Atherosclerosis 152 (2000) 239 – 248
The contribution of candidate genes to the response of plasma
lipids and lipoproteins to dietary challenge
Yechiel Friedlander
a, Eran Leitersdorf
b,*, Roni Vecsler
a, Harald Funke
c,
Jeremy Kark
aaThe Department of Social Medicine,The Hebrew Uni6ersity-Hadassah School of Public Health,Jerusalem, Israel bThe Department of Medicine,Hadassah Uni6ersity Hospital,PO Box12221,91120 Jerusalem, Israel
cInstitute of Clinical Chemistry and Laboratory Medicine,Uni6ersity of Munster,Munster, Germany
Received 7 June 1999; received in revised form 19 October 1999; accepted 2 November 1999
Abstract
The possible role of four candidate genes in lipid and lipoprotein response to diet was examined in 214 members of two large kibbutz settlements in Israel. Four site polymorphisms (signal peptide insertion/deletion, XbaI, EcoRI and MspI) of the apo B gene, the common apo E genotypes, three common mutations (T-93G, S447stop and N291S) of the LPL gene and the CETP I405V RFLP were determined. The average reduction induced by diet in participants with the absence of the EcoRI restriction site (L4154) of the apo B gene compared with those found to be homozygotes for the restriction site (G/G4154) were: 16.2 and 8.0 mg/dl for total cholesterol (TC) (P=0.01); and 15.6 and 6.2 mg/dl for LDL-C (P=0.007), respectively. TC and LDL-C baseline levels were significantly different among the apo-E genotypes, yet there were no significant effects on lipid and lipoprotein dietary response. Triglyceride baseline values were significantly lower (P=0.007) among subjects with the LPL S447stop mutation and HDL-C was significantly lower (P=0.008) among subjects found to be heterozygous for the LPL N291S mutation. A heterogeneous response for triglyceride was observed for individuals with the S291 allele as compared to those individuals who were found to be homozygous for the N291 allele. No differences in dietary responsiveness were observed among the apo E and CETP genotypes. In conclusion, our results suggest that sequence variation(s) in the coding region of the apo B gene linked to the EcoRI polymorphism are associated with total cholesterol and LDL-C responsiveness to dietary manipulation. In our study population, LPL mutations had a significant effect on TG and HDL-C baseline levels and on their response to diet. © 2000 Elsevier Science Ireland Ltd. All rights reserved.
Keywords:Lipids; Lipoproteins; Genetics; Diet
www.elsevier.com/locate/atherosclerosis
1. Introduction
Levels of plasma lipids and lipoproteins can be al-tered by diet and the magnitude of dietary effects varies between individuals [1 – 4]. These differences could re-sult from different dietary adherence or may be due to true inter-individual variation in the response [1,5]. Other factors that may influence the response include sex [6], age [7], BMI [8] and the basal level of plasma lipids [9].
It is well established that genetic variation
con-tributes to the basal plasma levels of lipids and lipo-proteins. Various studies have shown an association between lipid and lipoprotein levels and the apolipo-protein B (apo-B) RFLPs [10 – 14], however this has not been consistent [15,16]. In addition, studies have indi-cated that the apo E polymorphism influences plasma lipid levels [17]. Recent studies have shown that varia-tion at the lipoprotein lipase (LPL) and at the choles-terol ester transfer protein (CETP) loci also contribute to between-individual variation in plasma lipid and lipoprotein levels within the normal population [18 – 22].
It has been proposed that genetic variation may also contribute to the variability of these levels over time and in response to environmental exposure [23 – 26]. * Corresponding author. Tel.: +972-2-6778029; fax: +
972-2-6411136.
E-mail address:[email protected] (E. Leitersdorf).
Recent studies provided evidence that genetic variabil-ity at apolipoprotein gene loci may contribute to the variation in lipid and lipoprotein response to dietary manipulations [27 – 36].
The present study examined the association between genotypes at the apo B, apo E, LPL and CETP loci with the response of plasma lipids and lipoproteins to dietary manipulation. This was carried out in 214 indi-viduals who participated in dietary experiments in which fatty acid composition and cholesterol intake were modified while total energy intake remained constant.
2. Material and methods
The study was performed on healthy members of two large kibbutz settlements. Before the beginning of the study, all eligible subjects were assembled and the pur-pose of the study, its performance and requirements, on the part of the participants, were explained by the investigators. Signed consent was obtained from all subjects agreeing to participate.
Each subject underwent a medical examination and a routine biochemical screening. Subjects with endocrine or metabolic disturbances, such as diabetes mellitus, hypothyroidism, or those who reported any other cause of secondary dyslipoproteinemia were excluded. Also excluded were subjects who were found to consume more than 20% of their energy intake outside the kibbutz. Prior to the intervention, subjects were asked to record their entire food intake for several days. This enabled the investigators to calculate the energy
re-quirements of each participant and to plan his/her diet
accordingly.
Two different diets were administered in a crossover design. The first diet was characterized by a high
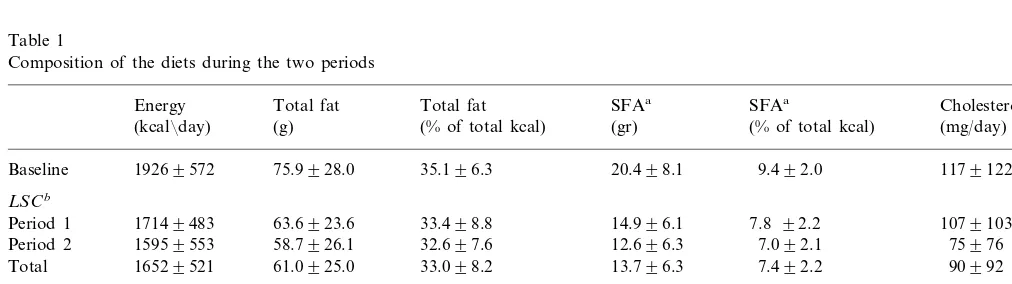
con-tent of saturated fatty acid and cholesterol (HSC) while the second diet consisted of low saturated fatty acid and low cholesterol content (LSC). The two diets were administered to the randomly allocated groups for a 4-week period (period 1) followed by a wash-out period of 4 weeks consisting of the participants’ regular home diet. Thereafter, subjects were given the other of the two diets for a second 4-week period (period 2). During the last week of each dietary period the participants were asked again to keep a record for several days a record of all foods eaten. The food records were coded by dieticians. Quantities of food were coded by fre-quencies, with one frequency representing the weight or volume of a standard serving or part of it. For several composite dishes, such as cakes or fillings, standard recipes were used to estimate their composition. A set of coding rules was used to estimate the amount of fat absorbed during cooking or frying and factors were calculated for converting raw materials to cooked and baked foods. The nutrient content of the codes was derived from several sources including local food pro-ducers and retailers, and local laboratory analyses for some items. For more general items, local food tables were applied [37]. These records were analyzed and the results are presented in Table 1. Total caloric intake was not significantly different between the two diets, but the SFA content and the amount of cholesterol intake did differ.
At the beginning and at the end of each period, fasting blood was drawn twice within 2 – 3 days for determination of lipids and lipoproteins. Participants’ weights were measured at several occasions throughout the study and an attempt was made to immediately identify all subjects who showed weight change. Such subjects where then advised by the dieticians how to adjust their energy intake so as to control the weight.
Table 1
Composition of the diets during the two periods
Energy Total fat Total fat SFAa SFAa Cholesterol
(kcal¯day) (g) (% of total kcal) (gr) (% of total kcal) (mg/day)
19269572 75.9928.0 35.196.3
Baseline 20.498.1 9.492.0 1179122
LSCb
17149483 63.6923.6 33.498.8
Period 1 14.996.1 7.8 92.2 1079103
15959553 58.7926.1 32.697.6
Period 2 12.696.3 7.092.1 75976
16529521 61.0925.0 33.098.2
Total 13.796.3 7.492.2 90992
HSCb
18659682
Period 1 78.6933.3 37.597.5 25.7912.6 12.193.3 2109164
81.5933.9 38.498.7 27.7912.0
Period 2 18839596 13.193.8 2649222
Total 18749637 80.1933.6 38.098.2 26.8912.3 12.693.6 2389199
aSFA, saturated fatty acids.
Y.Friedlander et al./Atherosclerosis152 (2000) 239 – 248 241
3. Laboratory methods
Plasma cholesterol and triglycerides were determined by an enzymatic procedure (Vitalab, Vital Scientific, Diesen, Netherlands). HDL-cholesterol (HDL-C) con-centrations were determined after precipitation of apo-B-containing lipoproteins with phosphotungstic acid. Low-density lipoprotein cholesterol (LDL-C) was esti-mated by the Friedewald equation [38], which has been validated in our population [39].
The apo B insertion/deletion site in exon 1, and the
XbaI, MspI, and EcoRI RFLPs at the 3%end of the apo
B gene were studied. The polymorphism at the 5%end of
the apo B gene involves the insertion/deletion of 9 bp in
exon 1 which results in the addition or deletion of the
three amino acids in the signal peptide [40]. The XbaI
RFLP involves the third base of threonine codon 2488 (ACCACT) without affecting the amino acid sequence,
the MspI RFLP changes codon 3611 from arginine
(CGG) to glutamine (CAG) and the EcoRI RFLP
changes codon 4154 from glutamic acid (GAA) to lysine (AAA) [41].
The three different alleles (o2,o3 ando4) of the apo
E gene were studied after PCR amplification of the targeted region within the apo E gene, digestion with
the HhaI restriction enzyme, electrophoretic separation
on 8% polyacrylamide gels, and staining with ethidium bromide [42]. The E2 and E4 isoforms differ from the E3 common allele by a single amino substitution at position 112 and 158.
Identification of carriers of the T-93G mutation in the promoter of the LPL gene was performed by PCR
using primers: 5%
-GGCAGGGTTGTTCCTCATTACT-GTT-3% (sense) and 5%-GACACTGTTT
TCACGC-CAAGGCTGC-3% (anti-sense) [43]. The reaction
mixture included 15 pmol of each primer, 0.5 (mg
genomic DNA, 0.2 mM of each dNTP, 10 mM Tris –
HCl; pH 9.0, 1.5 mM MgCl2, 50 mM KCl, 0.01% (w/v)
gelatin, 0.1% Triton X-100 and 1 unit Taq polymerase
in a total volume of 50ml. Amplification was performed
for 32 cycles of 30 s at 94°C, 30 s at 50°C, and 1 min at 72°C, with an initial denaturation period of 3 min.
Some 20 ml of PCR products was digested with the
restriction enzyme A6aII according to
recommenda-tions of the supplier (Pharmacia). Thereafter, fragments were separated on a 4% MP agarose gel (Boehringer Mannheim, FRG) and stained with ethidium bromide.
In the case of the mutant allele an A6aII restriction site
is abolished and therefore digestion of the PCR product will reveal one fragment of 189 bp for the mutant allele, and two fragments of 137 and 52 bp for the normal allele.
To identify carriers with the S447stop mutation at the LPL gene, PCR analysis was performed using
pri-mers: 5%-CATCCATTTTCTTCCACAGGG-3% (sense);
5%-GCCCAGAATGCTCACC AGACT-3% (anti-sense).
After amplification, the PCR product was digested with
HinfI, and subsequently fragments were separated on a
4% agarose gel. The 3%-primer was elongated with a 20
base-pair AT-tail for optimal visualization of the di-gested fragment. In the case of the S447stop mutation, two fragments could be detected.
The LPL mutation N291S was detected by the PCR method as previously described [44]. Exon 6 of the LPL
gene was amplified with a 5%-PCR primer located in
intron 5 near the 5% boundary of exon 6 (5%
-GCCGA-GATACAATCTTGGTG-3%). The 3% mismatch PCR
primer was located in exon 6 near the Asn291Ser
mutation (5%
-CTGCTTCTTTTGGCTCTGACTGTA-3%). PCR amplification reactions were performed; 20 ml
of the PCR product was digested with 10 U of Rsa1,
and the digested fragments were separated on 2% agarose gel.
Identification of the CETP I405V RFLP was per-formed as described previously [45]. This RFLP
in-volves an AG nucleotide substitution in exon 14
which does not result in the creation or disappearance of an enzyme restriction site. Therefore, a mismatch
promoter that introduced an RsaI restriction site in the
presence of the mutation, was used. Target sequences
were amplified with a 5%-PCR primer 5%
-CTGTTTC-CAACTTGACTGAG-3%). The 3% mismatch PCR
primer was 5%
-CGCCCGCCGCGCCCCGCGCCCGT-CCCGCCGCCCCCGCCC
CCATTGACTGCAGGA-AGCTCTGTA-3%). The amplification reaction mixture
included 10 mmol/l Tris – HCl; pH 9.0, 50 mmol/l KCl,
0.1% (w/v) gelatin, 1.5 mmol/l MgCl2, 1% Triton X-100
and 20 mg/dl BSA containing 0.1 – 0.5 (mg genomic
DNA and final concentrations of 100 – 200 mmol/l
dNTPs and 0.5 mmol/l primers in a total volume of 50
ml. After initial denaturation (10 min, 95°C), 0.3 – 0.5 U
thermostable DNA polymerase was added, followed by 30 amplification cycles at 95°C for 1 min, 60°C for 1 min, and 72°C for 1 min, with a final extension step of 10 min at 72°C. Some 20 ml of the PCR reaction product was digested with the restriction enzyme ac-cording to the instructions of the manufacturer in a
total volume of 20 ml for 2 h at 72°C. Thereafter,
fragments were separated on 1 – 2% agarose gel (Boehringer Mannheim, FRG) and stained with ethid-ium bromide. DNA restriction fragments were visual-ized and analyzed on transilluminator.
4. Statistical methods
Y
.
Friedlander
et
al
.
/
Atherosclerosis
152
(2000)
239
–
248
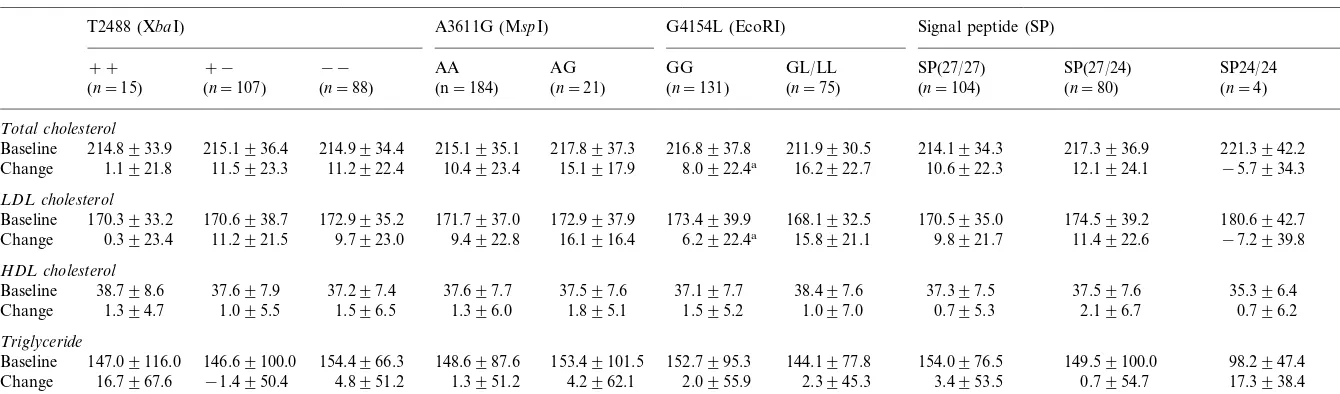
Table 2
Baseline values and lipid changes adjusted for sex and age by apo B polymorphic sites
Signal peptide (SP) G4154L (EcoRI)
A3611G (MspI) T2488 (XbaI)
GG
AA AG GL/LL SP(27/24)
−− SP24/24
+− SP(27/27)
++
(n=21)
(n=184) (n=131) (n=80) (n=4)
(n=88) (n=75)
(n=107)
(n=15) (n=104)
Total cholesterol
216.8937.8 211.9930.5 214.1934.3
215.1936.4 217.3936.9
214.8933.9 214.9934.4 215.1935.1 217.8937.3 221.3942.2
Baseline
15.1917.9
1.1921.8 11.5923.3 11.2922.4 10.4923.4 8.0922.4a 16.2922.7 10.6922.3 12.1924.1 −5.7934.3
Change
LDL cholesterol
172.9937.9
170.3933.2 170.6938.7 172.9935.2 171.7937.0 173.4939.9 168.1932.5 170.5935.0 174.5939.2 180.6942.7 Baseline
6.2922.4a 15.8921.1 9.8921.7 11.4922.6 −7.2939.8
16.1916.4 9.7923.0
Change 0.3923.4 11.2921.5 9.4922.8
HDL cholesterol
37.197.7 38.497.6 37.397.5
Baseline 38.798.6 37.697.9 37.297.4 37.697.7 37.597.6 37.597.6 35.396.4
1.595.2 1.097.0 0.795.3 2.196.7 0.796.2 1.895.1
Change 1.394.7 1.095.5 1.596.5 1.396.0
Triglyceride
152.7995.3 144.1977.8 154.0976.5 149.59100.0
154.4966.3 98.2947.4
146.69100.0 148.6987.6 153.49101.5 Baseline 147.09116.0
4.2962.1
16.7967.6 −1.4950.4 4.8951.2 1.3951.2 2.0955.9 2.3945.3 3.4953.5 0.7954.7 17.3938.4 Change
Y.Friedlander et al./Atherosclerosis152 (2000) 239 – 248 243
Table 3
Baseline values and lipid changes adjusted for sex and age by apo E genotypes
o3o3 (n=132) o3o4 (n=29) o2o3 (n=25)
Total cholesterol
Baseline 212.78932.52a 234.87939.15 204.48939.68 11.42921.60
Change 7.28926.60 10.06926.15
LDL cholesterol
Baseline 169.02932.72a 194.21942.42 159.14942.58 10.97923.48
Change 9.93921.31 8.52926.80
HDL cholesterol
Baseline 37.3597.63 35.7296.10 39.6198.96 −0.1696.24 1.0997.71 Change 1.6895.63
Triglyceride
Baseline 149.22990.01 171.83989.37 142.38972.78 Change 5.95955.68 −4.16946.82 −11.30945.89
aP50.01.
Table 5
Baseline values and lipid changes adjusted for sex and age by CETP I405 polymorphic site
V¯V (n=34) V¯I (n=88) I¯I (n=72)
Total cholesterol
219.23939.61
Baseline 216.15930.41 212.86938.35 10.48926.01
Change 10.88923.02 11.02920.99
LDL cholesterol
176.45937.64
Baseline 172.06933.26 170.00940.06 Change 9.28925.95 10.30921.11 10.08921.69
HDL cholesterol
158.14988.34 146.83985.07 152.68989.13 Baseline
Change 4.48951.41 −1.27953.37 4.00954.02
were performed on sex and age adjusted values. Our results from the crossover study indicate an overall significant effect of the diets on plasma cholesterol, LDL-C and triglyceride concentrations (data not shown). For all variables, no carry over effect was demonstrated, and the order of the given diets had no effect on the response of the lipid phenotypes. Due to these results, the mean values of the two determinations made at the end of the LSC diet compared with that determined at the end of the HSC diet, irrespective of the order of the diets, were used in order to test the null hypothesis that phenotypic change was not associated with the genetic variation at the gene locus under study (Tables 2 – 5).
The frequency of apoB polymorphisms is similar to those previously described in the Israeli population [14,35]. The frequency distribution of apoE genotype treatment and period in this basic two-period crossover
design were assessed by means of t-tests according to
Fleiss [46]. Analysis of variance was performed in order to examine the homogeneity of baseline levels and changes across the genotype groups.
5. Results
Study participants comprised 108 males and 106 females. The mean of age was 46.4 years (range 15 – 74) for men and 44.0 years (range 14 – 70) for women. The
means of BMI were 26.0 (93.1) and 25.7 (94.2) for
men and women, respectively. Since the variability of the change in lipids and lipoproteins was found to be associated with age and not with BMI, further analyses
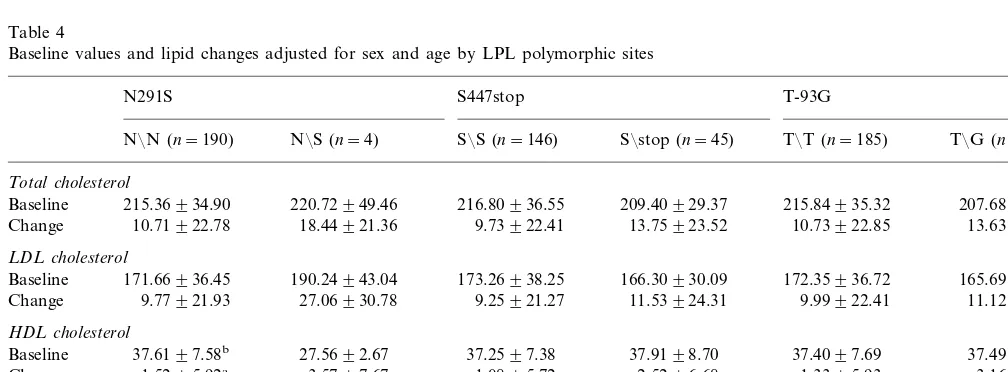
Table 4
Baseline values and lipid changes adjusted for sex and age by LPL polymorphic sites
S447stop
Baseline 215.36934.90 216.80936.55 209.40929.37 215.84935.32
13.63921.17 Change 10.71922.78 18.44921.36 9.73922.41 13.75923.52 10.73922.85
LDL cholesterol
171.66936.45 190.24943.04 173.26938.25
Baseline 166.30930.09 172.35936.72 165.69934.65
9.77921.93 11.12915.40
Change 27.06930.78 9.25921.27 11.53924.31 9.99922.41
HDL cholesterol
37.4996.94 37.6197.58b
was in Hardy – Weinberg equilibrium and in a relatively
uniform pattern (allele frequencies: o2=0.079, o3=
0.815, o4=0.105) similar to that previously described
in other populations [47]. All the LPL and CETP
polymorphisms were also in Hardy – Weinberg
equilibrium.
Table 2 presents the average lipid and lipoprotein levels at baseline and the average changes from the end of LSC diet to the end of the HSC diet for the different apo B genotypes. Subjects homozygous for the apo B
XbaI restriction site allele (designated as + +) showed
a smaller change in plasma cholesterol (1.1 mg/dl) and
LDL-C (0.3 mg/dl) levels compared to subjects with the
X-allele (11.4 mg/dl) and LDL-C (10.6 mg/dl).
How-ever, these differences in change between the two XbaI
genotype groups were not statistically significant for
total cholesterol (P=0.092) and for LDL-C (P=
0.098). Subjects heterozygous for the less frequent apo
B EcoRI allele (G/L4145) showed a significant change
in plasma cholesterol (16.2 mg/dl) and LDL-C (15.6
mg/dl) levels, compared with a smaller change for
cholesterol (8.0 mg/dl) and LDL-C (6.2 mg/dl) among
subjects homozygous for the apoB EcoRI restriction
site allele (G/G4154). These differences in change
be-tween the EcoRI genotype groups were statistically
significant (P=0.01 for total cholesterol andP=0.007
for LDL-C). The Apo B signal peptide polymorphism appeared to influence the response of HDL-C to the
intervention diet. HDL-C change among the SP27/24
(insertion/deletion alleles) heterozygotes was greater
than that observed in the SP27/27 genotype; however,
this difference was not significant (P=0.099).
Table 3 indicates that although TC and LDL-C baseline levels were significantly different among the apo-E genotypes, there were no significant apo E geno-type effects on lipid and lipoprotein changes.
Mean TG and HDL-C on the basal diet and the average response to diet were significantly different among LPL genotypes (Table 4). Triglyceride baseline
values were higher (P=0.092) and HDL-C significantly
lower (P=0.008) among subjects found to be
het-erozygous for the LPL N291S mutation. A
heteroge-neous response for HDL-C was observed for
individuals harboring the S291 allele compared to those homozygous for the N291 allele. Baseline triglyceride levels were also found to be significantly affected by the LPL S447stop mutation. Finally, we observed a
statisti-cally non-significant (P=0.1) influence of the LPL-93
mutation on the triglyceride response to the dietary
change; Triglyceride declined among the T/G
het-erozygotes compared to a slight increase among those
individuals with the T/T genotype.
In the present study, there were no significant effects of the CETP genotypes on lipid and lipoprotein base-line levels and on their response to dietary manipula-tions (Table 5).
6. Discussion
In this study of 214 Israeli subjects, conducted in a free-living setting, various polymorphisms at five major gene loci were determined. The response to diet was significantly greater in participants heterozygous for the
absence of the apo B EcoRI site (G/L4154) as
com-pared to those individuals with the genotype G/G4154.
The EcoRI L4154 allele, which occurs at frequency of
0.1 – 0.2, in most populations has been associated with CHD and with increased plasma cholesterol concentra-tion in some studies [15,48 – 50], but not in all [16,51].
The EcoRI site of the apo B gene has been also
suggested as a ‘variability’ gene affecting the apo B level in the plasma [52,53]. Moreover, in a study of
identical twins, the presence of the EcoRI cutting site
was found to be associated with a decreased co-twins difference in serum cholesterol [54]. In a study of fifty-one subjects who were classified as diet responsive or non-responsive, the responders more frequently had
the EcoRI cutting site absent then the non-responders
[29]. In a recent controlled dietary study conducted on 44 healthy middle-aged subjects, the increase of plasma
total and LDL cholesterol was greater in apoB G/
G4154 subjects compared with those individuals with the other genotypes [55]. Although in other studies this association was not evident [35,56], a meta-analysis of
all published dietary trials confirmed the role of EcoRI
locus [55].
The mechanism through which variation at the apo B
EcoRI locus may affect the response in cholesterol
levels to diet is not fully understood. This association could be explained by variation in the synthetic rate or in the catabolism of apo B and apo B containing
lipoproteins. The EcoRI RFLP reflects a single base
pair change in exon 29 that results in an amino acid change from glutamic acid to lysine [41]. This sequence variation is located in a region presumed to be near the two putative LDL receptor binding sites of apolipo-protein B [57]. Such variation in the receptor-binding site of apo B could lead to different affinity for the LDL receptor and thereby different catabolism.
How-ever, there was no effect of the EcoRI polymorphism
on the binding of LDL to LDL receptor [58], although this does not exclude other effects in vivo.
Several studies have indicated a significant role of apo E genotypes on cholesterol and LDL-C response to dietary treatment [59]. It has been suggested that the differences in cholesterol absorption [60], in apoB
pro-duction and cholesterol synthesis [61] and/or in affinity
to the B, E receptor [62] in subjects with varying apo E alleles may alter lipids response to diet. However, in other studies, such relationships were not demonstrated
[59]. In the present study sample, subjects with theo3o4
geno-Y.Friedlander et al./Atherosclerosis152 (2000) 239 – 248 245
types. However, no apo E effects on the response of lipids and lipoproteins to dietary challenge was ob-served. Ethnic differences, baseline dietary fat and cholesterol intake and baseline lipid levels are among other reasons why studies on the role of apoE in modulation of dietary response have given inconsistent results. Results from a meta-analysis which included 1015 normo- and hypercholesterolemic participants in 16 studies have suggested that an apoE effect may be more evident when the total amount of dietary fat is changed, than when compositional changes are made [63].
Lipoprotein lipase plays a central role in lipoprotein metabolism and genetic variations in the LPL locus have been associated with lipid levels and CHD risk [18,19]. In our study the N291S mutation was found to be associated both with level and variability in TG and HDL-C response to diet. Data from other studies have shown those with one or more alleles for the LPL S291 mutation had significantly higher TG levels, as well as a greater increase in plasma TG over a 3-year period compared to non-carriers [64]. In a group of young MI survivors and healthy matched controls, plasma TG concentrations following an oral fat load were consis-tently higher among carriers of the LPL S291 as com-pared to non-carriers, with the differences being more pronounced in the late postprandial period (8 – 12 h after the fat load) [65]. In the European Atherosclerosis Research Study (EARS), allelic frequencies for S291 mutation did not differ between subjects with parental history of MI (cases) and those without such a history [66]. Yet, among the cases, carriers of the S291 allele had higher TG levels 6 h post an oral fat tolerance test
(P50.04) than non-carriers. In vitro mutagenesis and
expression in COS cells demonstrated that the S291 allele resulted in an LPL protein with activity reduced to 30 – 50% compared to the wild-type protein and a decreased stability [64].
In our study, the Stop447 mutation in the LPL gene was found to be associated with levels of TG but did not alter lipid response to diet. In a study of MI patients and age-matched controls, no significant asso-ciation was found among the healthy subjects between lipid variables and genotypes of the S447stop mutation [67]. However, in monozygotic twins, the Stop447 mu-tation was associated with significantly smaller within-pair differences in plasma HDL-C, total cholesterol, and triglyceride levels [68]. This suggests that individu-als without this mutation are more susceptible to fluctu-ations in their lipid and lipoprotein levels in response to
environmental exposure. In addition, the HindIII
RFLP has also been found to be strongly associated with variability in lipid response to diet manipulations [63,69]. It the ECTIM control populations the S447stop mutation was in nearly complete disequilibrium with
the HindIII RFLP (D/Dmax=0.97, PB10
−4) [70]. It
has been shown that this mutation leads to a truncation of the two carboxyl-terminal amino acids of LPL and mutations affecting the carboxy region have been re-ported to alter the LPL-specific activity in vitro [71]. Yet these findings are inconsistent [72] and the mecha-nism through which this mutation exerts level gene and variability gene effects requires further investigation. Rare mutations in the CETP gene causing partial or full CETP deficiency, that markedly elevate HDL-C and markedly lower VLDL-C, have been described in various populations. Recently, the ECTIM study found evidence of a gene-environment interaction involving
the CETP TaqI B polymorphism, HDL-C, and alcohol
consumption, albeit very high levels of alcohol intake [29]. Our study did not find significant effects of the CETP genotypes on lipid and lipoprotein response to dietary manipulations and we are unaware of other studies that have examined the relationship between dietary response and the CETP gene.
In summary, the metabolic response to changes in dietary intake of saturated fatty acids and cholesterol is likely to be under multifactorial control. Our results suggest that sequence variation(s) in the coding region
of the apo B gene linked to the EcoRI polymorphism
may have a modifiable influence on total cholesterol and LDL-C changes. In our study population, TG and HDL-C response to dietary manipulations were af-fected by LPL mutations. However, other factors, in-cluding genes investigated here, may also be involved and should be re-examined in larger studies in order to uncover their possible role in plasma lipid response to diet, and so contribute to revealing the mechanisms involved in differential responsiveness.
Acknowledgements
The authors would like to thank Limor Ben Yaacov for her key role in the conduct of the dietary trial. This research was supported by the National Council for Research and Development, Israel and the BMFT Ger-many, and partially by the Israel Science Foundation administered by The Israel Academy of Sciences and Humanities.
References
[1] Katan M, Beynen AC, DeVries JH, Nobles AA. Existence of consistent hypo- and hyperresponders to dietary cholesterol in man. Am J Epidemiol 1986;123:221 – 34.
[2] Katan MB, Berns MA, Glatz JF, Knuiman JT, Nobels A, de Vries JH. Congruence of individual responsiveness to dietary cholesterol and to saturated fat in humans. J Lipid Res 1988;29:883 – 92.
cholesterol to fat-modified diets in man. Eur J Clin Invest 1988;18:644 – 7.
[4] Beynen AC, Katan MB, van Zutphen LF. Hypo- and hyperre-sponders: individual differences in the response of serum cholesterol concentration to changes in diet. Adv Lipid Res 1987;22:115 – 71.
[5] Beynen AC, Katan MB. Hypo- and hyperresponders to dietary cholesterol. Am J Clin Nutr 1986;43:974 – 87.
[6] Mensink RP, Katan MB. Effect of monounsaturated fatty acids versus complex carbohydrates on high density lipoprotein in healthy men and women. Lancet 1987;1:122 – 5.
[7] Cobb MM, Risch N. Low-density lipoprotein cholesterol re-sponsiveness to diet in normolipidemic subjects. Metabolism 1993;42:7 – 13.
[8] Clifton PM, Abbey M, Noakes M, Beltame S, Rumbelow N, Nestel PJ. Body fat distribution is a determinant of the high-density lipoprotein response to dietary fat and cholesterol in women. Arterioscler Thromb Vasc Biol 1995;15:1070 – 8. [9] Cobb MM, Teitlebaum H. Determinants of plasma cholesterol
responsiveness to diet. Br J Nutr 1994;71:271 – 84.
[10] Law A, Powell LM, Brunt H, Knott TJ, Altman DG, Rajput J, Wallis SC, Pease RJ, Priestley LM, Scott J, Miller GJ, Miller NE. Common DNA polymorphism within coding se-quence of apolipoprotein B gene associated with altered lipid levels. Lancet 1986;1:1301 – 3.
[11] Talmud PJ, Barni N, Kessling AM, Carlsson P, Darnfors C, Bjursell G, Galton DJ, Wynn V, Humphries SE. Apolipo-protein B gene variants are involved in the determination of serum cholesterol levels: a study in normo- and hyperlipi-daemic individuals. Atherosclerosis 1987;67:81 – 9.
[12] Xu C-F, Tikkanen MJ, Huttunen JK, Pietinen P, Butler R, Humphries S, Talmud P. Apolipoprotein B signal peptide in-sertion/deletion polymorphism is associated with Ag epitopes and involved in the determination of serum triglyceride levels. J Lipid Res 1990;31:1255 – 61.
[13] Boerwinkle E, Chen S-H, Visvikis S, Hanis CL, Siest G, Chan L. Signal peptide-length variation in human apolipoprotein B gene; molecular characteristics and association with plasma glucose levels. Diabetes 1991;40:1539 – 44.
[14] Friedlander Y, Kaufmann NA, Cedar H, Weinberg N, Kark JD. The role of XbaI polymorphism of the apolipoprotein B gene in determining levels and covariability of lipids and lipo-protein variables in a sample of Israeli offspring with family history of myocardial infarction. Atherosclerosis 1993;98:165 – 77.
[15] Hegele RA, Huang LS, Herbert PN, Blum CB, Buring JE, Hennekens CH, Breslow JL. Apolipoprotein B gene DNA polymorphisms associated with myocardial infarction. N Engl J Med 1986;315:1509 – 15.
[16] Deeb S, Failor A, Brown BG, Brunzell JD, Albers JJ, Motul-sky AG. Molecular genetics of apolipoproteins and coronary heart disease. Cold Spring Harbor Symp Quant Biol 1986;51:403 – 9.
[17] Sing CF, Davignon J. Role of apolipoprotein E polymorphism in determining normal plasma lipid and lipoprotein variation. Am J Hum Genet 1985;37:268 – 85.
[18] Jemaa R, Tuzet S, Portos C, Betoulle D, Apfelbaum M, Fumeron F. Lipoprotein lipase gene polymorphism: association with hypertriglyceridemia and body mass index in obese peo-ple. Int J Obesity 1995;19:270 – 4.
[19] Chen L, Pastch W, Boerwinkle E. HindIII DNA polymor-phism in the lipoprotein lipase gene and plasma lipid pheno-types and carotid artery atherosclerosis. Hum Genet 1996;98:551 – 66.
[20] Kondo I, Berg K, Drayana D, Lawn R. DNA polymorphism at the locus for cholesteryl ester transfer protein (CETP) is
associated with high density lipoprotein cholesterol and apolipoprotein levels. Clin Genet 1989;35:49 – 56.
[21] Freeman DJ, Packard CJ, Gaffney D. Polymorphisms in the gene coding for cholesteryl ester transfer protein are related to plasma high-density cholesterol and transfer protein activity. Clin Sci 1990;79:575 – 81.
[22] Fumeron F, Betoulle D, Luc G, Behague I, Ricard S, Poirier O, Jemaa R, Evans A, Arveiler D, Marques-Vidal P, et al. Alcohol intake modulates the effect of polymorphism of the cholesteryl ester transfer protein gene on plasma high density lipoprotein and the risk of myocardial infarction. J Clin Invest 1995;96:1664 – 71.
[23] Berg K. Variability gene effect on cholesterol at the Kidd blood group locus. Clin Genet 1988;33:102 – 7.
[24] Berg K, Kondo I, Drayna D, Lawn R. ‘Variability gene’ effect of cholesterol ester transfer protein (CETP) genes. Clin Genet 1989;35:437 – 45.
[25] Monsalve MV, Roninson D, Woolcock NE, Powell JT, Green-halgh RM, Humphries SE. Within-individual variation in serum cholesterol levels: association with DNA polymorphisms at the apolipoprotein B and AI-CIII-AIV loci in patients with peripheral arterial disease. Clin Genet 1991;39:260 – 73. [26] Friedlander Y, Austin MA, Newman B, Edwards K,
Mayer-Davis EL, King MC. Heritability of longitudinal changes in coronary heart disease risk factors in women twins. Am J Hum Genet 1996;60:1502 – 12.
[27] Tikkanen MJ, Xu C-F, Hamalainen T, Talmud P, Sarna S, Huttunen JK, Pietinen P, Humphries SE. XbaI polymorphism of the apolipoprotein B gene influences plasma lipid response to diet intervention. Clin Genet 1990;37:327 – 34.
[28] Xu C-F, Boerwinkle E, Tikkanen MJ, Humphries SE, Talmud PJ. Genetic variation at the apolipoprotein gene loci contribute to response of plasma lipids to dietary change. Genet Epi-demiol 1990;7:261 – 75.
[29] Abbey M, Belling B, Clifton P, Nestel P. Apolipoprotein B gene polymorphism associates with plasma cholesterol changes induced by dietary fat and cholesterol. Nutr Met Cardiovasc Dis 1991;1:10 – 2.
[30] Glatz JFC, Demacker PNM, Turner PR, Katan MB. Re-sponse of serum cholesterol to dietary cholesterol in relation to apolipoprotein E phenotype. NMCD 1991;1:13 – 7.
[31] Manttari M, Koskinen P, Ehnholm C, Huttunen JK, Manni-nen V. Apolipoprotein E polymorphism influences the serum cholesterol response to dietary intervention. Metabolism 1991;40:217 – 21.
[32] Gaddi A, Ciarrocci A, Matteucci A, Rimondi S, Ravaglia G, Descovish GC, Sirtori CR. Dietary treatment for familial hy-percholesterolemia — differential effects of dietary soy protein according to the apolipoprotein E phenotypes. Am J Clin Nutr 1991;53:1191 – 6.
[33] Xu CF, Angelico F, Del Ben M, Pannozzo F, Mazzarella B, Miller NE, Humphries SE, Talmud PJ. Polymorphisms at the apolipoprotein loci and response of plasma lipids to dietary change in Italian children. NMCD 1992;2:26 – 32.
[34] Friedlander Y, Kaufman NA, Cedar H, Kark JD. XbaI poly-morphism of the apolipoprotein B gene and plasma lipid and lipoprotein response to dietary fat and cholesterol: a clinical trial. Clin Genet 1993;43:223 – 31.
[35] Friedlander Y, Berry EM, Eisenberg S, Stein Y, Leitersdorf E. Plasma lipids and lipoproteins response to a dietary challenge: analysis of four candidate genes. Clin Genet 1995;47:1 – 12. [36] Jemaa R, Tuzet S, Betoulle D, Apfelbaum M, Fumeron F.
Y.Friedlander et al./Atherosclerosis152 (2000) 239 – 248 247
[37] Guggenheim JK, Kaufmann NA, Reshef A. Food composition tables, College of Nutrition and Home Economics, 6th edition. Jerusalem: Ministry of Education and Culture, Goverment of Israel, 1980.
[38] Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low density lipoprotein cholesterol in plasma-without use of preparative ultracentrifuge. Clin Chem 1972;18:449 – 502.
[39] Friedlander Y, Kark JD, Eisenberg S, Stein Y. Calculation of LDL-cholesterol from total cholesterol, triglyceride and HDL-cholesterol: A comparison of methods in the Jerusalem Lipid Research Clinic prevalence study. Isr J Med Sci 1982;18:1242 – 52.
[40] Boerwinkle E, Le SS, Butler R, Schumaker VN, Chan L. Rapid typing of apolipoprotein B polymorphisms by DNA amplification. Atherosclerosis 1990;81:225 – 32.
[41] Ludwig EH, Blackhart BD, Pierotti VR, Caiati L, Fortier C, Knott T, Scott J, Mahley RW, Levy-Wilson B, McCarthy BJ. DNA sequence of the human apolipoprotein B gene. DNA 1987;6:363 – 72.
[42] Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res 1990;31:545 – 8.
[43] Deeb SS, Peng RL. Structure of the human lipoprotein lipase gene [published erratum appears in Biochemistry 1989 Aug 8;28(16):6786]. Biochemistry 1989; 28:4131-4135.
[44] Reymer PWA, Gagne E, Groenemeyer BE, Zhang H, Forysyth I, Jansen H, Seidell JC, Kromhout D, Lie KE, Kastelein J, Hayden MR. A lipoprotein lipase mutation (Asn291Ser) is associated with reducd HDL cholesterol levels in premature athrosclerosis. Nat Genet 1995;10:28 – 34. [45] Funke H, Wiebusch H, Fuer L, Muntoni S, Schulte H,
Ass-mann G. Identification of mutations in the cholesterol ester transfer protein in Europeans with elavated high density lipo-protein cholesterol. Circulation 1994;90:I241.
[46] Fleiss JL. The Design and Analysis of Clinical Experiments. New York: Wiley, 1985.
[47] Hallman DM, Boerwinke E, Saha N, Sandholzer C, Menzel HJ, Csazar A, Utermann G. The apolipoprotein E polymor-phism: a comparison of allele frequencies and effects in nine populations. Am J Hum Genet 1991;49:338 – 89.
[48] Myant NB, Gallagher J, Babir M, et al. Restriction fragment length polymorphism in the apoB gene in relation to coronary artery disease. Atherosclerosis 1989;77:193 – 201.
[49] Paulweber B, Friedl W, Krempler F, et al. Association of DNA polymorphism at the apolipoprotein B gene locus with coronary heart disease and serum very low density lipoprotein levels. Atherosclerosis 1990;10:17 – 24.
[50] Genest JJ, Ordovas JM, McNamara JR, et al. DNA polymor-phisms of the apolipoprotein B gene in patients with prema-ture coronary heart disease. Atherosclerosis 1990;82:7 – 17. [51] Ferns GA, Robinson D, Galton DJ. DNA haplotypes of the
human apoprotein B gene in coronary atherosclerosis, Hum Genet 1988;81:8176 – 80.
[52] Berg K, Powell LM, Wallis SC, Pease R, Knoott TJ, Scott J. Genetic linkage between antigenic group (Ag) variation and the apolipoprotein B Gene:assignment of the Ag locus. Proc Natl Acad Sci USA 1986;83:7367 – 70.
[53] Berg K. Role of genetic factors in atherosclerotic disease. Am J Clin Nutr 1989;49:1025 – 9.
[54] Berg K. Twin studies of coronary heart disease and its risk factors. Acta Genet Med Gemellol (Roma) 1987;36:439 – 53. [55] Rantala M, Rantala TT, Savolainen M, Friedlander Y,
Ke-saniemi YA. Apolipoprotein B gene polymorphisms and serum lipids- meta-analysis of the role of genetic variation on diet responsiveness, Am J Hum Nutr, 2000 (in press).
[56] Abbey M, Hirata F, Chen GC, et al. Restriction fragment length polymorphism of the apolipoprotein B gene and re-sponse to dietary fat and cholesterol. Can J Cardiol Suppl G 1995;11:79 – 85.
[57] Knott TJ, Pease RJ, Powell LM, Wallis SC, Rall SC Jr., Innerarity TL, Blackhart B, Taylor WH, Marcel YL, Milne RW, Johnson D, Fuller M, Lusis AJ, McCarthy BJ, Mahley RW, Levy-Wilson B, Scott J. Complete protein sequence and identification of structural domains of human apolipoprotein B. Nature 1986;323:734 – 8.
[58] Gallagher JJ, Myant NB. Does the EcoRI polymorphism in the apolipoprotein B gene affect the binding of low density lipoprotein to the low density lipoprotein receptor. Arthe-rioscler Thromb 1992;12:256 – 60.
[59] Tikkanen MJ. Apolipoprotein E polymorphism and plasma cholesterol response to dietary change. In: Simopoulos AP, editor. Genetic Variation and Dietary Response. Basel, Karger: World Rev Nutr Diet, 1977:15 – 21.
[60] Miettinen TA, Gylling H, Vanhanen H, Ollus A. Cholesterol absorption, elimination, and synthesis related to LDL kinetics during varying fat intake in men with different apoprotein E phenotypes. Atheroscler Thromb 1992;12:1044 – 52.
[61] Nikkila M, Solakivi T, Lehtimaki T, Koivula T, Laippala P, Astrom B. Postprandial plasma lipoprotein changes in relation to apolipoprotein E phenotypes and low density lipoprotein size in men with and without coronary artery disease. Atherosclerosis 1994;106:149 – 57.
[62] Davignon JR, Gregg E, Sing CF. Apolipoprotein E polymor-phism and atherosclerosis. Arteriosclerosis 1988;8:1 – 21. [63] Ordovas MJ, Lopez-Miranda J, Mata P, Perez-Jimenez F,
Lichtenstein AH, Schaefer EJ. Gene-diet interaction in deter-mining plasma lipid response to dietary intervention. Atherosclerosis Suppl 1995;118:S11 – 27.
[64] Fisher RM, Mailly F, Peacock RE, Hamsten A, Seed M, Yud-kin JS, Beisiegel U, Feussner G, Miller G, Humphries SE, et al. Interaction of the lipoprotein lipase asparagine 291serine mutation with body mass index determines elevated plasma triacylglycerol concentrations: a study in hyperlipidemic sub-jects, myocardial infarction survivors, and healthy adults. J Lipid Res 1995;36:2104 – 12.
[65] Karpe F, Steiner G, Olivercrona T, Carlson LA, Hamsten A. Metabolism of triglyceride-rich lipoproteins during alimentary lipemia. J Clin Invest 1993;91:748 – 58.
[66] Gerdes C, Fisher RM, Nicaud V, Boer J, Humphries SE, Tal-mud PJ, Faergeman O. Lipoprotein lipase variants D9N and N291S are associated with increased plasma triglyceride and lower high-density lipoprotein cholesterol concentrations: stud-ies in the fasting and postprandial states: the European Atherosclerosis Research Studies. Circulation 1997;96:733 – 40. [67] Peacock R, Hamsten A, Nilsson-Ehle P, Humphries SE.
Asso-ciations between lipoprotein lipase polymorphisms and plasma correlations of lipids, lipoproteins and lipase activities in young myocardial infarction survivors and age-matched healthy individuals from Sweden. Atherosclerosis 1992;97:171 – 85.
[68] Thorn JA, Needham WA, Stocks J, Galton DJ. The Ser447-Ter mutation of the lipoprotein lipase gene relates to variabil-ity of serum lipid and lipoprotein levels in monozygotic twins. J Lipid Res 1998;39:437 – 41.
[69] Jemaa R, Tuzet S, Betoulle D, Apfelbaum M, Fumeron F. Hind III polymorphism of the lipoprotein lipase gene and plasma lipid response to low calorie diet. Int J Obes 1997;21:280 – 3.
[71] Kozaki K, Gotoda T, Kawamura M, Shimano H, Yazaki Y, Ouchi Y, Orimo H, Yamada N. Mutational analysis of human lipoprotein lipase by carboxy-terminal truncation. J Lipid Res 1993;34:1765 – 72.
[72] Zhang H, Henderson H, Gagne SE, Clee SM, Miao L, Liu GQ, Hayden MR. Common sequence variants of lipoprotein lipase: standardized studies of in vitro expression and catalytic function. Biochim Biophys Acta 1996;1302:159 – 66.