 1 Pembuktian empiris untuk 1000 reads dengan minimum overlap 30
22
Lampiran 1 Lanjutan
24
Lampiran 1 Lanjutan
26
Lampiran 1 Lanjutan
28
Lampiran 1 Lanjutan
30
Lampiran 1 Lanjutan
32
RIWAYAT HIDUP
Penulis dilahirkan di Jakarta pada tanggal 27 Oktober 1990 dari ayah Michel Kamaludin dan ibu Heniati Sugiarto. Penulis merupakan putra pertama dari dua bersaudara. Pada tahun 2009 penulis lulus dari SMA Regina Pacis Jakarta dan pada tahun yang sama penulis masuk ke Institut Pertanian Bogor melalui jalur SNMPTN dan diterima di Departemen Ilmu Komputer, Fakultas Matematika dan Ilmu Pengetahuan Alam.