www.elsevier.com / locate / bres
Interactive report
21 21
L-type Ca
channel-mediated Zn
toxicity and modulation
1
by ZnT-1 in PC12 cells
a a a a b
Albert H. Kim , Christian T. Sheline , Min Tian , Toshio Higashi , Robert J. McMahon ,
b a ,
*
Robert J. Cousins , Dennis W. Choi
a
Department of Neurology and Center for the Study of Nervous System Injury, Washington University School of Medicine, 660 S. Euclid Ave.,
St. Louis, MO 63110, USA b
Food Science and Human Nutrition Department and Center for Nutritional Sciences, University of Florida, Gainesville, FL 32611, USA Accepted 22 September 2000
Abstract
21 21
In view of evidence that Zn neurotoxicity contributes to some forms of pathological neuronal death, we developed a model of Zn neurotoxicity in a cell line amenable to genetic manipulations. Exposure to 500mM ZnCl for 15 min under depolarizing conditions2
21
resulted in modest levels of PC12 cell death, that was reduced by the L-type Ca channel antagonist, nimodipine, and increased by the
21 21 21
L-type Ca channel opener, S(2)-Bay K 8644. At lower insult levels (200 mM Zn 1Bay K 8644), Zn -induced death appeared
apoptotic under electron microscopy and was sensitive to the caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-CH F (Z-VAD); at higher2 insult levels (1000mM1Bay K 8644), cells underwent necrosis insensitive to Z-VAD. To test the hypothesis that the plasma membrane
21
transporter, ZnT-1, modulates Zn neurotoxicity, we generated stable PC12 cell lines overexpressing wild type or dominant negative
21
forms of rat ZnT-1 (rZnT-1). Clones T9 and T23 overexpressing wild type rZnT-1 exhibited enhanced Zn efflux and reduced
21
vulnerability to Zn -induced death compared to the parental line, whereas clones D5 and D16 expressing dominant negative rZnT-1 exhibited the opposite characteristics. 2000 Elsevier Science B.V. All rights reserved.
Theme: Disorders of the nervous system
Topic: Neurotoxicity
Keywords: Zinc; Cell death; PC12; Bay K 8644; ZnT-1
1. Introduction inhibits GABA and NMDA receptor currents [37,55],
1 21
blocks voltage-gated Na and Ca channels, and poten-The transition metal zinc is a normal constituent of tiates AMPA, glycine, and P2x receptor currents [16,38].
21
transcription factors and metalloenzymes [31,51]. In the Given its lack of intrinsic redox activity, Zn has been central nervous system, an additional pool of chelatable considered to be relatively nontoxic [3,46]. However, the
21
Zn exists in a subpopulation of glutamatergic synaptic toxic translocation of presynaptic zinc into postsynaptic 21
vesicles [5,8,13,35] and can be released in a Ca -depen- neurons has now been implicated in the pathogenesis of dent manner by electrical or chemical stimulation [17], selective neuronal death following transient global is-possibly reaching synaptic concentrations in the hundred chemia, prolonged seizures, and trauma [23,42,45,48]. The
21
micromolar range [2]. Zn can modify the behavior of cytotoxicity of exogenously applied zinc has been demon-several membrane receptors and channels [6,15,43]; it strated in vitro and in vivo. In neuronal as well as nonneuronal cells such as thymocytes, prolonged exposure
21
to even 20–80 mM Zn induces cell death [12,26,41]. 1
Published on the World Wide Web on 12 October 2000. 21
Short exposures to 150–600 mM Zn destroys cultured *Corresponding author. Tel.: 11-314-362-9460; fax: 1
1-314-362-cortical [58] or cerebellar granule neurons [28], and 9462.
E-mail address: [email protected] (D.W. Choi). intraparenchymal injection of ZnCl2 induces neuronal
necrosis in the rat hippocampus [25]. With concurrent Val-Ala-Asp-CH F (Z-VAD) was purchased from Enzyme2 21
depolarization, the toxicity of extracellular Zn is en- Systems Products. PC12 cells were transfected using hanced such that short exposure to low micromolar con- FuGene 6 (Boehringer Mannheim), and stable transfectants
21
centrations of Zn becomes neurotoxic [54]. This de- were selected in medium containing 500mg / ml G418. polarization-induced enhancement likely reflects
preferen-21
tial entry of zinc through L-type voltage-gated Ca 2.2. Constructs channels, leading to toxic elevations of intracellular free
21 21
Zn ([Zn ] ) in the range of 300–500 nM [4,40].i Wild type rZnT-1 cDNA (kindly provided by Dr. R.D. 21
Palmiter) and dominant negative rZnT-1 cDNA (an 83 bp Likely opposing such toxic elevations of [Zn ] arei
EagI fragment deletion; [34]) were subcloned into a CMV several mechanisms responsible for maintaining
intracellu-21
promoter-driven mammalian expression vector. After lar Zn homeostasis, including binding to
metallothio-mutating the terminal stop codon in both rZnT-1 forms, a neins [1,10,50,53], and export across the plasma membrane
fragment encoding a hexameric myc epitope tag (gener-mediated by the ubiquitously expressed zinc transporter,
ously provided by Dr. R. Kopan) was inserted in-frame ZnT-1 [34]. Related zinc transporters appear to bear
21
distal to the altered stop codon. responsibility for transporting Zn into endosomes
(ZnT-2) [32] or synaptic vesicles (ZnT-3) [33]. Baby hamster
2.3. Toxicity experiments kidney (BHK) cells [34] and N2A cells [49]
overexpres-sing ZnT-1 exhibit resistance to death induced by pro-21
Immediately before toxic exposures, cells were washed longed Zn exposure. Following transient global ischemia
twice using a HEPES-buffered salt solution (HSS) with the in gerbils, ZnT-1 transcription is upregulated in CA1
1 1
following composition: 130 mM Na , 5.4 mM K , 0.8 pyramidal neurons, consistent with the possibility that
21 21 2
mM Mg , 1.8 mM Ca , 131 mM Cl , 20 mM HEPES ZnT-1 induction may be a cellular strategy to counter
21
(pH 7.4 at 258C), 15 mM glucose. Exposures of 15 min to ischemia-induced toxic Zn influx [49].
21
toxic solutions were conducted at room temperature in We set out to develop a model of Zn influx-induced
1 1
HSS with equimolar substitution of K for Na in cell death in a neuronal cell line. We chose PC12 cells
1
solutions with elevated [K ]. To terminate toxic expo-because they have been widely used for investigating
sures, solutions were washed twice with media stock (MS), multiple aspects of neurobiology, including neuronal
dif-which consists of Eagle’s minimal essential medium plus ferentiation, intracellular signaling pathways, and cell
21
21 mM glucose, and then replaced with MS containing 1% survival [14,56], and specifically express L-type Ca
21
serum (2:1, horse serum:fetal bovine serum) before being channels [18,47], the primary route of toxic Zn entry
21
returned to a 378C incubator. A23187 exposures were into neurons. There is one previous report of Zn -induced
conducted at 378C in MS after sham washes. Z-VAD was death of PC12 cells, utilizing prolonged (24 h) exposure to
21
added to the final replacement solution (MS and 1% Zn in the absence of serum, thought to be due to direct
21
serum) following exposures and washes. inactivation of NGF by Zn [39]. To eliminate this death
21
mechanism and focus on toxic Zn entry, we utilized 21
2.4. Cell death assays brief (15 min) exposure to Zn , followed by return to
serum-containing medium, as the presence of serum abro-21
Cell death was assessed morphologically by phase-con-gated Zn -mediated neurotrophin deprivation-induced
trast microscopy and propidium iodide fluorescence. For death [39].
propidium iodide staining, cells were incubated for 30 min at 378C with propidium iodide solution (5mg / ml) (Molec-ular Probes), and dead cells (cells with compromised
2. Materials and methods membrane integrity) were visualized by excitation at 488
nm. Cell death was quantified by measuring lactate dehy-2.1. Cell culture, drugs, and stable transfection drogenase (LDH) released by injured cells into the cell medium [21]. LDH values were normalized by subtracting PC12 cells were grown in Dulbecco’s modified Eagle’s the background LDH released by sham-washed cells from medium (DMEM) supplemented with 10% heat-inacti- treated cells and scaling to the signal associated with vated horse serum, 5% fetal bovine serum, and 2 mM complete cell death (5100), induced by 24 h exposure to L-glutamine (GIBCO BRL) in a humidified incubator at 30mM A23187.
378C and 5% CO . Twenty-four hours prior to experi-2
ments, cells were plated onto poly-L-lysine-coated 24-well 2.5. Electron microscopy 5
(16 mm diameter) plates at a density of 1.25310 cells /
were then fixed and embedded. After cutting and staining 100mM GdCl ), and the cultures were placed on ice. After3 sections with lead citrate and uranyl acetate, cells were 10 min, the quench solution was aspirated, and the cells examined and photographed with a JEOL 100 CS electron were lysed by addition of 100ml hot 0.2% SDS. The lysate
microscope. was then dotted onto filter paper (Whatman 3MM,
What-man, Inc.), dried by heating lamp, and counted in a gamma
2.6. Immunofluorescence counter (Beckman 4000, Beckman Instruments, Inc.).
Other cultures from the same plating were used to de-Plated cells were fixed with cold methanol and immuno- termine protein concentrations for each sample.
stained with a monoclonal antibody against the myc epitope (Calbiochem). After incubating cells with a
fluorescence-conjugated anti-mouse Alexa 488 secondary 3. Results antibody (Molecular Probes), myc epitope-positive cells
21
were visualized by fluorescence microscopy (excitation: To approximate acute cellular Zn loading followed by
495 nm). later death, we exposed PC12 cells to 500mM ZnCl in2
1
the presence of 90 mM K for 15 min. Many cells had lost 2.7. Western blot analysis brightness, 12–24 h later, under phase-contrast microscopy and no longer excluded propidium iodide (Fig. 1A); some To isolate membrane proteins, cells were harvested in had detached from the dish. Because of this detachment, lysis buffer (20 mM TrisCl (pH 7.5), 0.32 M sucrose, 0.2 we quantified cell death by LDH release to the bathing mM EDTA, and 0.5 mM EGTA with protease inhibitors (1 medium and determined that about a fourth of the total mM dithiothreitol, 1 mM phenyl-methyl-sulfonylfluoride,
20 mg / ml aprotinin, 20 mg / ml leupeptin) and sonicated. After centrifugation of samples at 30,000 g for 30 min at 48C, membrane pellets were resuspended in a buffer consisting of 20% glycerol, 10 mM TrisCl (pH 8), 1 mM EDTA, and protease inhibitors, and total membrane protein was quantified by BCA measurement (Pierce). Membrane proteins were resolved by SDS–PAGE and transferred onto nitrocellulose. To confirm equal loading, blots were stained with Ponceau-S (Sigma). To detect endogenous rZnT-1 expression, blots were blocked in Tris-buffered saline plus Tween-20 (TBS-T) (10 mM TrisCl (pH 7.4), 137 mM NaCl, 0.05% Tween-20) containing 5% milk. After apply-ing affinity-purified rabbit anti-rZnT-1 antibody (50 mg / ml) [29], followed by anti-rabbit horseradish peroxidase (HRP)-conjugated antibody (Vector), protein was visual-ized by enhanced chemiluminscence (ECL) (Pierce). To detect myc epitope-tagged rZnT-1 expression, blots were blocked in phosphate buffered-saline (PBS) containing 10% milk, and immunoblotted with anti-myc monoclonal antibody (kindly provided by R. Kopan). Blots were incubated with anti-mouse HRP-conjugated secondary antibody (Vector), and proteins were detected by ECL.
21
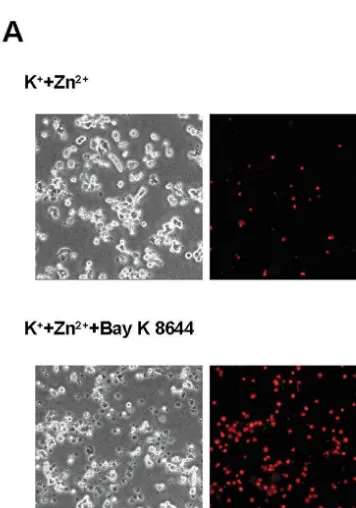
Fig. 1. Bay K 8644 potentiates Zn -induced death in depolarized PC12 65
2.8. Zn efflux assay cells. (A) Phase-contrast (left column) and corresponding propidium iodide fluorescence photomicrographs (right column) of PC12 cells, 24 h
21 1
following 15 min exposure to 500mM Zn 190 mM K , or 500 mM PC12 cultures were washed with MS, allowed to sit for 21 1
Zn 190 mM K 11mM Bay K 8644. (B) PC12 cell death 24 h after 15
5 min, and washed with HSS. Then HSS containing 1
min treatment to the indicated combinations of 90 mM K , 1mM Bay K 65 21
Zn (2 mCi / ml, 1–5 Ci / g, DuPont NEN Research 21 31
8644, 500 mM Zn , 1 mM nimodipine, 10 mM Gd , and 2 mM
21 21
Products) plus cold Zn ([total Zn ]520 mM) was pyruvate as assessed by LDH efflux to the bathing media, scaled to the applied to the cells for loading. After the 90-min loading near-complete cell death induced by 24 h exposure to 30 mM A23187 (5100). All bars depict mean6S.E.M. from three to four independent period, the solution was washed three times with HSS, and
experiments (n512–16 cultures per condition). * indicates significant then cells were returned to HSS plus 5% FBS for efflux. 21
difference from Zn alone at P,0.05.[indicates significant difference
After indicated efflux periods, cells were washed twice 1 21 1
from K 1Zn at P,0.05. $ indicates significant difference from K 1 21
Fig. 1. (continued )
cells had died (Fig. 1B). This level of cell death remained stable over the next 24 h (data not shown).
Consistent with earlier studies with primary cortical 21
neurons, blockade of L-type voltage-gated Ca channels 21
with 1mM nimodipine markedly attenuated Zn -induced PC12 cell death but only under depolarizing conditions,
21
and enhancement of L-type Ca channel opening with 1 mM Bay K 8644 increased cell death such that about half of the total population was killed. This Bay K 8644-enhanced cell death was still sensitive to the broad
21 31
spectrum voltage-gated Ca channel blocker, Gd (10 mM), and was attenuated by pyruvate as was shown recently in cortical neuronal cultures [41]. Bay K 8644,
1 31
K , Gd , or nimodipine by themselves were not toxic in 21 21 1
Fig. 2. Bay K 8644-enhanced Zn toxicity is both Zn and K -these exposure conditions (data not shown). Death was
dependent. (A) Cell death 24 h after 15 min exposure to 1mM Bay K 21
1 21
induced by Zn concentrations over the range 30–1000 8644, 90 mM K , and the indicated concentrations of Zn
1
mM and extracellular K concentrations over the range (mean6S.E.M. from three independent experiments, n512 cultures per condition). Cell death was assessed by measuring LDH activity in the 20–90 mM (Fig. 2A,B).
media, scaled to the near-complete cell death induced by 24 h exposure to We examined the type of death — apoptosis or necrosis
30mM A23187 (5100). (B) Cell death 24 h after 15 min exposure to 1
— induced by this protocol. Transmission electron mi- 21 1
mM Bay K 8644, 500mM Zn , and the indicated concentrations of K crographs of PC12 cells 12 h after a 15 min exposure to (mean6S.E.M.). Cell death was assessed by measuring LDH activity in
21
200mM Zn in the presence of 1mM Bay K 8644 and 90 the media as in (A).
1
mM K revealed hallmarks of apoptosis, including apo-ptotic body formation, preservation of plasma membranes,
condensation of chromatin, and mitochondrial integrity To test the hypothesis that PC12 cells exposed to
21 21 21
(Fig. 3A). In contrast, cells exposed to 1000 mM Zn extracellular Zn die due to toxic elevations in [Zn ] ,i 21
exhibited necrosis, with mitochondrial swelling and disrup- we stably expressed the plasma membrane Zn transpor-tion of plasma membranes (Fig. 3B). A mixed morphologi- ter, ZnT-1 [34]. We first determined if PC12 cells
en-21
cal profile was observed in cells exposed to 500mM Zn , dogenously express rZnT-1 by immunoblot analysis using with certain cells appearing necrotic and others apoptotic, a previously characterized polyclonal affinity-purified anti-and some cells exhibiting a mixture of features (Fig. 3C). body (Fig. 5A) [29]. To test if rZnT-1 activity can regulate
21
Consistent with the morphology, addition of 100 mM zinc homeostasis in our Zn toxicity paradigm, we Z-VAD attenuated cell death at lower (200–500mM) but generated stable PC12 lines expressing wild type and
21
not higher (1000 mM) Zn concentrations (Fig. 4). The dominant negative rat ZnT-1 constructs. To distinguish 21
cell death induced by 500 mM Zn in the presence of transfected from endogenous ZnT-1 protein, we subcloned
1
only K was also apoptotic in nature, with Z-VAD a hexameric myc epitope tag distal to the 39 end of the reducing this death from 24.161.5% to 5.164.8%, n59 rZnT-1 cDNA.
wild-21 1 Fig. 3. Transmission electron micrographs. (A) Neuronal cell apoptosis 12 h after 15 min exposure to 200mM Zn 11mM Bay K 8644190 mM K . Bar52mm. The arrow points to apoptotic bodies. A portion of a relatively intact cell can also be seen at the right edge of the micrograph. (B) Same as A
21 21
but with 1000mM Zn . Necrosis associated with plasma membrane disruption and swollen mitochondria. (C) Same as A but with 500mM Zn . A cell with a mixed death phenotype (arrow) can be seen adjacent to a necrotic cell (arrowhead). Although the former cell demonstrates some features typical of apoptotic death — an intact plasma membrane, a shrunken cell body, and compact chromatin, the cell also exhibits swollen mitochondria.
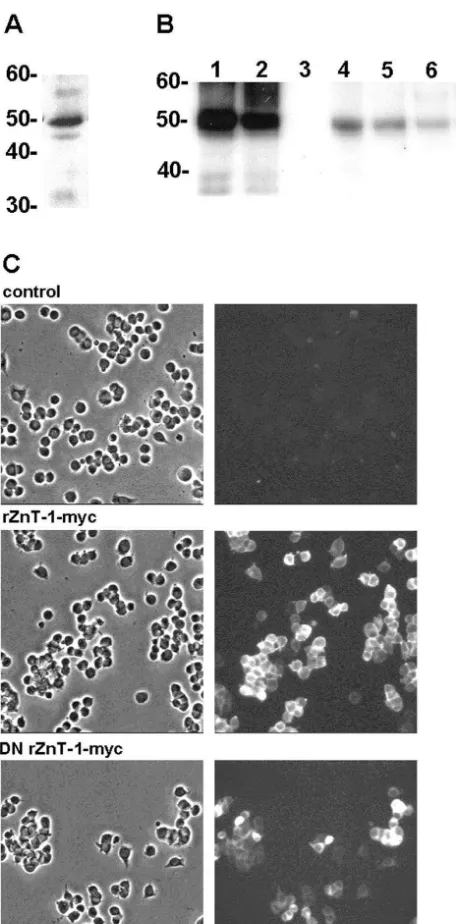
type (rZnT-1-myc) or dominant negative (DN rZnT-1-myc) To confirm the predicted function of overexpressed transporter predominantly at the plasma membrane, al- rZnT-1 proteins, we assessed the ability of lines stably though, consistent with the work of Palmiter and Findley expressing rZnT-1-myc or DN rZnT-1-myc to export
65 21
[34], some punctate staining over the entire cell was also preloaded Zn . Cells were preloaded by 90 min expo-21 65 21
apparent (Fig. 5C and data not shown). The selected lines sure to cold Zn plus Zn (20mM total); over the next were then examined by immunoblotting to select clones 2 h, the rZnT-1-myc line, T9, demonstrated enhanced
65 21
expressing high levels of myc-tagged rZnT-1 proteins. Zn extrusion, and the DN rZnT-1-myc cell line, D5, 65 21
Similar to endogenous ZnT-1, both rZnT-1-myc and DN demonstrated reduced Zn extrusion, compared to the rZnT-1-myc migrated at about 50 kDa in stably transfected parental line (Fig. 6). These lines also exhibited altered
21
PC12 cells. N2A neuroblastoma cells transiently transfect- vulnerability to Zn -induced death. Over a range of tested 21
ed with these constructs were included as positive controls Zn concentrations between 200 and 1000mM, the rZnT-21
(Fig. 5B). 1-myc line exhibited reduced vulnerability to Zn
-in-duced death, and the DN rZnT-1-myc cell line exhibited enhanced vulnerability, compared to the parental line (Fig. 7). To control for potential integration-related effects, we observed similar results with additional lines stably ex-pressing each construct (T23 and D16, data not shown).
4. Discussion
21
We describe here a model system for investigating Zn influx-induced neurotoxicity utilizing PC12 cells. This
21
PC12 cell system exhibited five key features of Zn neurotoxicity as previously delineated with primary
corti-21
cal neurons: (1) cell death was induced by brief Zn 21
exposure (15 min); (2) the extent of death was Zn -21
concentration dependent; (3) Zn -induced death was
21 enhanced by concurrent depolarization and largely
depen-Fig. 4. Lower levels of Zn -induced death are sensitive to inhibition by
21
Z-VAD. Cell death 24 h after 15 min exposure to 1mM Bay K 8644, 90 dent upon L-type voltage-gated Ca channel activation;
1 21
mM K , and the indicated concentrations of Zn , in the presence or (4) lower levels of toxic zinc exposure induced apoptosis, absence of 100mM Z-VAD (mean6S.E.M. from three to four independent whereas higher levels induced necrosis; and (5) death was experiments, n512–16 cultures per condition). * indicates a significant
reduced by the addition of 2 mM pyruvate. difference between Z-VAD treated and untreated cell death at the same
21 As noted above, Ross et al. have reported that prolonged
Zn concentration at P,0.05. Statistical analysis was performed using a
21
inac-Fig. 6. Effects of wild type and dominant negative rZnT-1-myc over-65
expression on Zn efflux from PC12 cells. Cells were preloaded for 90
65 21 21
min with Zn (2mCi / ml, total Zn concentration 20mM). After the efflux period, cells were washed, lysed, and harvested for quantitation of radioactivity by gamma counter (mean6S.E.M. from two to three independent experiments, n58–12 cultures per condition). Sister cultures were used to determine the protein concentration of each sample. * indicates significant difference compared to the parental line at P,0.05, using two-way ANOVA followed by a Bonferroni t-test.
21
since Zn exposure was brief (15 min), and serum-containing media was restored after exposure termination (as noted above, serum prevented death by this neuro-trophin inactivation mechanism). Also, while neuronal death in our paradigm was likewise apoptotic, death was markedly reduced by nimodipine (see Fig. 1B) and in-fluenced by the expression of ZnT-1, consistent with
21 21
mediation by Zn -influx through L-type Ca channels 21
and consequent toxic elevation in intracellular free Zn , as occurs in primary neurons.
The vulnerability of PC12 cells to death induced by
Fig. 5. Endogenous rZnT-1, and rZnT-1-myc protein expression in PC12 stable lines. (A) Membrane proteins prepared from the parental line were examined for expression of rZnT-1 using a rabbit polyclonal antibody. (B) Membrane proteins prepared from neuroblastoma N2A cells tran-siently transfected with rZnT-1-myc (lane 1) and DN rZnT-1-myc (lane 2) constructs, the parental PC12 line (lane 3), PC12 clones T9 and T23 expressing rZnT-1-myc (lane 4 and 5), and clones D5 and D16 express-ing DN rZnT-1-myc (lanes 6 and 7) were assessed for expression of transfected rZnT-1 constructs using a monoclonal antibody against the myc epitope. (C) Phase-contrast (left column) and corresponding immunofluorescence (right column) photomicrographs of PC12 lines stably transfected with rZnT-1-myc or DN rZnT-1-myc. Control cells were not transfected. In the right column, cells were immunostained with
Fig. 7. Effects of wild type and dominant negative rZnT-1-myc expres-monoclonal antibody against the myc epitope. 21
sion on Zn -induced death. Cell death 24 h after 15 min exposure to 1
1 21
21 21
exogenously applied extracellular Zn is somewhat less metabolic effects of Zn exposure on neurons could be than that of primary central neurons [28,54]. Most likely, reversed by the addition of pyruvate, presumably due to
1
this quantitative difference reflects the relative paucity of regeneration of NAD upon conversion to lactate. We 21
membrane L-type Ca channels on the former relative to postulate that similar effects account for the protective the latter [24] (and unpublished results), although differ- effects of pyruvate on PC12 cells presented here.
21 21
ences in Zn homeostasis mechanisms or downstream Finally, we showed that Zn -induced death in this toxicity cascades might also contribute. We therefore neuronal system can be influenced by altering the function
21
increased the contribution of the existing L-type channels of the plasma membrane Zn transporter, ZnT-1. Our
1
by increasing extracellular K to 90 mM, and adding S(2) findings extend previous observations regarding the ability Bay K 8644, which acts at the dihydropyridine binding site of transfected ZnT-1 or a dominant negative ZnT-1 to
21
to increase L-type Ca channel open time [11]. Although modify the vulnerability of BHK or N2A cells to pro-L-type channels have been implicated as the predominant longed zinc exposure [34,49]. Our results suggest that both
21 21
route of neurotoxic Zn influx [22,40], other routes endogenous and ectopic ZnT-1 contribute to critical Zn
21 21
probably also participated, including other Ca channels, homeostasis after acute Zn exposure under depolarizing 21
NMDA receptors, Ca -permeable AMPA / kainate recep- conditions. Molecular or pharmacological manipulation of
1 21
tors, and the Na / Ca exchanger [40,57]. These other ZnT-1 function may constitute a useful neuroprotective routes appeared more prominent under non-depolarizing strategy in certain disease settings such as global brain
1
conditions, whereas elevation of extracellular K shifted ischemia. 21
the route of influx to favor L-type Ca channels (Fig. 1B). While membrane depolarization would be expected to
21
reduce the electromotive gradient driving Zn entry by all Acknowledgements routes, presumably the ability of depolarization to increase
21 21
Zn entry through the opening of L-type Ca channels This investigation was supported by National Institutes more than compensated for this reduced driving force. of Health Grants NS 30337 (DWC) and DK 31127 (RJC)
21
The present observation that lower levels of Zn and Individual National Research Service Award DK exposure induce PC12 cells to undergo apoptosis fits with 09628 (RJM), and Boston Family Endowment funds of the earlier studies in cerebellar granule and cortical neurons, University of Florida (RJC, RJM).
21
which when exposed to lower levels of toxic Zn , undergo apoptosis associated with DNA breakdown [20,28] and sensitivity to Z-VAD and bax gene deletion
References [26], but necrosis when exposed to higher levels of toxic
21
Zn [7,19,20,26]. These studies are in general agreement
[1] M. Aschner, M.G. Cherian, C.D. Klaassen, R.D. Palmiter, J.C. with our PC12 results where we also demonstrated a mixed
Erickson, A.I. Bush, Metallothioneins in brain — the role in death with the apoptotic component predominating at physiology and pathology, Toxicol. Appl. Pharmacol. 142 (1997)
21
lower levels of Zn exposure as defined by ultrastructure 229–242.
21
[2] S.Y. Assaf, S.H. Chung, Release of endogenous Zn from brain and sensitivity to Z-VAD.
21 tissue during activity, Nature 308 (1984) 734–736.
The observation that Zn induced apoptosis of primary
[3] J.M. Berg, Y. Shi, The galvanization of biology: a growing apprecia-neurons or PC12 cells add support to the hypothesis that
tion for the roles of zinc, Science 271 (1996) 1081–1085. 21
Zn is an important mediator of selective neuronal death [4] L.M. Canzoniero, D.M. Turetsky, D.W. Choi, Measurement of following transient global ischemia in vivo, as the latter intracellular free zinc concentrations accompanying zinc-induced likewise exhibits morphological [30] and biochemical [27] neuronal death, J. Neurosci. (Online) 19 (1999) RC31.
[5] G. Charton, C. Rovira, Y. Ben-Ari, V. Leviel, Spontaneous and
features of apoptosis, although some conflicting results 21
evoked release of endogenous Zn in the hippocampal mossy fiber have been reported [9,52]
zone of the rat in situ, Exp. Brain Res. 58 (1985) 202–205. Further studies will be needed to delineate the links [6] D.W. Choi, J.Y. Koh, Zinc and brain injury, Annu. Rev. Neurosci. 21 between elevated intracellular zinc and activation of (1998) 347–375.
apoptosis mechanisms in PC12 cells and central neurons. [7] D.W. Choi, M. Yokoyama, J. Koh, Zinc neurotoxicity in cortical cell culture, Neuroscience 24 (1988) 67–79.
That apoptosis can occur at all is somewhat surprising,
21 [8] G. Danscher, G. Howell, J. Perez-Clausell, N. Hertel, The dithizone,
given that Zn inhibits the activity of multiple caspase
Timm’s sulphide silver and the selenium methods demonstrate a family members in lysates [36,44]. We hypothesize that the chelatable pool of zinc in CNS. A proton activation (PIXE) analysis
21
intracellular Zn levels achieved in the present experi- of carbon tetrachloride extracts from rat brains and spinal cords ments is sufficient to initiate steps leading towards cellular intravitally treated with dithizone, Histochemistry 83 (1985) 419–
422. apoptosis but not sufficient to shut off late caspase
21 [9] J. Deshpande, K. Bergstedt, T. Linden, H. Kalimo, T. Wieloch,
activation. Excessive Zn influx also leads to ATP
Ultrastructural changes in the hippocampal CA1 region following depletion in near-pure cortical neuronal cultures, largely transient cerebral ischemia: evidence against programmed cell death,
1
due to cellular loss of NAD and consequent inhibition of Exp. Brain Res. 88 (1992) 91–105.
Palmiter, Disruption of the metallothionein-III gene in mice: analy- [31] T.V. O’Halloran, Transition metals in control of gene expression [see sis of brain zinc, behavior, and neuron vulnerability to metals, aging, comments], Science 261 (1993) 715–725.
and seizures, J. Neurosci. 17 (1997) 1271–1281. [32] R.D. Palmiter, T.B. Cole, S.D. Findley, ZnT-2, a mammalian protein [11] J. Ferrante, E. Luchowski, A. Rutledge, D.J. Triggle, Binding of A that confers resistance to zinc by facilitating vesicular sequestration,
1,4-dihydropyridine calcium channel activator, (2) S Bay K 8644, Embo J. 15 (1996) 1784–1791.
to cardiac preparations, Biochem. Biophys. Res. Commun. 158 [33] R.D. Palmiter, T.B. Cole, C.J. Quaife, S.D. Findley, ZnT-3, a (1989) 149–154. putative transporter of zinc into synaptic vesicles, Proc. Natl. Acad. [12] P.J. Fraker, W.G. Telford, A reappraisal of the role of zinc in life and Sci. USA 93 (1996) 14934–14939.
death decisions of cells, Proc. Soc. Exp. Biol. Med. 215 (1997) [34] R.D. Palmiter, S.D. Findley, Cloning and functional characterization 229–236. of a mammalian zinc transporter that confers resistance to zinc, [13] C.J. Frederickson, Neurobiology of zinc and zinc-containing neu- Embo J. 14 (1995) 639–649.
rons, Int. Rev. Neurobiol. 31 (1989) 145–238. [35] J. Perez-Clausell, Distribution of terminal fields stained for zinc in [14] L.A. Greene, A.S. Tischler, Establishment of a noradrenergic clonal the neocortex of the rat, J. Chem. Neuroanat. 11 (1996) 99–111.
line of rat adrenal pheochromocytoma cells which respond to nerve [36] D.K. Perry, M.J. Smyth, H.R. Stennicke, G.S. Salvesen, P. Duriez, growth factor, Proc. Natl. Acad. Sci. USA 73 (1976) 2424–2428. G.G. Poirier, Y.A. Hannun, Zinc is a potent inhibitor of the apoptotic
21
[15] N.L. Harrison, S.J. Gibbons, Zn : an endogenous modulator of protease, caspase-3. A novel target for zinc in the inhibition of ligand- and voltage-gated ion channels, Neuropharmacology 33 apoptosis, J. Biol. Chem. 272 (1997) 18530–18533.
(1994) 935–952. [37] S. Peters, J. Koh, D.W. Choi, Zinc selectively blocks the action of [16] N. Hori, T. Galeno, D.O. Carpenter, Responses of pyriform cortex N-methyl-D-aspartate on cortical neurons, Science 236 (1987) 589–
neurons to excitatory amino acids: voltage dependence, conductance 593.
changes, and effects of divalent cations, Cell Mol. Neurobiol. 7 [38] F.A. Rassendren, P. Lory, J.P. Pin, J. Nargeot, Zinc has opposite (1987) 73–90. effects on NMDA and non-NMDA receptors expressed in Xenopus [17] G.A. Howell, M.G. Welch, C.J. Frederickson, Stimulation-induced oocytes, Neuron 4 (1990) 733–740.
uptake and release of zinc in hippocampal slices, Nature 308 (1984) [39] G.M. Ross, I.L. Shamovsky, G. Lawrance, M. Solc, S.M. Dostaler, 736–738. S.L. Jimmo, D.F. Weaver, R.J. Riopelle, Zinc alters conformation [18] D. Janigro, G. Maccaferri, J. Meldolesi, Calcium channels in and inhibits biological activities of nerve growth factor and related
undifferentiated PC12 rat pheochromocytoma cells, FEBS Lett. 255 neurotrophins, Nat. Med. 3 (1997) 872–878.
(1989) 398–400. [40] S.L. Sensi, L.M. Canzoniero, S.P. Yu, H.S. Ying, J.Y. Koh, G.A. 21
[19] E.Y. Kim, J.Y. Koh, Y.H. Kim, S. Sohn, E. Joe, B.J. Gwag, Zn Kerchner, D.W. Choi, Measurement of intracellular free zinc in entry produces oxidative neuronal necrosis in cortical cell cultures, living cortical neurons: routes of entry, J. Neurosci. 17 (1997) Eur. J. Neurosci. 11 (1999) 327–334. 9554–9564.
[20] Y.H. Kim, E.Y. Kim, B.J. Gwag, S. Sohn, J.Y. Koh, Zinc-induced [41] C.T. Sheline, M.M. Behrens, D.W. Choi, Zinc-induced cortical cortical neuronal death with features of apoptosis and necrosis: neuronal death: contribution of energy failure attributable to loss of mediation by free radicals, Neuroscience 89 (1999) 175–182. NAD(1) and inhibition of glycolysis, J. Neurosci. 20 (2000) 3139– [21] J.Y. Koh, D.W. Choi, Quantitative determination of glutamate 3146.
mediated cortical neuronal injury in cell culture by lactate dehydro- [42] R.S. Sloviter, A selective loss of hippocampal mossy fiber Timm genase efflux assay, J. Neurosci. Methods 20 (1987) 83–90. stain accompanies granule cell seizure activity induced by perforant [22] J.Y. Koh, D.W. Choi, Zinc toxicity on cultured cortical neurons: path stimulation, Brain Res. 330 (1985) 150–153.
involvement of N-methyl-D-aspartate receptors, Neuroscience 60 [43] T.G. Smart, X. Xie, B.J. Krishek, Modulation of inhibitory and (1994) 1049–1057. excitatory amino acid receptor ion channels by zinc, Prog. Neuro-[23] J.Y. Koh, S.W. Suh, B.J. Gwag, Y.Y. He, C.Y. Hsu, D.W. Choi, The biol. 42 (1994) 393–441.
role of zinc in selective neuronal death after transient global cerebral [44] H.R. Stennicke, G.S. Salvesen, Biochemical characteristics of ischemia, Science 272 (1996) 1013–1016. caspases-3, -6, -7, and -8, J. Biol. Chem. 272 (1997) 25719–25723. [24] S. Kongsamut, R.J. Miller, Nerve growth factor modulates the drug [45] S.W. Suh, J.W. Chen, M. Motamedi, B. Bell, K. Listiak, N.F. Pons, sensitivity of neurotransmitter release from PC-12 cells, Proc. Natl. G. Danscher, C.J. Frederickson, Evidence that synaptically-released Acad. Sci. USA 83 (1986) 2243–2247. zinc contributes to neuronal injury after traumatic brain injury, Brain [25] G.J. Lees, A. Lehmann, M. Sandberg, A. Hamberger, The neuro- Res. 852 (2000) 268–273.
toxicity of zinc in the rat hippocampus, Neurosci. Lett. 120 (1990) [46] F.W. Sunderman Jr., The influence of zinc on apoptosis, Ann. Clin.
155–158. Lab. Sci. 25 (1995) 134–142.
[26] D. Lobner, L.M. Canzoniero, P. Manzerra, F. Gottron, H. Ying, M. [47] L. Toll, Calcium antagonists high-affinity binding and inhibition of Knudson, M. Tian, L.L. Dugan, G.A. Kerchner, C.T. Sheline, S.J. calcium transport in a clonal cell line, J. Biol. Chem. 257 (1982) Korsmeyer, D.W. Choi, Zinc-induced neuronal death in cortical 13189–13192.
neurons [In Process Citation], Cell Mol. Biol. (Noisy-Le-Grand) 46 [48] N. Tonder, F.F. Johansen, C.J. Frederickson, J. Zimmer, N.H. (2000) 797–806. Diemer, Possible role of zinc in the selective degeneration of dentate [27] J.P. MacManus, A.M. Buchan, I.E. Hill, I. Rasquinha, E. Preston, hilar neurons after cerebral ischemia in the adult rat, Neurosci. Lett.
Global ischemia can cause DNA fragmentation indicative of apop- 109 (1990) 247–252.
tosis in rat brain, Neurosci. Lett. 164 (1993) 89–92. [49] M. Tsuda, K. Imaizumi, T. Katayama, K. Kitagawa, A. Wanaka, M. [28] H. Manev, E. Kharlamov, T. Uz, R.P. Mason, C.M. Cagnoli, Tohyama, T. Takagi, Expression of zinc transporter gene, ZnT-1, is Characterization of zinc-induced neuronal death in primary cultures induced after transient forebrain ischemia in the gerbil, J. Neurosci. of rat cerebellar granule cells, Exp. Neurol. 146 (1997) 171–178. 17 (1997) 6678–6684.
[29] R.J. McMahon, R.J. Cousins, Regulation of the zinc transporter [50] B.L. Vallee, The function of metallothionein, Neurochem. Int. 27 ZnT-1 by dietary zinc, Proc. Natl. Acad. Sci. USA 95 (1998) (1995) 23–33.
4841–4846. [51] B.L. Vallee, K.H. Falchuk, The biochemical basis of zinc physi-[30] T. Nitatori, N. Sato, S. Waguri, Y. Karasawa, H. Araki, K. Shibanai, ology, Physiol. Rev. 73 (1993) 79–118.
[53] M. van Lookeren Campagne, H. Thibodeaux, N. van Bruggen, B. [56] H. Yano, M.V. Chao, Neurotrophin receptor structure and interactions Cairns, R. Gerlai, J.T. Palmer, S.P. Williams, D.G. Lowe, Evidence [In Process Citation], Pharm. Acta Helv. 74 (2000) 253–260.
21 21
for a protective role of metallothionein-1 in focal cerebral ischemia, [57] H.Z. Yin, J.H. Weiss, Zn permeates Ca permeable AMPA / Proc. Natl. Acad. Sci. USA 96 (1999) 12870–12875. kainate channels and triggers selective neural injury, Neuroreport 6 [54] J.H. Weiss, D.M. Hartley, J.Y. Koh, D.W. Choi, AMPA receptor (1995) 2553–2556.
activation potentiates zinc neurotoxicity, Neuron 10 (1993) 43–49. [58] M. Yokoyama, J. Koh, D.W. Choi, Brief exposure to zinc is toxic to 21
[55] G.L. Westbrook, M.L. Mayer, Micromolar concentrations of Zn cortical neurons, Neurosci. Lett. 71 (1986) 351–355. antagonize NMDA and GABA responses of hippocampal neurons,