www.elsevier.com / locate / bres
Interactive report
1
The AMPAR subunit GluR2: still front and center-stage
*
Hidenobu Tanaka, Sonja Y. Grooms, Michael V.L. Bennett, R. Suzanne Zukin
Department of Neuroscience, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461, USA Accepted 26 September 2000
Abstract
21
Abnormal influx of Ca through AMPA-type glutamate receptors (AMPARs) is thought to contribute to the neuronal death associated
21 21
with a number of brain disorders. AMPARs exist as both Ca -impermeable and Ca -permeable channels. AMPARs are encoded by four genes designated GluR1 (GluR-A) through GluR4 (GluR-D). The presence of the GluR2 subunit renders heteromeric AMPA receptor
21
assemblies Ca -impermeable. Molecular diversity of AMPARs under physiological and pathological conditions is generated by differential spatio-temporal patterns of GluR expression, by alternative RNA splicing and editing and by targeting and trafficking of receptor subunits at dendritic spines. The GluR2 gene is under transcriptional control by the RE1 element specific transcription factor, a gene silencing factor which renders it neuron-specific. GluR2 transcripts are edited by ADAR2 (double-stranded RNA-specific editase 1). AMPAR targeting and trafficking to spines are regulated by synaptic activity and are critical to synaptic plasticity. Recent studies involving animal models of transient forebrain ischemia and epilepsy show that GluR2 mRNA and GluR2 subunit expression are
21
downregulated in vulnerable neurons prior to cell death. Ca imaging and electrical recording from individual pyramidal neurons in hippocampal slices reveal changes in AMPAR functional properties after ischemia. In slices from post-ischemia animals, CA1 neurons
21
with robust action potentials exhibit greatly enhanced AMPA-elicited rises in intracellular Ca . Excitatory postsynaptic currents in
21 21
post-ischemic CA1 exhibit an enhanced Ca -dependent component that appears to be mediated by Ca -permeable AMPARs. These
21
studies provide evidence for Ca influx through AMPARs in neurons destined to die. To examine whether acute GluR2 downregulation, even in the absence of a neurological insult, can induce neuronal death, we performed knockdown experiments in rats and gerbils with antisense oligonucleotides targeted to GluR2 mRNA. GluR2 antisense oligonucleotide induced neuronal cell death of pyramidal neurons
21
and enhanced pathogenicity of brief ischemic episodes. These observations provide evidence for Ca influx through AMPARs in neurons
21
destined to die and implicate Ca -permeable AMPARs in the pathogenesis of ischemia-induced neuronal death. 2000 Elsevier Science B.V. All rights reserved.
Theme: Neurotransmitters, modulators, transporters, and receptors
Topic: Excitatory amino acid receptors: ligand-gated channels
Keywords: Excitatory amino acid receptor; AMPA receptor; Calcium; Neurotoxicity; Ischemia; Seizure
1. Introduction
mission and play a critical role in synaptogenesis and
formation of neuronal circuitry, as well as in synaptic
Glutamate receptors mediate excitatory neurotrans-
plasticity including long-term potentiation and long-term
depression. Excessive activation of glutamate receptors is
thought to contribute to the neurodegeneration following a
1
Published on the World Wide Web on 30 October 2000.
wide range of neurological insults including ischemia,
Abbreviations: ADAR, Double-stranded RNA adenosine deaminase;
trauma, hypoglycemia and epileptic seizures. Chronic
AMPA, a-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid;AM-neurodegenerative disorders such as Alzheimer’s disease,
PARs, AMPA receptors; EAA, excitatory amino acids; EPSCs, excitatorypostsynaptic currents; NBQX, 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo
Huntington’s chorea, AIDS encephalopathy, and
amyot-(f)quinoxiline; PMSF, phenylmethylsulfonyl fluoride; NSF, N-methyl-rophic lateral sclerosis may also involve glutamate-induced
maleimide sensitive fusion protein; NRSF, neural restrictive silencingneuronal cell death (for reviews, see [28,73]).
factor; REST, RE1 element specific transcription factor
Considerable interest has focused on the molecular
*Corresponding author. Tel.: 11-718-430-2160; fax: 11-718-430-mechanisms underlying glutamate receptor-mediated
neu-8932.E-mail address: [email protected] (R.S. Zukin).
ronal death. Glutamate induces neuronal death by eliciting
21
a rise in intracellular free Ca
, which activates a number
[42,52] voltage-dependent block by polyamines [13,32]
of proteases, phospholipases and endoncleases, by gene-
and single channel conductance [141] of recombinant
ration of free radicals that destroy cellular membranes by
AMPARs expressed in Xenopus oocytes and mammalian
lipid peroxidations (for reviews, see [27,29,122,135]) and
cells.
by induction of apoptosis [29]. Possible mechanisms by
AMPARs are ligand-gated channels and, by analogy to
21
which glutamate could elicit a rise in intracellular Ca
the nicotinic acetylcholine receptor, are thought to be
21
include: (1) activation of Ca
-permeable AMPA (
a
-
tetrameric or pentameric assemblies arranged around a
amino-3-hydroxy-5-methyl-4-isoazole-proprionic
acid)-
central aqueous pore.
type glutamate receptors (AMPARs) [146,148]; (2) activa-
The current model of GluR subunit topology in the
tion
of
group
1
metabotropic
glutamate
receptors
membrane includes: (1) a large extracellular
amino-termi-(mGluRs), which are positively linked to inositol phos-
nal domain; (2) three transmembrane-spanning domains
21
phates; (3) activation of voltage-sensitive Ca
channels;
(TM1, TM3 and TM4); (3) a fourth amphipathic segment
and / or (4) de-activation of extrusion and / or sequestration
(TM2) that forms a channel-lining reentrant hairpin loop,
1
systems [99].
similar in structure to the pore-forming region of K
21
Until recently, AMPARs were thought to be Ca
-
channels [158]; (4) a binding domain for agonists formed
impermeable. It is now well established that the presence
from segments of the amino-terminal domain and
extracel-of the GluR2 subunit in heteromeric AMPAR assemblies
lular loop [138]; and (5) an intracellular C-terminal
21 21
governs the permeability of AMPARs to Ca
and Zn
.
domain. The dominance of the GluR2 subunit in
determin-21
In the adult mammalian central nervous system under
ing permeability to Ca
and other divalent ions is
physiological conditions, the vast majority of cells and
attributed to the presence of a positively charged arginine
21
tissues express GluR2-containing, Ca
-impermeable AM-
(R) in place of a glutamine (Q) residue within TM2, which
PARs. Thus, a change in the level of GluR2 expression
forms the selectivity filter of AMPARs [21,57]. Although
would be expected to have significant physiological conse-
most
hippocampal
neurons
express
predominantly
quences. The relative expression of GluR2 subunit mRNA
heteromeric AMPARs, they may also express GluR1
and protein in neurons is not static but is regulated in a
homomers [80,157].
cell-specific manner during development [108] and may be
AMPARs are differentially expressed throughout
neu-remodeled after seizures [38,113,114] or ischemic insult
rons of the mammalian central nervous system. Studies
[38,44,111,114] and by administration of anti-psychotics
involving patch-clamp recording combined with RT-PCR
21
[35] drugs of abuse [36,102] or corticosteroids [93]. Ca
-
(reverse transcriptase-polymerase chain reaction)
demon-21
permeable AMPARs are implicated in synaptogenesis and
strate that AMPAR permeability to Ca
varies inversely
formation of neuronal circuitry, particularly at times and in
with abundance of GluR2 mRNA in a wide range of cell
21
cells in which NMDAR expression is low. Ca
influx via
types (Table 1). Excitatory principal neurons such as
21
Ca
-permeable AMPARs is thought to play a critical role
hippocampal [12] and neocortical [60] pyramidal cells and
in growth cone movement and experience-dependent prun-
dentate gyrus granule cells [42] express abundant GluR2
21
ing of synaptic connections during early development [81].
mRNA and exhibit low AMPAR Ca
-permeability. Thus,
This article reviews recent studies that address transcrip-
a change in GluR2 expression would be expected to have
tional and translation regulation and targeting and traffick-
significant physiological consequences.
ing of the AMPAR subunit GluR2 under physiological and
pathological conditions, with a particular emphasis on
2.1. GluR2 RNA editing
transcriptional regulation of GluR2 in ischemia and status
epilepticus.
The functionally critical arginine (R) residue within
TM2 is not encoded by the GluR2 gene, but rather arises
by adenosine-specific RNA editing of the double-stranded
2. The AMPAR gene family
pre-mRNA, which converts an adenosine residue in the
glutamine codon to an inosine [50,136] (Fig. 1). RNA
AMPARs mediate fast excitatory synaptic transmission
editing, the process by which genomically encoded
in-in the vertebrate central nervous system. AMPARs are
formation is enzymatically modified, is an important mode
assemblies of four subunits, GluR1-4 (or GluRA-D),
of receptor regulation [8,78,97]. To date, three
structurally-encoded by separate genes which are differentially ex-
related RNA-editing enzymes with adenosine deaminase
pressed throughout the CNS (for review, see [53,130]).
activity have been identified in mammalian tissues:
AMPARs assembled from combinations of GluR1, GluR3
ADAR1 (dsRNA-specific adenosine deaminase, dsRAD or
and / or GluR4 subunits (lacking the GluR2 subunit) are
DRADA [63,74,98,107]; ADAR2 (dsRNA-specific editase
21 21
permeable to Ca
and Zn
and have doubly rectifying
1, RED-1 [43,69,82,85,97]; and RED2 (dsRNA-specific
current–voltage relations due to voltage-dependent block
editase 2) [82]). Each of the editing enzymes contains two
by intracellular polyamines [21,52,152]. The presence of
to three dsRNA binding domains within the
amino-termi-21
carboxy-Table 1
a
Studies supporting the GluR2 hypothesis
Condition Cell type Refs
Suppression of GluR2 gene expression
Developing brain various [108,111]
Transient global ischemia CA1 pyramidal cells [3,44,49,109–111,113]
Oxygen–glucose dissociated hippocampal [159]
deprivation neurons
Status epilepticus CA3 pyramidal cells [36–38,45,70,71,111,113,114]
Mutant spastic rats cerebellar Purkinje and [79]
granule cells
Schizophrenia parahippocampal [34]
pyramidal cells
Amyotrophic lateral spinal motor neurons [153]
sclerosis
Alzheimer’s disease entorhinal cortex [144]
GluR2 knockdown-induced neuronal death
Adult rat and gerbil hippocampal pyramidal [100]
neurons
Developing brain hippocampal pyramidal [39]
neurons
21
Expression of Ca -permeable AMPARs
Developing brain retinal ganglion cells [120,139]
Transient global ischemia CA1 pyramidal cells [44,146,147]
Oxygen–glucose deprivation dissociated hippocampal [159]
neurons
Editing-deficient GluR2 mice CA3 pyramidal cells [16]
21
Neurotoxicity mediated by Ca -permeable AMPARs
Primary cultures cerebellar Purkinje cells [15]
Primary cultures neocortical neurons [77,151,156]
primary cultures spinal motor neurons [5]
cell line oligodendroglial lineage
Oxygen–glucose dissociated hippocampal [159]
deprivation neurons
Editing-deficient GluR2 mice CA3 pyramidal cells [16]
a
Modified from [109].
terminal domain [6]. Whereas ADAR1 and ADAR2 are
at the R / G site and splicing of the flip / flop cassettes of
expressed in most mammalian tissue; RED2 is expressed
GluR1-4 are developmentally regulated. During early
exclusively in the brain [63,82,97].
postnatal life, mammalian neurons express only GluR flip
ADAR2 is an extremely efficient and specific double-
splice forms; expression of GluR flop forms occurs at a
stranded RNA editase. In neurons of mammalian brain,
later stage of development, leading to a reduction in the
ADAR2 edits primary transcripts encoding glutamate
steady-state phase of glutamate-evoked currents [87,88].
receptor subunits at the ‘Q / R’ site, thereby altering the
gating and ionic permeability properties of the transmitter-
2.3. Transcriptional regulation of GluR2
activated channel [83,124,131]. The other main target of
ADAR2 is the serotonin 5HT-2C receptor, a member of
Recent studies indicate the presence of a functional
the large superfamily of G protein-coupled receptors [23].
repressor element 1 (RE1-like silencer) in the proximal
ADAR2 edits the 5HT-2C pre-mRNA at a site near its 5
9
promoter of the GluR2 gene [92] and show that under
end to convert a genomically-encoded isoleucine residue to
physiological conditions, the GluR2 gene is under
tran-valine. Fully edited versions of the serotonin 5HT-2C
scriptional control by REST (RE1 element specific
tran-receptor couple with less efficiency to G proteins.
scription factor, also known as NRSF or XBR), a gene
In neonatal and adult rat brain, virtually 100% of GluR2
silencing factor which renders it highly neuron selective
mRNA is edited at the Q / R site to yield GluR2 subunits
[56,92] (Fig. 2). Recombinant REST represses
transcrip-21
that form Ca
-impermeable AMPARs. The unedited form
tion of the GluR2 gene by recruiting the co-repressors
is detectable only in prenatal life (E14 to P0) but never
Sin3A and histone deacetylase to the RE1 site of the
exceeds 1% of total GluR2 mRNA [21]. In human brain
GluR2 promoter [56].
[96], Q / R editing is somewhat less in substantia nigra
REST, a gene silencing factor which binds the RE1
(72%), striatum (89%) and fetal tissue (96%), suggesting
regulatory element, is thought to serve a critical role in
that editing efficiency may vary among species and / or
differentiation by repression of a subset of neuron-specific
developmental stages.
genes (Fig. 2). Known target genes including NaCh II
In the AMPAR family, desensitization kinetics are
[17,51], calbindin I [1], synaptotagmin IV [10], neuronal
controlled by editing of GluR2 through GluR4 at a site
nicotinic ACh receptor
a
2[154] and AMPAR subunit
preceding the putative fourth transmembrane region [75].
GluR2 [56,90]. REST, a member of the Kruppel family of
Editing at the ‘R / G’ site is specific for GluR2, -3 and -4,
zinc finger transcription factor proteins, contains two
and is about 80–90% complete in adult rat brain. The
repressor motifs within the N- and C-terminal domains and
degree of RNA editing at the R / G site increases with age
nine zinc finger motifs (contained within the central
through the embryonic and postnatal periods in a subunit-
portion of the protein). Molecular diversity of REST is
and splice variant-specific manner [75].
conferred by alternative RNA splicing which generates at
least six splice variants, many of which are truncated and
2.2. GluR2 RNA splicing
exhibit reduced trans-repressor activity and DNA binding
capacity [104]. The structure of the REST gene and
In addition to RNA editing, further molecular diversity
regulation by alternative RNA splicing are conserved in
of AMPAR subunits is conferred by alternative RNA
human, mouse and rat.
splicing of GluR1-4. AMPAR subunits exist as either of
In early embryogenesis, REST is expressed at high
two distinct isoforms termed ‘flip’ and ‘flop’, which are
levels specifically in non-neural tissues and in
undifferen-generated by alternative splicing of a 114 bp region
tiated neural precursor cells [30,127,129] and is thought to
immediately adjacent to the R / G editing site. Alternative
serve a critical role by repression of a subset of
neuron-splicing introduces one of two functionally critical casset-
specific genes [30,127,128]. The pattern of REST
expres-tes of 38 amino acids (flip or flop) [135] into the
sion in the immature nervous system is inversely correlated
extracellular loop of the GluR subunit. RNA editing at the
with patterns of known target genes. In adult rat brain
R / G site and splicing at the flip / flop site act cooperatively
under physiological conditions, REST is expressed by
to control the desensitization and recovery rates of
neuronal cells, but less abundantly than in non-neuronal
AMPAR
responses
[131].
The
allosteric
modulator
cells. Highest levels of REST expression occur in neurons
cyclothiazide strongly attenuates desensitization in flip but
of the hippocampus, pons / medulla and midbrain. Although
not in flop splice variants of recombinant AMPARs [105].
all splice forms are expressed in neuronal cells, two are
Consistent with their extracellular position, neither the
specific. These splice forms contain short
neuron-21
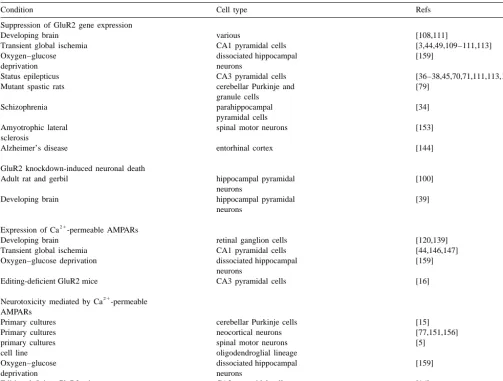
Fig. 2. Models of neuron restrictive silencer element (NRSE), RE-1-silencing trancription factor (REST), REST function and GluR2 promoter. (A) DNA sequence of the neuron restrictive silencer element (NRSE) consensus, which serves as a recognition sequence for REST. (B) Schematic representation of the repressor element 1(RE-1)-silencing transcription factor (REST) protein. (C) Function of NRSF / REST in non-neuronal cells and neuronal cells. (D) Schematic showing the location of the silencer in the GluR2 promoter showing silencer element. A,B, reprinted from [61]; C, reprinted from [127]; D, reprinted from [92].
Under pathological conditions, REST expression is
mRNAs encoding them are localized to the spines of
markedly upregulated in hippocampal neurons. Kainic
hippocampal neurons. A number of proteins implicated in
acid-induced status epilepticus induces a dramatic, but
synaptic plasticity and the mRNAs encoding them are
transient upregulation [56] of REST in dentate granule
localized to spines of hippocampal neurons. These include
cells and a pronounced, long-term (24 h or longer)
microtubule-associated protein 2 (MAP2) [40,65], the
a
21
upregulation of REST in hippocampal pyramidal neurons
subunit of Ca
/ calmodulin-dependent protein kinase II
a
following severe limbic seizures [104]. These observations
(CamKII
a
) [19], the activity-related cytokseletal protein
raise the possibility that other neuronal insults as, for
ArC [19], the RNA binding protein, CREB [31], RNA
example, global ischemia, also induce REST expression in
polymerase III transcript BC1 [145], and AMPAR and
neurons destined to die.
NMDAR mRNAs [41,86] (for review, see [7]). Thus,
translational regulation of specific transcripts within
de-2.4. Translational regulation of the GluR2 transcript
ndrites would be expected to provide fine control over the
temporal and spatial extent of gene expression, including
Recent interest in translational control of synaptic
those for glutamate receptor subunits.
which regulate the initiation and elongation of mature
this mechanism comes from the finding of pools of
transcripts, and (2) translational repressor and enhancer
AMPARs within spines [118], the high concentration of
factors. In addition, translational efficiency is influenced by
NSF in the hippocampus [54,116], its high localization
transcript-specific structural motifs, which serve as recog-
within hippocampal PSDs [155], and its accumulation in
nition sequences for the RNA binding proteins. These
PSDs following transient cerebral ischemia [55].
include: (1) sequences residing in the 5
9
(and 3
9
) UTR; (2)
alternate or additional 5
9
-UTR AUG codons (or their
cognate short open reading frames); (3) consensus se-
4. Regulation of GluR2 expression in
quences for RNA binding proteins; and / or (4) the exact
neurodegenerative disorders
nucleotide context of the initiator AUG [58].
The GluR2 transcript appears to be under negative
4.1. Ischemia
translational control. The presence of the 5
9
-UTR region in
the GluR2 transcript suppresses translation of GluR2 in
4.1.1. Global ischemia-induced suppression of GluR2
Xenopus oocytes and in cell-free reticulocyte lysates [92].
mRNA and subunit expression
The longest GluR2 transcripts identified in vivo, beginning
Global ischemia during cardiac arrest affects 150,000
481 bases downstream from the AUG, exhibit greatly
Americans each year and in many cases results in delayed
reduced translational efficiency (by about 40- to 50-fold)
onset of neurological deficits [112]. In addition, open heart
relative to the GluR2 transcript containing only a seven-
surgery can cause brain ischemia [14,119,140]. Transient,
base leader when assayed in reticulocyte lysates [67,92].
severe global or forebrain ischemia, observed in patients
The region with the greatest suppressor activity (
|
20-fold)
during cardiorespiratory arrest, cardiac surgery or
ex-overlaps all identified transcription initiation sites and
perimentally in animals, induces selective and delayed
includes two of the five upstream AUG codons and a
neuronal death [14,119,140]. Pyramidal neurons in the
40-nucleotide imperfect GU repeat. Although the mecha-
CA1 region of the hippocampus are particularly
vulner-nism for translational suppression by the GluR2 5
9
-UTR
able. Histological evidence of degeneration, exhibiting the
has yet to be identified, the upstream AUGs on their own
hallmarks of apoptosis, is not observed until two to three
exert only minor suppression of translation [91].
days after induction of ischemia in rats [25,64,115,121].
The delayed cell death after ischemia requires an early rise
21 21
in Ca
in CA1 and initial translocation of Zn
from
3. GluR2 receptor targeting and trafficking
presynaptic neurons at CA1 synapses. During the ischemic
21
episode cells depolarize, exhibit a rise in intracellular Ca
Trafficking and targeting of specific mRNAs and poly-
and become inexcitable. Following reperfusion, cells
ap-ribosomes to dendritic spines are thought to play an
pear morphologically normal, exhibit normal intracellular
21
important role in the modification of synaptic strength
Ca
and regain the ability to generate action potentials for
during synaptogenesis and in synaptic plasticity (for
24 to 72 h after the ischemic insult. Ultimately,
intracellu-21 21
review, see [7]). New protein synthesis is essential to
lar Ca
and / or Zn
rises in vulnerable neurons and cell
long-lasting modifications of synaptic strength [4] and the
death ensues.
maintenance phase of LTP [80]. Moreover, targeting of
This delayed neurodegeneration, which may have
clini-mRNAs to dendrites and dendritic spines can be regulated
cal relevance, is thought to be triggered by a transient rise
by synaptic transmission and plasticity [47,90,134,143].
in glutamate release during the ischemic episode [142],
21
Individual spines along a single dendrite may express
followed by late and excessive Ca
influx through
AMPARs which differ in subunit composition and func-
glutamate receptor channels [29,135]. Although NMDARs
21 21
tional properties including conductance, kinetics and Ca
are highly Ca
-permeable, there is now general consensus
permeability [70,123]. Recent studies support an NSF(N-
that antagonists of AMPARs, like the quinoxalinedione
methyl-maleimide
sensitive
fusion
protein)-dependent
NBQX, appear to be much more effective than NMDA
mechanism for insertion of GluR2-containing AMPARs
antagonists in preventing CA1 cell death following severe
and reveal a rapid recycling of functional AMPARs at CA1
global ischemia, even when given as late as 24 h after
synapses under physiological conditions (Fig. 3). Immuno-
ischemia [18,94,133]. These observations indicate that
precipitation experiments demonstrate that GluR2 exists
AMPARs are critical mediators of ischemia-induced
neuro-together
with
NSF
in
a
complex
in
hippocampus
nal death and that neuronal damage is not irreversible for
[103,137]. Yeast two-hybrid studies reveal that GluR2 (and
24 h after the insult.
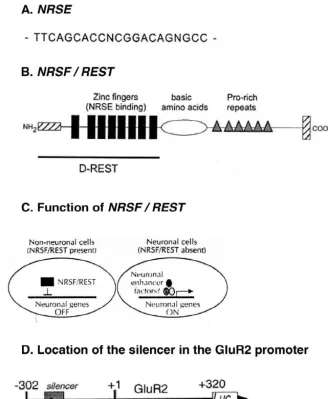
Fig. 3. Model of NSF modulation of AMPA receptor function. (A) Regulation of receptor insertion. Interaction of NSF with C terminus of the GluR2 or GluR4 subunit in subsynaptic vesicles (1) may regulate the association or ‘docking’ (2) of the vesicles with the postsynaptic membrane fusion machinery and promote the insertion of AMPA receptors into the synaptic plasma membrane (3). (B) Regulation of receptor function or clustering. Interaction of NSF with the C terminus of the GluR2 or GluR4 subunit in the synaptic plasma membrane (1) regulates the conformation (2) of the GlurR subunits and modulates channel function and / or the association of the receptors with synaptic proteins involved in synaptic targeting. Reprinted from [137].
mRNA downregulation directly precedes neural damage,
lated into changes in subunit abundance. Studies involving
immunofluorescent labeling of brain sections and Western
and does not occur in the CA3 or dentate gyrus regions
blot analysis of microdissected hippocampal subfields
which are resistant to ischemia-induced damage. GluR3
indicate that global ischemia triggers a pronounced and
mRNA levels also decrease, but to a lesser degree (to
cell-specific reduction in GluR2 subunit abundance in CA1
about 50% of control values); GluR1 and NR1 levels
pyramidal neurons [101]. At 72 h after ischemia, virtually
remain constant. The GluR1 / GluR2 ratio is markedly
all CA1 pyramidal neurons exhibited greatly reduced
increased in CA1 at 24 h, but not in CA3 or dentate gyrus.
GluR2 immunolabeling throughout their somata and
de-Because the presence of the edited GluR2 subunit greatly
21
ndritic processes (Fig. 5). GluR2 immunolabeling was
reduces the Ca
permeability of AMPA channels, these
21
unchanged in pyramidal cells of the CA3 and granule cells
findings predict expression of functional of Ca
-perme-of the dentate gyrus, regions resistant to ischemia-induced
able AMPARs in CA1 neurons after global ischemia, but at
damage. In contrast, immunolabeling of the AMPAR
times preceding neuronal death. Moreover, timing of the
GluR1 was unchanged in CA1, CA3 and DG. Western
switch is consistent with a causal role for
AMPAR-me-21
analysis indicated that GluR2 subunit abundance was
diated Ca
influx in ischemia-induced damage. Since
markedly reduced in CA1 at 60 and 72 h after the ischemic
editing of GluR2 mRNA at the Q / R site is unaltered after
insult, times prior to the onset of neuronal death; GluR1
global ischemia [106,125,159], the change in AMPAR
abundance was unchanged in all subfields at all times
functional properties would appear to be a direct
conse-examined. These findings, together with our previous
quence of altered gene expression.
21
observation of enhanced AMPA-elicited Ca
influx in
Because AMPAR subunit expression is known to be
post-ischemic CA1 neurons [44] demonstrate expression of
under translational control, an important question was
21
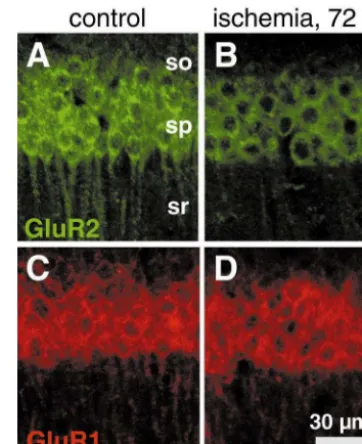
Fig. 5. Global ischemia induces downregulation of GluR2, but not GluR1, immunolabeling in CA1 pyramidal neurons. GluR1 and GluR2 immunoreactivity in brain sections from (A, C) a control (sham-operated) and (B, D) an experimental animal 72 h after ischemia. (B) GluR2 immunoreactivity decreased in CA1 pyramidal cells after ischemia, although (D) GluR1 immunoreactivity was unchanged. so: stratum oriens; sp: stratum pyramidale; sr: stratum radiatum. Similar results were observed in experimental and control animals.
21
acetyl spermine [147], channel blockers selective for Ca
-permeable AMPARs [11,48]. These findings are consistent
with a mechanism whereby post-ischemic CA1 neurons
21
generate slow EPSCs mediated by newly expressed Ca
-permeable AMPARs. Many post-ischemic CA1 pyramidal
Fig. 4. Expression of GluR2 mRNA is reduced specifically in CA1 afterneurons are irreversibly depolarized by prolonged low
ischemia. Photomicrographs of autoradiograms of GluR1, GluR2, a NR1frequency stimulation of the Schaffer collateral /
commisur-mRNA in situ hybridization in coronal sections of gerbil brain at the levelal input [149]. Moreover, post-ischemic neurons fail to
of the dorsal hippocampus from control animals (A, C, E) and fromexhibit long-term potentiation following tetanic stimulation
experimental animals 72 h after ischemia (B, D, F) are shown. GluR2[64]. Hippocampal neurons in culture subjected to
subleth-mRNA was dramatically reduced in the pyramidal cell layer of thevulnerable CA1 but not in the pyramdial cell layer of CA3 or in the
al oxygen–glucose deprivation exhibit increased
AMPA-21
granule cell layer of the dentate gyrus, areas that do not undergo
or kainate-induced Ca
accumulation, increased
sensitivi-neurodegeneration. GluR1 and NR1 mRNAs were somewhat reduced inty of AMPARs to Joro spider toxin, and increased
vul-CA1 72 h after ischemia. Reprinted from [44].nerability to AMPAR-mediated excitotoxity, suggesting
21
increased formation of GluR2-lacking, Ca
-permeable
AMPARs [159].
vulnerable neurons. Thus, the present study provides an
To determine whether downregulation of GluR2
expres-21
important link in the causal chain between global ischemia
sion translates into enhanced Ca
influx through
AM-and delayed death of CA1 pyramidal neurons.
PARs into CA1 neurons, we used a combination of
electrophysiological intracellular recording and optical
21
4.1.2. Ischemia-induced changes in glutamate receptor
imaging with the Ca
indicator dye fura-2 [44]. In
function
hippocampal slices from gerbils 48 h after ischemia,
21
Global ischemia induces a number of functional changes
AMPA elicits only a small rise in [Ca
] , which is not
i 21in the hippocampal CA1 indicative of increased expression
significantly different from Ca
rises in control neurons.
21
of Ca
-permeable AMPARs. AMPAR-mediated excitat-
At 72 h after the ischemic insult individual CA1 neurons
ory postsynaptic currents (EPSCs) at the CA1 / Schaffer
that retain the ability to fire action potentials exhibit a
21
collateral synapse are prolonged after ischemia and exhibit
greatly enhanced AMPA-elicited rise in [Ca
] . Basal
i 21pyramidal cells (Fig. 6). These findings indicate that
AMPAR antagonist, 6-cyano-7-nitro-quinoxiline-2,3-dione
21
AMPAR functional responses are altered following global
disodium, or of the Ca
permeable AMPAR channel
21
ischemia and provide direct evidence for Ca
-influx
blocker, 1-naphthyl acetyl spermine, afforded protection
directly through AMPARs in vulnerable CA1 neurons at
against antisense-induced cell death. This finding indicates
21
times after brief global ischemia and preceding obvious
that antisense-induced cell death is mediated by Ca
cell loss.
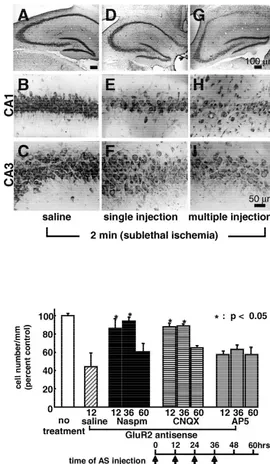
permeable AMPARs. GluR2 antisense and brief sublethal
To examine whether acute downregulation of the GluR2
global ischemia acted synergistically to cause degeneration
subunit, even in the absence of a neurological insult, can
of pyramidal neurons, consistent with a common
mecha-cause neuronal cell death, we performed GluR2 ‘knoc-
nism of cell death. These findings demonstrate that
dow-kdown’ experiments in rats and gerbils with antisense
nregulation of GluR2 is sufficient to induce delayed death
oligonucleotides targeted to GluR2 mRNA [100].
of specific neuronal populations.
21
Knockdown of receptor subunits has proven valuable in
Recent evidence implicates Zn
entry as a critical
investigation of receptor function in vivo and in vitro.
component of neuronal death after transient global
is-21
GluR2 antisense induced delayed death of pyramidal
chemia in rats [66]. After a brief ischemic insult, Zn
is
neurons in CA1 and CA3. Antisense-induced neurodegene-
translocated from presynaptic terminals and accumulates in
ration was preceded by a reduction in GluR2 mRNA, as
degenerating neurons in CA1 [66]. This accumulation
indicated by in situ hybridization analysis, and in GluR2
precedes the onset of neurodegeneration and is prevented
21
protein, as indicated by Western blot analysis. GluR2
by intraventricular administration of the Zn
chelator
antisense suppressed GluR2 mRNA in the dentate gyrus,
Ca-EDTA just prior to induction of ischemia. Ca-EDTA
but did not cause cell death (Fig. 7). Administration of the
administration 1 h after reperfusion is not neuroprotective,
21
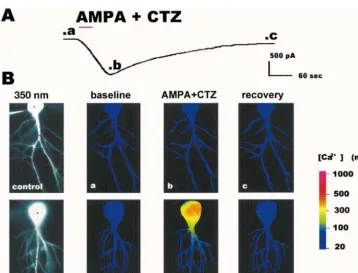
Fig. 6. AMPA-elicited inward current and rise in intracellular free Ca concentration in a CA1 pyramidal neuron after ischemia. (A) Inward current elicited by AMPA (30mM with 10mM cyclothiazide (CTZ)) in a CA1 pyramidal neuron in a hippocampal slice from an animal 72 h after ischemia.
21 1
AMPA and CTZ were bath applied for 30 s (red bar) in saline containing NMDA, Ca and Na channel blockers. Then the AMPA was washed out with
21 1
saline containing the NMDA, Ca and Na channel blockers. After|5 min, CNQX (20mM) was added to the other blockers to cause more rapid recovery. In the control neuron illustrated in B, the AMPA-elicited inward current in the presence of 30mM CTZ was of somewhat lower amplitude but similar in time course. (B) Optical imaging Optical imaging (excitation wavelength, 350 nm) of individual CA1 pyramidal neurons injected with fura-2 in hippocampal slices from a control animal (upper row) and an experimental animal 72 h after ischemia (lower row, same cell as in A). (a) Image taken before bath application of agonist (time indicated in current trace above); (b) image taken at peak inward current after application of AMPA (30mM with
21
10mM CTZ); (c) image taken after recovery to near baseline current. Color represents Ca concentration determined from the ratio of fluorescence
21
obtained at two excitation wavelengths (350 nm and 380 nm); calibration at right. AMPA elicited little change in intracellular free Ca in the control
21
neuron. In contrast, AMPA elicited a rise in Ca in the soma of the postischemic neuron and a smaller increase in its proximal dendrites. Red circles in the
21
21
indicating that Zn
entry during or immediately after
ischemia is toxic. Moreover, ischemia induces mRNA
expression of the transporter gene ZNT-1 in CA1 neurons
that are destined to die, presumably in response to the
21 21
Zn
accumulation, since Zn
induces expression of the
gene in cultured neurons [150]. Although one can interpret
the induction as a homeostatic response, it does not prevent
cell death. Since GluR2-lacking AMPARs are permeable to
21 21
Zn
[132], delayed entry of Zn
may contribute to
degeneration. If so, Ca-EDTA treatment delayed by several
days after the ischemic insult would also be
neuroprotec-tive.
21
Finally, evidence in support of a role for Ca
-perme-able AMPARs in ischemia-induced damage comes from
studies involving dissociated primary cultures of cortical
21
neurons [24]. This study reveals that activation of Ca
-permeable AMPARs, in addition to activation of NMDA
channels, leads to the generation of oxygen radicals, a
well-established cause of cell damage.
Together, these studies provide substantial evidence that
suppression of GluR2 expression in CA1 after transient
global ischemia leads to assembly and insertion of
GluR2-lacking AMPARs at CA1 synapses. The change in subunit
composition is consonant with observed changes in
AMPAR functional properties and increased influx of toxic
21 21
Ca
and Zn
[9,109].
4.2. Status epilepticus
Kainic acid-induced status epilepticus leads to delayed,
selective death of pyramidal neurons of the hippocampus.
Status epilepticus in adult rats triggers a pronounced
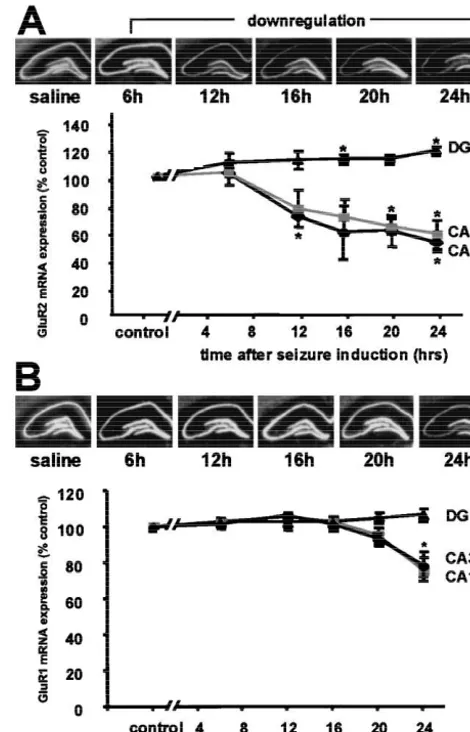
Fig. 8. Status epilepticus induces downregulation of GluR2, but not
suppression of GluR2 mRNA in vulnerable CA1 and CA3
GluR1 mRNA in hippocampal CA1 and CA3. (A) GluR2 mRNA
pyramidal neurons prior to the onset of neuronal death, as
expression in rat hippocampus after status epilepticus. (Upper) Filmassessed by in situ hybridization [38,113] (Fig. 8). To
autoradiograms of GluR2 mRNA expression in the hippocampus of status epilepticus and control rats. Downregulation of GluR2 mRNA expressionexamine whether the observed changes in GluR2 mRNA
is first detected at 12 h after KA injection within CA1 and CA3, but
are translated into changes in subunit expression, we
remains stable in DG. (Lower) Time-course of GluR2 mRNA expression
performed immunolabeling and Western blot analysis [45]
after onset of status epilepticus. Mean densities in hippocampal CA1 and
(Fig. 9). Double immunolabeling revealed individual CA1
CA3 decreased and increased in DG. (B) GluR1 mRNA expression in ratand CA3 pyramidal neurons expressing abundant GluR1
hippocampus after status epilepticus. (Upper) Film autoradiograms of GluR1 mRNA expression at 6, 12, 16, 20, 24 h after KA-induced statusand greatly reduced GluR2. GluR2 immunolabeling was
epilepticus and in saline controls. GluR1 mRNA expression remains
enhanced in granule cells of the dentate gyrus, a region
stable at all time points assayed, although there is a modest decrease in
resistant to seizure-induced damage. Quantitative Western
CA1 and CA3 at 24 h. (Lower) Mean densities of GluR1 mRNA remain
blot analysis revealed a reduction in GluR2 subunit
unchanged. Values are expressed as a percentage of saline injectedabundance by about 15% in CA1 and by about 30% in
controls. Error bars represent the standard error of the mean. *P,0.05,Student’s unpaired t test. Reprinted from [45].
CA3 at 20 h, with no change in dentate gyrus. Thus,
GluR2 subunit abundance was regulated in a cell-specific
manner. GluR1 subunit abundance was unchanged in all
subfields at all times examined. These findings indicate
induces status epilepticus, followed by delayed
neurode-that AMPAR subunit composition is remodeled in response
generation of pyramidal neurons in the hippocampal CA3.
21
to neuronal injury and support a role for Ca
-permeable
Status epilepticus triggered suppression of GluR2 mRNA,
21
AMPAR as critical mediators in the neuronal death associ-
consistent with expression of GluR2-lacking, Ca
-perme-ated with status epilepticus.
able AMPARs in neurons destined to die. In pup rats,
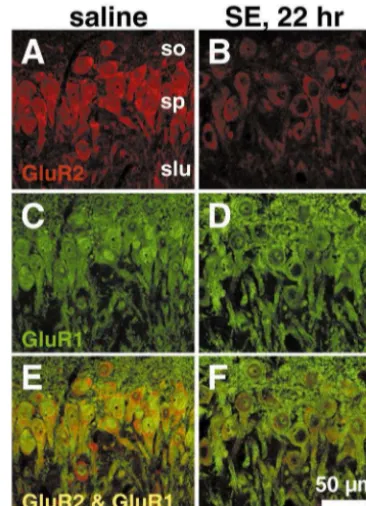
Fig. 9. Status epilepticus downregulates GluR2 subunit expression in CA3. (Upper) Co-localization of GluR1 and GluR2 immunoreactivity in CA3 pyramidal neurons. Saline injected control sections stained for GluR1 (A) and GluR2 (C). GluR2 immunoreactivity decreased in CA3 pyramidal cells 22h after kainate administration (B), although GluR1 immunoreactivity was unchanged (D). Co-localization of GluR1 and GluR2 occurred primarily in stratum pyramidale (E,F). (Lower) Status epilepticus decreases GluR2 but not GluR1 protein levels in CA1 and CA3. Quantitative Western blot analysis of GluR1 and GluR2 protein expression in microdissected hippocampal subfields (see Insert in A) after status epilepticus. (A) GluR2 protein levels decreased in CA3a–b at 16 h after KA injections, and further declined at 20 and 24 h. Similar but smaller changes were seen in CA1, but were not significant until 24 h. No changes in the level of GluR2 protein were observed in DG-CA3c. The plotted ratios are the means of the ratios for individual animals. Error bars represent SEMs. **P,0.001; *P,0.05; Student’s upaired t test. Reprinted from [45].
subunit expression vary with development: GluR2 dow-
in synaptic plasticity during brain development, and failure
21
nregulation occur only at those ages at which seizure-
to decrease Ca
-permeability of AMPARs at later stages
induced damage is observed. These findings provide
may lead to aberrant development.
21
evidence for Ca
-permeable AMPARs in the causal chain
Why do neurons which express little or no GluR2 (e.g.,
of events between severe limbic seizures and delayed
hippocampal interneurons) and hippocampal pyramidal
neuronal death.
neurons of the GluR2(
2
/
2
) knockout mouse [59] survive?
Viability of the GluR2 knock-out mice in contrast to that
of the GluR2 editing deficient mice could occur for any of
5. Genetic approaches to the role of GluR2 in
a number of reasons. These include (1) compensatory
21
neuronal cell death
increases in Ca
buffering and extrusion (as, for example,
21
enhanced expression of Ca
-binding proteins [68,117]),
Gene inactivation (knockout) and antisense (knoc-
(2) reduced AMPAR currents, due to slowed receptor
kdown) approaches have proven useful in determining the
assembly or reduced expression of GluR1 and GluR3,
function of a particular protein under physiological and
and / or (3) expression of receptors with altered properties,
pathological conditions. Altogether four animal models
such as number, localization and interaction of AMPARs
have been developed using genetic techniques: (1) the
with signaling and / or anchoring proteins and enhanced
Q / R editing deficient mouse (lacking intron 11 of the
desensitization [42]. The viability of these neurons suggest
GluR2 gene); (2) the GluR2 knock-out mouse; (3) the
that acute knockdown of GluR2 in neurons that normally
GluR2-flip over expressing mouse; and (4) the gerbil
express high levels of GluR2 (and relatively rapid changes
21
acutely treated with GluR2 antisense oligonucleotides
in AMPAR Ca
permeability) may be necessary to induce
(knockdown). This section briefly reviews the conse-
neuronal death. It should, however, be noted that the
quences of these genetic manipulations on brain develop-
GluR2 (
2
/
2
) knockout was made on a 129 / SvEMS
3
ment and susceptibility to excitotoxic cell death.
C57BL / 6 hybrid, a strain with high resistance to
gluta-Heterozygous transgenic mice engineered for a Q / R
mate-induced excitotoxicity [42,126]. Moreover, Jia et al.
editing deficient GluR2 allele express AMPARs with
(1996) [59] did not evaluate the susceptibility of the
21
increased Ca
-permeability, particularly in hippocampal
corresponding wild-type mice to excitotoxic cell death.
and neocortical principal neurons [16]. The primary conse-
Modifications other than downregulation of GluR2
quence is the onset of spontaneous and recurrent seizures
expression can increase excitotoxicity. The presence of the
[16]. The mice develop recurrent seizures and die within
GluR2-flip splice-variant subunit in heteromeric AMPAR
the first three weeks of life, with cell loss in the hippocam-
leads to a larger current flow through these channels
pus. In these animals, unedited GluR2 may contribute to
[42,89] Transgenic mice that over-express GluR2-flip
21
the formation of a greater number of Ca
-permeable
show enhanced susceptibility to excitotoxic
glutamate-AMPARs than in the GluR2 knockout mice (see below).
mediated damage after permanent middle cerebral artery
Transgenic mice with targeted disruption of the GluR2
occlusion, and glutamate excitotoxicity is increased
rela-gene (‘GluR2 knock-out mice’) differ considerably from
tive to that of wild-type in neurons cultured from the
GluR2Q / R editing deficient mice; the knock-out mice are
transgenic mice [72]. Excitotoxicity may be caused by
21 21
viable and fertile. The knock-out mice exhibit a nine-fold
increased Ca
influx through voltage-gated Ca
chan-21
increase in kainate-elicited Ca
-influx into individual
nels and through NMDAR channels [26,73,84] following
CA1 pyramidal neurons and increased inward rectification
increased depolarization mediated by AMPARs containing
of kainate-elicited responses and of the AMPAR com-
primarily the flip isoform of GluR2 [42,89].
ponent of EPSCs [59]. The passive membrane properties at
GluR2 knockdown as a technique offers the advantage
the resting potential are unchanged, except that input
of examining the effects of GluR2 suppression in an
resistance is increased, perhaps due to reduced cell size.
animal that has developed under normal conditions.
In-EPSCs are little changed in amplitude and the decay rate is
jection of antisense oligonucleotides directly into the brain
unchanged (unlike the EPSCs in post ischemic gerbil
of gerbils and rats has been used to demonstrate a probable
[146]. The AMPAR-mediated component of the EPSCs is
causal relationship between downregulation of GluR2
reduced relative to the NMDAR-mediated component,
expression and delayed neuronal cell death [100]. This
possibly due to reduction in AMPAR density as a result of
study demonstrates that knockdown of GluR2 by
intraven-inefficient receptor assembly. LTP in GluR2 knock-out
tricular injection of specific antisense oligonucleotides
animals is increased and has a substantial NMDAR-in-
leads to death of CA1 and CA3 neurons. Scrambled
dependent component. These data strongly suggest that
antisense administered under the same conditions is
with-21 21
LTP can be mediated by Ca
influx through Ca
-
out effect. Since the induced neurotoxicity is blocked by
permeable AMPARs (see also [46]). The GluR2 knockout
1-naphthyl acetyl spermine, the cause of death is likely to
21 21
neuro-nal damage) leads to greater cell death than is observed for
References
antisense administration to control animals.
[1] H. Abe, M. Watanabe, T. Yamakuni, R. Kuwano, Y. Takahashi, H.
Given the prominent role of GluR2 in normal
physi-Kondo, Localization of gene expression of calbindin in the brain of
ology, brain development, and excitotoxicity, an important
adult rats, Neurosci. Lett. 138 (1992) 211–215.
future direction would be to apply spatial and temporal
[2] S. Akbarian, M.A. Smith, E.G. Jones, Editing for an AMPA receptorrestrictions to the regulation of GluR2 expression as, for
subunit RNA in prefrontal cortex and striatum in Alzheimer’sexample, by conditional knockout and / or knockin ap-
disease, Huntington’s disease and schizophrenia, Brain Res. 699(1995) 297–304.
proaches. Such studies would be expected to aid in our
[3] E.M. Aronica, J.A. Gorter, S. Grooms, J.A. Kessler, M.V. Bennett,
understanding of the role of the GluR2 subunit during
R.S. Zukin, D.M. Rosenbaum, Aurintricarboxylic acid prevents
synaptogenesis and formation of neuronal circuitry and in
GLUR2 mRNA down-regulation and delayed neurodegeneration inthe neurodegeneration associated with stroke and epilepsy.
hippocampal CA1 neurons of gerbil after global ischemia, Proc.Natl. Acad. Sci. USA 95 (1998) 7115–7120.
[4] C.H. Bailey, P. Montarolo, M. Chen, E.R. Kandel, S. Schacher, Inhibitors of protein and RNA synthesis block structural changes that accompany long-term heterosynaptic plasticity in Aplysia,
6. Concluding remarks
Neuron 9 (1992) 749–758.
[5] O. Bar-Peled, R.J. O’Brien, J.H. Morrison, J.D. Rothstein, Cultured
This article reviewed the regulation of AMPAR expres-
motor neurons possess calcium-permeable AMPA / kainate receptors,sion at CA1 synapses, with particular emphasis on RNA
Neuroreport 10 (1999) 855–859.transcription, editing and splicing, translational regulation,
[6] B.L. Bass, RNA editing and hypermutation by adenosine deamina-tion, Trends. Biochem. Sci. 22 (7) (1997) 157–162, Publishedreceptor targeting and trafficking, cell surface expression
erratum appears in Trends. Biochem. Sci. 22 (7) (1997) 278.
and anchoring. In addition, this article reviewed evidence
[7] G.J. Bassell, Y. Oleynikov, R.H. Singer, The travels of mRNAs
that severe neurological insults such as global ischemia
through all cells large and small [see comments], FASEB J. 13and limbic seizures trigger a ‘molecular switch’ that shuts
(1999) 447–454.off GluR2 subunit expression in neurons destined to die. In
[8] R. Benne, RNA editing. The long and the short of it [news;21 comment], Nature 380 (1996) 391–392.
neurons that normally express Ca
-impermeable
chan-21 [9] M.V.L. Bennett, D.E. Pellegrini-Giampietro, J.A. Gorter, E. Aronica,
nels, Ca
entry via GluR2-lacking AMPARs could
con-J.A. Connor, R.S. Zukin, The GluR2 hypothesis: Ca(11
)-perme-tribute significantly to delayed cell death. In addition to
able AMPA receptors in delayed neurodegeneration, Cold Spring21
their role in neurological disorders, Ca
-permeable AM-
Harb. Symp. Quant. Biol. 61 (1996) 373–384.PARs are thought to serve a number of physiological
[10] F. Berton, C. Iborra, J.A. Boudier, M.J. Seagar, B. Marqueze, Developmental regulation of synaptotagmin I, II, III, and IV mRNAsfunctions, including strengthening of synaptic transmission
in the rat CNS, J. Neurosci. 17 (1997) 1206–1216.
in certain spinal neurons [46], induction of long-term
[11] M. Blaschke, B.U. Keller, R. Rivosecchi, M. Hollmann, S.
potentiation as observed in the GluR2 knockout mouse
Heinemann, A. Konnerth, A single amino acid determines the21 1
[59], and possibly activation of Ca
-dependent K
chan-
subunit-specific spider toxin block ofalpha-amino-3-hydroxy-5-nels as well as inactivation of NMDARs [22,60]. Increased
methylisoxazole-4-propionate / kainate receptor channels, Proc. Natl.21 Acad. Sci. USA 90 (1993) 6528–6532.
Ca
permeability through AMPARs may also be involved
[12] P. Bochet, E. Audinat, B. Lambolez, F. Crepel, J. Rossier, M. Iino,
in the development of kindled seizures [114]. The
underly-K. Tsuzuki, S. Ozawa, Subunit composition at the single-cell level
21
ing mechanism by which AMPAR Ca
permeability
explains functional properties of a glutamate-gated channel, Neuronincreases under physiological or pathological conditions
12 (1994) 383–388.[13] D. Bowie, M.L. Mayer, Inward rectification of both AMPA and
reported to date involves a suppression of GluR2 gene
kainate subtype glutamate receptors generated by
polyamine-me-expression, rather than a reduction in Q / R editing
diated ion channel block, Neuron 15 (1995) 453–462.
[2,21,62,106]. Reduction in GluR2 expression has also
[14] J. Brillman, Central nervous system complications in coronary artery
been observed in other vulnerable neuronal populations
bypass graft surgery, Neurol. Clin. 11 (1993) 475–495.21
including cerebellar Purkinje and granule cells in mutant
[15] J.R. Brorson, P.A. Manzolillo, R.J. Miller, Ca entry via AMPA / KA receptors and excitotoxicity in cultured cerebellar Purkinje cells,spastic rats [79], pyramidal cells of the parahippocampal
J. Neurosci. 14 (1994) 187–197.
gyrus in schizophrenia [33], and spinal motor neurons in
[16] R. Brusa, F. Zimmermann, D.S. Koh, D. Feldmeyer, P. Gass, P.H.
amyotrophic lateral sclerosis [153] (Table 1). These
exam-Seeburg, R. Sprengel, Early-onset epilepsy and postnatal lethality
ples indicate the more general applicability of the GluR2
associated with an editing-deficient GluR-B allele in mice, Sciencehypothesis to neuropsychiatric diseases and disorders
270 (1995) 1677–1680.[17] W. Brysch, O.D. Creutzfeldt, K. Luno, R. Schlingensiepen, K.H.
involving glutamate-induced cell death.
Schlingensiepen, Regional and temporal expression of sodium channel messenger RNAs in the rat brain during development, Exp. Brain Res. 86 (1991) 562–567.
[18] A.M. Buchan, H. Li, S. Cho, W.A. Pulsinelli, Blockade of the
Acknowledgements
AMPA receptor prevents CA1 hippocampal injury following severe but transient forebrain ischemia in adult rats, Neurosci. Lett. 132
This work was supported by NIH grants no. NS 20752
(1991) 255–258.and NS 31282 (to R.S.Z.). M.V.L.B. is the Sylvia and
[19] K.E. Burgin, M.N. Waxham, S. Rickling, S.A. Westgate, W.C.calmodulin-dependent protein kinase in developing rat brain, J. Bennett, S.L. Moshe, R.S. Zukin, Kainate-induced status epilepticus alters glutamate and GABAA receptor gene expression in adult rat Neurosci. 10 (1990) 1788–1798.
hippocampus: an in situ hybridization study, J. Neurosci. 14 (1994) [20] N. Burnashev, H. Monyer, P.H. Seeburg, B. Sakmann, Divalent ion
2697–2707. permeability of AMPA receptor channels is dominated by the edited
[39] L.K. Friedman, J. Veliskova, GluR2 hippocampal knockdown re-form of a single subunit, Neuron 8 (1992) 189–198.
veals developmental regulation of epileptogenicity and neurodegene-[21] N. Burnashev, R. Schoepfer, H. Monyer, J.P. Ruppersberg, W.
ration, Brain Res. Mol. Brain Res. 61 (1998) 224–231. Gunther, P.H. Seeburg, B. Sakmann, Control by asparagine residues
[40] C.C. Garner, R.P. Tucker, A. Matus, Selective localization of of calcium permeability and magnesium blockade in the NMDA
messenger RNA for cytoskeletal protein MAP2 in dendrites, Nature receptor, Science 257 (1992) 1415–1419.
336 (1988) 674–677. [22] N. Burnashev, A. Villarroel, B. Sakmann, Dimensions and ion
[41] A.H. Gazzaley, D.L. Benson, G.W. Huntley, J.H. Morrison, Dif-selectivity of recombinant AMPA and kainate receptor channels and
ferential subcellular regulation of NMDAR1 protein and mRNA in their dependence on Q / R site residues, J. Physiol. (Lond.) 496
dendrites of dentate gyrus granule cells after perforant path transec-(1996) 165–173.
tion, J. Neurosci. 17 (1997) 2006–2017. [23] C.M. Burns, H. Chu, S.M. Rueter, L.K. Hutchinson, H. Canton, E.
[42] J.R. Geiger, T. Melcher, D.S. Koh, B. Sakmann, P.H. Seeburg, P. Sanders-Bush, R.B. Emeson, Regulation of serotonin-2C receptor
Jonas, H. Monyer, Relative abundance of subunit mRNAs de-G-protein coupling by RNA editing, Nature 387 (1997) 303–308,
termines gating and Ca21 permeability of AMPA receptors in See comments.
principal neurons and interneurons in rat CNS, Neuron 15 (1995)
21
[24] S.G. Carriedo, H.Z. Yin, S.L. Sensi, J.H. Weiss, Rapid Ca entry 193–204.
21
through Ca -permeable AMPA / Kainate channels triggers marked [43] A. Gerber, M.A. O’Connell, W. Keller, Two forms of human intracellular Ca21rises and consequent oxygen radical production, double-stranded RNA-specific editase 1 (hRED1) generated by the J. Neurosci. 18 (1998) 7727–7738. insertion of an Alu cassette, RNA 3 (1997) 453–463.
[25] J. Chen, R.L. Zhu, M. Nakayama, K. Kawaguchi, K. Jin, R.A. [44] J.A. Gorter, J.J. Petrozzino, E.M. Aronica, D.M. Rosenbaum, T. Stetler, R.P. Simon, S.H. Graham, Expression of the apoptosis- Opitz, M.V. Bennett, J.A. Connor, R.S. Zukin, Global ischemia effector gene, Bax, is up-regulated in vulnerable hippocampal CA1 induces downregulation of Glur2 mRNA and increases AMPA neurons following global ischemia, J. Neurochem. 67 (1996) 64–71. receptor-mediated Ca21 influx in hippocampal CA1 neurons of [26] D.W. Choi, Calcium-mediated neurotoxicity: relationship to specific gerbil, J. Neurosci. 17 (1997) 6179–6188.
channel types and role in ischemic damage, Trends Neurosci. 11 [45] S.Y. Grooms, T. Opitz, M.V. Bennett, R.S. Zukin, Status epilepticus
(1988) 465–469. decreases glutamate receptor 2 mRNA and protein expression in
[27] D.W. Choi, Cerebral hypoxia: some new approaches and unanswered hippocampal pyramidal cells before neuronal death, Proc. Natl. questions, J. Neurosci. 10 (1990) 2493–2501. Acad. Sci. USA 97 (2000) 3631–3636.
[28] D.W. Choi, Excitotoxic cell death, J. Neurobiol. 23 (1992) 1261– [46] J.G. Gu, C. Albuquerque, C.J. Lee, A.B. MacDermott, Synaptic
21
1276. strengthening through activation of Ca -permeable AMPA
recep-[29] D.W. Choi, Calcium: still center-stage in hypoxic-ischemic neuronal tors, Nature 381 (1996) 793–796.
death, Trends Neurosci. 18 (1995) 58–60. [47] Y. Hayashi, S.H. Shi, J.A. Esteban, A. Piccini, J.C. Poncer, R. [30] J.A. Chong, J. Tapia-Ramirez, S. Kim, J.J. Toledo-Aral, Y. Zheng, Malinow, Driving AMPA receptors into synapses by LTP and M.C. Boutros, Y.M. Altshuller, M.A. Frohman, S.D. Kraner, G. CaMKII: requirement for GluR1 and PDZ domain interaction, Mandel, REST: a mammalian silencer protein that restricts sodium Science 287 (2000) 2262–2267.
channel gene expression to neurons, Cell 80 (1995) 949–957. [48] S. Herlitze, M. Raditsch, J.P. Ruppersberg, W. Jahn, H. Monyer, R. [31] P. Crino, K. Khodakhah, K. Becker, S. Ginsberg, S. Hemby, J. Schoepfer, V. Witzemann, Argiotoxin detects molecular differences
Eberwine, Presence and phosphorylation of transcription factors in in AMPA receptor channels, Neuron 10 (1993) 1131–1140. developing dendrites, Proc. Natl. Acad. Sci. USA 95 (1998) 2313– [49] C. Heurteaux, I. Lauritzen, C. Widmann, M. Lazdunski, Essential
1
2318. role of adenosine, adenosine A1 receptors, and ATP-sensitive K
[32] S.D. Donevan, M.A. Rogawski, Intracellular polyamines mediate channels in cerebral ischemic preconditioning, Proc. Natl. Acad. Sci.
21
inward rectification of Ca -permeable alpha-amino-3-hydroxy-5- USA 92 (1995) 4666–4670.
methyl-4-isoxazolepropionic acid receptors, Proc. Natl. Acad. Sci. [50] M. Higuchi, F.N. Single, M. Kohler, B. Sommer, R. Sprengel, P.H.
USA 92 (1995) 9298–9302. Seeburg, RNA editing of AMPA receptor subunit GluR-B: a
base-[33] S.L. Eastwood, P.J. Harrison, Decreased synaptophysin in the medial paired intron-exon structure determines position and efficiency, Cell temporal lobe in schizophrenia demonstrated using immuno- 75 (1993) 1361–1370.
autoradiography, Neuroscience 69 (1995) 339–343. [51] T. Himi, T. Okazaki, H. Wang, T.H. McNeill, N. Mori, Differential [34] S.L. Eastwood, B. McDonald, P.W. Burnet, J.P. Beckwith, R.W. localization of SCG10 and p19 / stathmin messenger RNAs in adult Kerwin, P.J. Harrison, Decreased expression of mRNAs encoding rat brain indicates distinct roles for these growth-associated proteins, non-NMDA glutamate receptors GluR1 and GluR2 in medial Neuroscience 60 (1994) 907–926.
21
temporal lobe neurons in schizophrenia, Brain Res. Mol. Brain Res. [52] M. Hollmann, M. Hartley, S. Heinemann, Ca permeability of
29 (1995) 211–223. KA-AMPA — gated glutamate receptor channels depends on
[35] L.W. Fitzgerald, A.Y. Deutch, G. Gasic, S.F. Heinemann, E.J. subunit composition, Science 252 (1991) 851–853.
Nestler, Regulation of cortical and subcortical glutamate receptor [53] M. Hollmann, S. Heinemann, Cloned glutamate receptors, Annu. subunit expression by antipsychotic drugs, J. Neurosci. 15 (1995) Rev. Neurosci. 17 (1994) 31–108.
2453–2461. [54] R.M. Hong, H. Mori, T. Fukui, Y. Moriyama, M. Futai, A.
[36] L.W. Fitzgerald, J. Ortiz, A.G. Hamedani, E.J. Nestler, Drugs of Yamamoto, Y. Tashiro, M. Tagaya, Association of
N-abuse and stress increase the expression of GluR1 and NMDAR1 ethylmaleimide-sensitive factor with synaptic vesicles, FEBS Lett. glutamate receptor subunits in the rat ventral tegmental area: 350 (1994) 253–257.
common adaptations among cross-sensitizing agents, J. Neurosci. 16 [55] B.R. Hu, M. Park, M.E. Martone, W.H. Fischer, M.H. Ellisman, J.A.
(1996) 274–282. Zivin, Assembly of proteins to postsynaptic densities after transient
[37] L.K. Friedman, A.R. Koudinov, Unilateral GluR2(B) hippocampal cerebral ischemia, J. Neurosci. 18 (1998) 625–633.
[57] R.I. Hume, R. Dingledine, S.F. Heinemann, Identification of a site in Kuner, H. Monyer, M. Higuchi, A. Bach, P.H. Seeburg, Control of glutamate receptor subunits that controls calcium permeability, kinetic properties of AMPA receptor channels by nuclear RNA
Science 253 (1991) 1028–1031. editing, Science 266 (1994) 1709–1713.
[58] R.J. Jackson, M. Wickens, Translational controls impinging on the [76] H. Lomeli, J. Mosbacher, T. Melcher, T. Hoger, J.R. Geiger, T. 59-untranslated region and initiation factor proteins, Curr. Opin. Kuner, H. Monyer, M. Higuchi, A. Bach, P.H. Seeburg, Control of
Genet. Dev. 7 (1997) 233–241. kinetic properties of AMPA receptor channels by nuclear RNA
editing, Science 266 (1994) 1709–1713. [59] Z. Jia, N. Agopyan, P. Miu, Z. Xiong, J. Henderson, R. Gerlai, F.A.
Taverna, A. Velumian, J. MacDonald, P. Carlen, W. Abramow- [77] Y.M. Lu, H.Z. Yin, J. Chiang, J.H. Weiss, Ca(21)-permeable
21 Newerly, J. Roder, Enhanced LTP in mice deficient in the AMPA AMPA / kainate and NMDA channels: high rate of Ca influx receptor GluR2, Neuron 17 (1996) 945–956. underlies potent induction of injury, J. Neurosci. 16 (1996) 5457–
5465. [60] P. Jonas, C. Racca, B. Sakmann, P.H. Seeburg, H. Monyer,
21
Differences in Ca permeability of AMPA-type glutamate receptor [78] S. Maas, T. Melcher, P.H. Seeburg, Mammalian RNA-dependent channels in neocortical neurons caused by differential GluR-B deaminases and edited mRNAs, Curr. Opin. Cell Biol. 9 (1997) subunit expression, Neuron 12 (1994) 1281–1289. 343–349.
[61] F.S. Jones, R. Meech, Knockout of REST / NRSF shows that the [79] J.E. Margulies, R.W. Cohen, M.S. Levine, J.B. Watson, Decreased protein is a potent repressor of neuronally expressed genes in GluR2(B) receptor subunit mRNA expression in cerebellar neurons non-neural tissues, Bioessays 21 (1999) 372–376. at risk for degeneration, Dev. Neurosci. 15 (1993) 110–120. [62] W. Kamphuis, F.E. de Leeuw, D.S.F. Lopes, Ischaemia does not [80] K.C. Martin, A. Casadio, H. Zhu, J.C. Rose, M. Chen, C.H. Bailey,
alter the editing status at the Q / R site of glutamate receptor-A, -B, E.R. Kandel, Synapse-specific, long-term facilitation of aplysia -5 and -6 subunit mRNA, Neuroreport 6 (1995) 1133–1136. sensory to motor synapses: a function for local protein synthesis in
memory storage, Cell 91 (1997) 927–938. [63] U. Kim, Y. Wang, T. Sanford, Y. Zeng, K. Nishikura, Molecular
cloning of cDNA for double-stranded RNA adenosine deaminase, a [81] J.W. McDonald, M.V. Johnston, Physiological and pathophysiologi-candidate enzyme for nuclear RNA editing, Proc. Natl. Acad. Sci. cal roles of excitatory amino acids during central nervous system
USA 91 (1994) 11457–11461. development, Brain Res. Brain Res. Rev. 15 (1990) 41–70.
[64] T. Kirino, Delayed neuronal death in the gerbil hippocampus [82] T. Melcher, S. Maas, A. Herb, R. Sprengel, M. Higuchi, P.H. following ischemia, Brain Res. 239 (1982) 57–69. Seeburg, RED2, a brain-specific member of the RNA-specific adenosine deaminase family, J. Biol. Chem. 271 (1996) 31795– [65] R. Kleiman, G. Banker, O. Steward, Differential subcellular
locali-31798. zation of particular mRNAs in hippocampal neurons in culture,
Neuron 5 (1990) 821–830. [83] T. Melcher, S. Maas, M. Higuchi, W. Keller, P.H. Seeburg, Editing [66] J.Y. Koh, S.W. Suh, B.J. Gwag, Y.Y. He, C.Y. Hsu, D.W. Choi, The of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid re-role of zinc in selective neuronal death after transient global cerebral ceptor GluR-B pre-mRNA in vitro reveals site-selective adenosine ischemia, Science 272 (1996) 1013–1016. to inosine conversion, J. Biol. Chem. 270 (1995) 8566–8570. [67] M. Kohler, H.C. Kornau, P.H. Seeburg, The organization of the gene [84] B. Meldrum, J. Garthwaite, Excitatory amino acid neurotoxicity and
for the functionally dominant alpha-amino-3-hydroxy-5-methylisox- neurodegenerative disease, Trends Pharmacol. Sci. 11 (1990) 379– azole-4-propionic acid receptor subunit GluR-B, J. Biol. Chem. 269 387.
(1994) 17367–17370. [85] L. Mittaz, H.S. Scott, C. Rossier, P.H. Seeburg, M. Higuchi, S.E. [68] M. Kondo, R. Sumino, H. Okado, Combinations of AMPA receptor Antonarakis, Cloning of a human RNA editing deaminase subunit expression in individual cortical neurons correlate with (ADARB1) of glutamate receptors that maps to chromosome expression of specific calcium-binding proteins, J. Neurosci. 17 21q22.3, Genomics 41 (1997) 210–217.
(1997) 1570–1581. [86] K. Miyashiro, M. Dichter, J. Eberwine, On the nature and
differen-[69] F. Lai, C.X. Chen, K.C. Carter, K. Nishikura, Editing of glutamate tial distribution of mRNAs in hippocampal neurites: implications for receptor B subunit ion channel RNAs by four alternatively spliced neuronal functioning, Proc. Natl. Acad. Sci. USA 91 (1994) 10800– DRADA2 double-stranded RNA adenosine deaminases, Mol. Cell 10804, See comments.
Biol. 17 (1997) 2413–2424. [87] H. Monyer, N. Burnashev, D.J. Laurie, B. Sakmann, P.H. Seeburg, [70] A.S. Landsend, M. Amiry-Moghaddam, A. Matsubara, L. Developmental and regional expression in the rat brain and func-Bergersen, S. Usami, R.J. Wenthold, O.P. Ottersen, Differential tional properties of four NMDA receptors, Neuron 12 (1994) 529– localization of delta glutamate receptors in the rat cerebellum: 540.
coexpression with AMPA receptors in parallel fiber-spine synapses [88] H. Monyer, P.H. Seeburg, W. Wisden, Glutamate-operated channels: and absence from climbing fiber-spine synapses, J. Neurosci. 17 developmentally early and mature forms arise by alternative
splic-(1997) 834–842. ing, Neuron 6 (1991) 799–810.
[71] W. Lason, J. Turchan, B. Przewlocka, D. Labuz, J. Mika, R. [89] J. Mosbacher, R. Schoepfer, H. Monyer, N. Burnashev, P.H. Przewlocki, Seizure-related changes in the glutamate R2 and R5 Seeburg, J.P. Ruppersberg, A molecular determinant for submil-receptor genes expression in the rat hippocampal formation, J. lisecond desensitization in glutamate receptors, Science 266 (1994)
Neural Transm. 104 (1997) 125–133. 1059–1062.
[72] D. Le, S. Das, Y.F. Wang, T. Yoshizawa, Y.F. Sasaki, M. Takasu, A. [90] I.A. Muslimov, E. Santi, P. Homel, S. Perini, D. Higgins, H. Tiedge, Nemes, M. Mendelsohn, P. Dikkes, S.A. Lipton, N. Nakanishi, RNA transport in dendrites: a cis-acting targeting element is Enhanced neuronal death from focal ischemia in AMPA-receptor contained within neuronal BC1 RNA, J. Neurosci. 17 (1997) 4722– transgenic mice, Brain Res. Mol. Brain Res. 52 (1997) 235–241. 4733.
[73] S.A. Lipton, P.A. Rosenberg, Excitatory amino acids as a final [91] S.J. Myers, R. Dingledine, K. Borges, Genetic regulation of common pathway for neurologic disorders [see comments], N. Engl. glutamate receptor ion channels, Annu. Rev. Pharmacol. Toxicol. 39
J. Med. 330 (1994) 613–622. (1999) 221–241.
[74] Y. Liu, C.X. George, J.B. Patterson, C.E. Samuel, Functionally [92] S.J. Myers, J. Peters, Y. Huang, M.B. Comer, F. Barthel, R. distinct double-stranded RNA-binding domains associated with Dingledine, Transcriptional regulation of the GluR2 gene: neural-alternative splice site variants of the interferon-inducible double- specific expression, multiple promoters, and regulatory elements, J. stranded RNA-specific adenosine deaminase, J. Biol. Chem. 272 Neurosci. 18 (1998) 6723–6739.
(1997) 4419–4428. [93] S.M. Nair, T.R. Werkman, J. Craig, R. Finnell, M. Joels, J.H.