125
Motor Neuron Disease
Lisa S. Krivickas, MD
DEFINITIONThe term motor neuron disease refers to a progressive neuromuscular disorder in which upper or lower motor neurons degenerate. The most common form of motor neuron disease is amyotrophic lateral sclerosis (ALS), which is the primary focus of this chapter. Management principles for other forms of motor neuron disease are similar. To meet diagnostic criteria for ALS, an individual must have both upper and lower motor neuron dysfunction. If only lower motor neuron dysfunction is present, the disease is called progressive muscular atrophy; if only upper motor neuron dysfunction is present, it is called primary lateral sclerosis. If only bulbar dysfunction is present, the disease is called progressive bulbar palsy. Most patients initially diagnosed as having progressive muscular atrophy, primary lateral sclerosis, or progressive bulbar palsy eventually have full-blown ALS. Those who do not convert to ALS have a slower rate of disease progression.
Most cases of ALS are idiopathic. However, 5% to 10% of patients have a familial form, usually transmitted in autosomal dominant fashion. In approximately 20% of these familial cases, mutations in SOD1 (superoxide dismutase) can be identified. Other rare forms of inherited adult motor neuron disease are Kennedy disease (X-linked recessive) and adult spinal muscular atrophy (autosomal recessive), which both have only lower motor neuron dysfunction.
ALS rapidly produces skeletal muscle weakness, eventually leading to the requirement for ventilator support or death from respiratory failure. The onset of weakness may be in any limb, the bulbar muscles, or in the respiratory muscles. The extraocular muscles and bowel and bladder function are generally spared. Mean survival, without tracheostomy, is 3 years from symptom onset, but the range may be less than 1 year to more than 20 years. One explanation for the extreme variability in rate of disease progression is that ALS is probably a heterogenous group of diseases rather than a single disease.1 The mean age at onset is in the
mid-50s, but ALS may develop in adults of any age. The cause of the disease is unknown, but leading theories concerning pathogenesis implicate glutamate excitotoxicity, oxidative stress, neuroinflammation, microglial cell activation, apoptosis, and mitochondrial dysfunction.
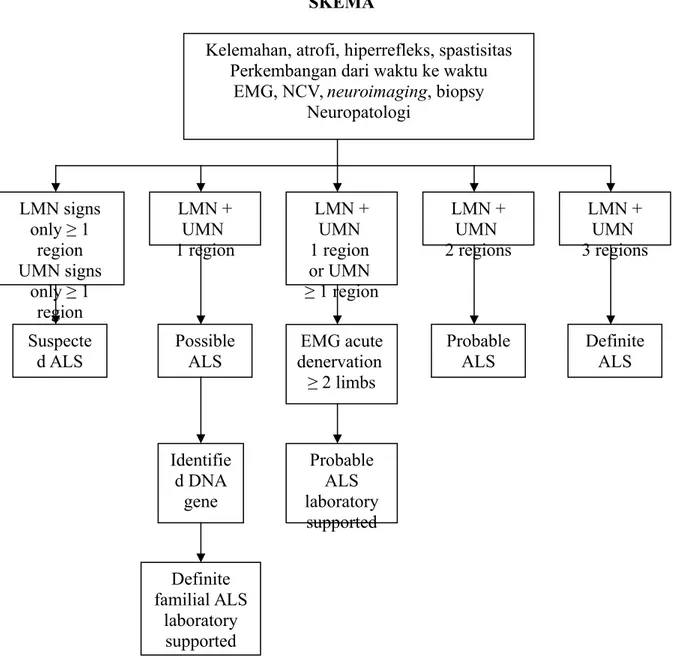
Figure 125-1. El Escorial criteria for the diagnosis of ALS. EMG, electromyography; NCV, nerve conduction velocity; LMN, lower motor neuron; UMN, upper motor neuron.
SYMPTOMS
Patients with upper motor neuron syndrome may present with spasticity, loss of dexterity, stiffness, weakness due to spasticity, and loss of voluntary motor control.
Patients with lower motor neuron syndrome may present with weakness, fasciculations, muscle atrophy, and muscle cramps.
Bulbar symptoms include dysarthria, dysphagia, sialorrhea (drooling), aspiration, and pseudobulbar affect (laughter or crying discordant with mood).
LMN signs only ≥ 1 region UMN signs only ≥ 1 region LMN + UMN 1 region Suspecte d ALS Possible ALS Identifie d DNA gene Definite familial ALS laboratory supported LMN + UMN 1 region or UMN ≥ 1 region Probable ALS laboratory supported EMG acute denervation ≥ 2 limbs LMN + UMN 2 regions Probable ALS Definite ALS LMN + UMN 3 regions SKEMA
Kelemahan, atrofi, hiperrefleks, spastisitas Perkembangan dari waktu ke waktu
EMG, NCV, neuroimaging, biopsy
Respiratory failure and constitutional symptoms of weight loss and generalized fatigue may be present. Cognitive symptoms include behavioral or executive dysfunction and frontotemporal dementia in a minority of patients.
PHYSICAL EXAMINATION
The emphasis of the physical examination of a patient with suspected or diagnosed motor neuron disease is on the neurologic, musculoskeletal, and cardiorespiratory systems. On neurologic examination, one is looking for evidence of upper and lower motor neuron dysfunction. The mental status, non-motor cranial nerve function, sensory examination, and cerebellar examination findings are usually normal. The “gold standard” for the diagnosis of upper motor neuron disease is the presence of pathologic reflexes-the Babinski sign, Hoffmann sign, and brisk jaw jerk. Patients with ALS may be hyperreflexic or hyporeflexic, depending on the stage at which they are in the disease process and whether they have a predominance of upper or lower motor neuron disease. Evidence of lower motor neuron disease includes muscle weakness, atrophy, hypotonia, hyporeflexia, and fasciculations. Atrophy often appears first in hand intrinsic muscle. Although fasciculations are not a necessary criterion for the diagnosis of ALS, one should question the diagnosis when none are observed. The tongue is examined for fasciculations and atrophy, and tongue strength and range of motion are assessed. The musculoskeletal examination focuses on assessment of range of motion and evaluation of painful joints or soft tissue structures. Because progressive respiratory failure develops, the cardiorespiratory system should be assessed at each visit. Forced vital capacity (FVC) can be measured with a hand-held spirometer in the office setting.
FUNCTIONAL LIMITATIONS
The majority of the functional limitations that develop in patients with ALS are the direct or indirect of muscle weakness. As the disease progresses, patients have impaired mobility and difficulties with performance of even the most basic activities of daily living, such as feeding themselves. Bulbar muscle weakness produces dysarthria (difficulty speaking) and dysphagia (difficulty swallowing); eventually, some patients become anarthric and unable to swallow even their own saliva. Reactive depression, generalized fatigue, and musculoskeletal pain may further limit function.
DIAGNOSTIC STUDIES
The diagnosis of ALS is based on appropriate physical examination and electrodiagnostic findings and the use of neuroimaging and clinical laboratory studies to exclude other conditions that may mimic ALS. All patients should undergo electrodiagnostic testing. The revised El Escorial criteria are currently used to diagnose ALS.2 They classify the certainty
level of the diagnosis into one of five categories: definite, probable, probable with laboratory support, possible, and suspected (Fig. 125-1).
The motor system is divided into four regions: bulbar, cervical, thoracic, and lumbosacral. Clinical evidence of upper and lower motor neuron disease is sought in each region. The certainty level of diagnosis depends on how many regions reveal upper motor neuron or lower motor neuron disease. Electrophysiologic findings can be used both to confirm lower motor neuron dysfunction in clinically affected regions and to detect lower motor neuron dysfunction in clinically uninvolved regions.
Imaging studies are used to exclude possibilities other than motor neuron disease from the differential diagnosis. Magnetic resonance imaging is the primary imaging modality in the evaluation of patients with suspected ALS. Almost all patients should have magnetic resonance imaging of the cervical spine to exclude cord compression, syrinx, or other spinal cord disease. The location of symptoms will dictate whether other regions of the spinal cord should be imaged. In those presenting with bulbar symptoms, brain magnetic resonance imaging should be preformed to exclude stroke, tumor, syringobulbia, and other pathologic processes.
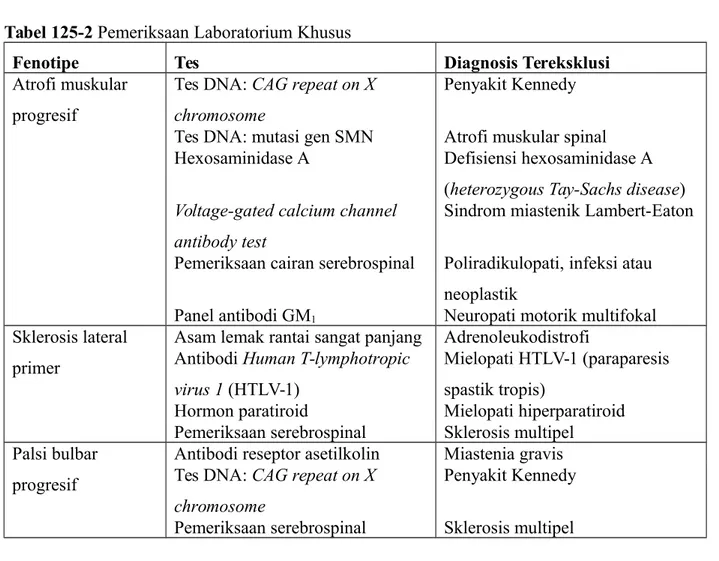
In most neuromuscular clinics, a routine panel of laboratory tests is performed for all patients thought to have ALS. A suggested set of such tests is provided in Table 125-1. The rationale behind the performance of this extensive battery of tests is to assess the general health of the patient and to exclude treatable conditions. The differential diagnosis, developed after the history and physical examination, may suggest that more specialized testing be performed. Table 125-2 suggests additional tests that may warranted when the presentation is with the progressive muscular atrophy, primary lateral sclerosis, or progressive bulbar palsy
phenotype. When there is a family history of motor neuron disease, SOD1 testing may be
considered.
Hematology
Complete blood countngkap Sedimentation rate
Chemistry
Electrolyte, blood urea nitrogen, creatinine Glucose Hemoglobin A Calcium Phosphorus Magnesium Creatine kinase Liver function tests Serum lead levelum Urine heavy metal screen Vitamin B12 Folate Endocrine Thyroxine Thyroid-stimulating hormone Immunology Serum immunoelectrophoresis Urine assay for Bence Jones proteins Antinuclear antibody Rheumatoid factor Microbiology Lyme titer VDRL test Optional
Human immunodeficiency virus test (if risk factors are present) Anti-Hu antibody (if malignant disease is suspected)
DNA test for SOD1 mutation (with family history)
Table 125-2 Specialized Laboratory Testing
Phenotype Test Diagnosis Excluded
Progressive muscular atrophy
DNA test: CAG repeat on X chromosome
Kennedy disease
DNA test: SMN gene mutation Spinal muscular atrophy
Hexosaminidase A Hexosaminidase A deficiency
(heterozygous Tay-Sachs disease)
Voltage-gated calcium channel antibody test
Lambert-Eaton myasthenic syndrome
Cerebrospinal fluid examination Polyradiculopathy, infection or
neoplastic
GM1 antibody panel Multifocal motor neuropathy
Primary lateral sclerosis
Very long chain fatty acids Adrenoleukodystrophy
Human T-lymphotropic virus 1 (HTLV-1) antibody
HTLV-1 myelopathy (tropical spastic paraparesis)
Parathyroid hormone Hyperparathyroid myelopathy
Cerebrospinal fluid examination Multiple sclerosis
Progressive bulbar palsy
Acetylcholine receptor antibodies Myasthenia gravis
DNA test: CAG repeat on X chromosome
Kennedy disease
Cerebrospinal fluid examination Multiple sclerosis
Differential Diagnosis
Differential diagnosis depends on whether the presentation is primarily lower motor neuron, upper motor neuron, bulbar, or mixed lower and upper motor neuron.
Subacute combined systems degeneration Hereditary spastic paraparesis
Myelopathy Syringomyelia
Lower Motor Neuron Only Bulbar
Progressive muscular atrophy Progressive bulbar palsy
Spinal muscular atrophy Myasthenia gravis
Kennedy disease Multiple sclerosis
Polymyelitis, post-poliomyelitis syndrome Foramen magnum tumor
Benign monomelic amyotrophy Brainstem glioma
Hxosaminidase A deficiency Stroke
Polyradiculopathy Syringobulbia
Multifocal motor neuropathy with conduction block
Head and neck cancer Polymyositis
Chronic inflammatory demyelinating polyneuropathy
Oculopharyngeal muscular dystrophy Kennedy disease
Motor neuropathy or neuronopathy
Lambert-Eaton syndrome Motor Neuron Atas dan Bawah
Plexopathy ALS
Benign fasciculations Cervical myelopathy with radiculopathy
Upper Motor Neuron Only
Primary lateral sclerosis
Spinal cord tumor or arteriovenous malformation Multiple sclerosis Adrenoleukodystrophy Lyme disease TREATMENT INITIAL PHARMACOLOGIC
Although there is not yet a cure for ALS, significant research advances are being made in an attempt to identify drugs that will slow disease progression. Offering patients pharmacologic treatment of their disease has psychological benefits that may outweigh the actual slowing of disease progression that currently can be achieved. Riluzole (Rilutek) is the only drug approved by the Food and Drug Administration (FDA) specifically for treatment of ALS. However, many neuromuscular experts recommend that their patents also take a combination of antioxidant vitamins and supplements.
Riluzole, an antiglutamate agent, was approved by the FDA for treatment of ALS in 1995 after two clinical trials showed that the drug slowed disease progression.3,4 A Cochrane
review reporting a meta-analysis of riluzole clinical trials showed a survival benefit of approximately 2 months.5 Unfortunately, no functional benefit was derived as strength
declined at a rate similar to in those taking riluzole and placebo. The recommended dose of riluzole is 50 mg twice daily. The most common adverse side effects are fatigue, nausea, and elevation of hepatic enzymes. Riluzole is contraindicated in those with hepatic enzyme activity of more than five times the upper limit of normal.
Because oxidative stress due to excessive free radical production in motor neurons is one of the proposed pathogenic factors in ALS, many physicians recommend a variety of antioxidants; vitamin E, vitamin C, and coenzyme Q10 are the most frequently used. No double-blind, placebo-controlled trials have proved the efficacy of these treatments. Safe recommended daily doses are 1000 to 3000 mg of vitamin C, 400 IU of vitamin E, and up to 1200 mg of coenzyme Q10.
Recent negative ALS clinical trials have tested a number of growth factors, creatine, glutamate antagonists, anti-inflammatory agents (celecoxib), calcium channel blockers, and amino acids.6 At present, trials of neurotrophic factors, antioxidants, glutamate antagonists,
coenzyme Q10, and tamoxifen are ongoing. In the future, a cocktail approach to slowing of disease progression may be the ideal treatment strategy.7
A number of drugs are useful for treatment of spasticity, sialorrhea, pseudobulbar affect, depression, and anxiety.
Spasticity requires treatment only if it interferes with function. Nonpharmacologic management involves teaching patients stretching exercises and positioning techniques that decrease muscle tone. Baclofen (Lioresal) is the most effective pharmacologic agent, followed by tizanidine (Zanaflex). Diazepam (Valium) should be avoided because it may suppress respiration, and dantrolene (Dantrium) is not recommended because it causes excessive muscle weakness. In general, pharmacologic management of spasticity is less successful in ALS than in multiple sclerosis or spinal cord injury because the lower motor neuron component of ALS makes patients extremely susceptible to the development of excessive weakness.
Patients with bulbar dysfunction experience sialorrhea because they have difficulty swallowing and managing the oral secretions they normally produce. A variety of anticholinergic drugs may be used to dry the mouth. Tricyclic antidepressants are often tried first but may not be tolerated because of adverse side effects (dry mouth, somnolence, urinary retention). One benefit of the tricylcic antidepressants is that they may treat other ALS-related symptoms, such as pseudobulbar affect (see next paragraph for definition), insomnia, and pain. Tricyclic antidepressants are contraindicated in patients with cardiac arrhythmia or conduction disorder. When tricyclic antidepressants are not tolerated, a scopolamine patch (Transderm Scop) or glycopyrrolate (Robinul) may be helpful. If these treatments are inadequate, patients may benefit from injection of botulinum toxin into the salivary glands.8
Pseudobulbar affect, also sometimes called emotional incontinence, refers to the patient’s inability to accurately portray emotions being experienced. Patients laugh or cry when they are experiencing sadness or happiness, respectively. They also may have an exaggerated response to situationally appropriate feelings. Amitriptyline (Elavil), a combination of dextromethorphan (Robitussin, Drixoral) and quinidine, or fluvoxamine (Luvox) may help blunt the intensity of these inappropriate or exaggerated reactions. A combination preparation of dextromethorphan and quinidine was found to be effective in a large randomized clinical trial,9 but this combination has not yet been approved by the FDA.
Reactive depression and anxiety are both normal responses to a diagnosis of ALS.10 Patients
and their families may benefit from individual counseling and participation in ALS support groups. Anxiety may be treated with benzodiazepines as long as the patient does not have significant reduction of vital capacity. Undetected hypoventilation may produce or contribute to preexisting feelings of anxiety. Depression should be treated pharmacologically because not treating it may have a significant negative impact on the quality of life remaining.11,12
Selective serotonin reuptake inhibitors (SSRIs) are good first choices because of their minimal side effects. However, the tricyclic antidepressants may be preferred if they are also needed to treat other symptoms, such as sialorrhea or pseudobulbar affect.
REHABILITATION
Skeletal muscle weakness is the primary impairment in ALS and causes the majority of the clinical problems. In the early stages of ALS, patients often inquire about the role of exercise in preventing or forestalling the development of weakness. Later in the disease, rehabilitation strategies must be used to maintain function and to compensate for muscle weakness.
Three forms of exercise training are relevant to patients with ALS: flexibility, strengthening, and aerobic exercise. Flexibility training helps prevent the development of painful contractures, nonpharmacologically decreases spasticity, and aborts painful muscle spasms. Traditionally, physicians have been reluctant to recommend strengthening exercises because they fear that overuse weakness will accelerate disability. This philosophy promotes the development of disuse weakness and muscle deconditioning, which may compound the weakness produced by ALS itself. The literature supporting the development of overwork weakness in neuromuscular patients is anecdotal, and overuse weakness has not been demonstrated in any controlled prospective studies. A single study of strength training has been published in ALS. It demonstrated a slowing of decline in physical function and no adverse effects.13 Studies of patients with more slowly progressive neuromuscular diseases
and post-poliomyelitis syndrome suggest that muscles only mildly affected by the disease process can be strengthened by a moderate resistance strengthening program.14-16
High-resistance eccentric exercise should be avoided because it may damage muscle.
I recommend that interested ALS patients begin or continue with a strengthening program to maximize the strength of unaffected or mildly affected muscles in an attempt to delay the time at which function becomes impaired. Weight training should be performed with a weight that can be lifted 20 times, and the individual should be instructed to perform submaximal sets of 10 to 15 repetitions; this ensures that the training is performed at a low to moderate
level of resistance. If an exercise regimen consistently produces muscle soreness or fatigue lasting longer than a half an hour after exercise, it is too strenuous.
Aerobic exercise helps maintain cardiorespiratory fitness. A study of moderate aerobic activity in patients with ALS demonstrated a short-term positive effect on disability, patients who exercised remained in a milder disease state longer.17 In addition, studies in a transgenic
mouse model of ALS have shown that aerobic exercise prolongs survival.18-20 Given the lack
of any apparent contraindication, aerobic exercise training is recommended for patients with ALS as long as it can be performed safely without a risk of falling or injury. In addition to the physical benefist, strengthening and aerobic exercise may have a beneficial effect on mood, psychological well-being, bone health, appetite, and sleep.
As ALS progresses, the rehabilitation focus shifts from exercise to maintenance of independent mobility and function for as long as possible. Interventions include assistive devices such as canes, walkers, braces, hand splints, wheelchairs, and scooters; home equipment such as dressing aids, adapted utensils, grab bars, raised toilet seats, shower benches, and lifts; home modifications (ramps, wide doorways); and automobile adaptations, such as hand controls, environmental control units, and voice-activated software. These rehabilitation interventions are best provided by a multidisciplinary team that includes a physiatrists, physical therapists, occupational therapists, and orthotists.
Patients frequently develop musculoskeletal pain syndromes, such as adhesive capsulitis (frozen shoulder), low back pain and neck pain due to muscle weakness and inability to change positions. Measures to prevent adhesive capsulitis include range of motion exercises and support of the arm as much as possible rather than allowing it to dangle at the side. Low back pain can be triggered by an uncomfortable seating system. Preventive measures include a lumbar support for the wheelchair, a good cushion on a solid seat, encouragement of frequent weight shifts, and a reclining back or tilt-in-space wheelchair. Neck pain associated with head drop is one of the most difficult musculoskeletal pain issues to remedy. A variety of cervical collars may be tried. A head support on the wheelchair or a reclining lounge chair may be more comfortable than a collar. Nonpharmacologic measures such as massage and transcutaneous electrical nerve stimulation can also be used for pain control. Acetaminophen, nonsteroidal anti-inflammatory drugs, Lidoderm patches, and, if necessary, opioids should be used to alleviate musculoskeletal pain. Trancutaneous electrical nerve stimulation can also be used. The major concerns with opiate use are respiratory depression and constipation. The respiratory depression may be acceptable in the late stages of the disease, in fact, morphine is good way to relieve air hunger in the terminal stage.
Adequate swallowing function is necessary to maintain the nutritional status of the patient with ALS (unless a feeding tube is in place). If nutritional status is not properly maintained, patients tend to use muscle as fuel and thus lose muscle mass and strength earlier than they would otherwise.21 Swallowing dysfunction may also precipitate aspiration pneumonia or
respiratory failure. Early signs and symptoms of dysphagia are drooling, a wet voice, coughing during or after drinking thin liquids, nasal regurgitations, and requiring an excessive amount of time to complete meals. Patients should be referred to a speech pathologist when the first signs of dysphagia develop. Those with mild swallowing difficulties can be taught compensatory techniques to reduce the risk of aspiration and
choking.22 Recommendations may be given concerning modification of food consistencies.
The development of aspiration pneumona, loss of more than 10% of body weight, and requiring an excessive amount of time to eat such that quality of life is impaired are indications for feeding tube placement.
Early or mild dysarthria may be managed by having a speech therapist teach patients adaptive strategies, such as overarticulation and slowing of the speaking rate. In patients with hypernasal speech caused by palatal weakness and primarily lower motor neuron dysfunction, a palatal lift or augmentation prosthesis may improve speech clarity.23 As dysarthria worsens,
patients require alternative forms of communication. Writing is a good alternative while hand function is intact. For those unable to write, Low-technology interventions include letter and word boards for written communication in tlose with good hand function. Higher technology solutions are augmentative communication devices that may have a voice synthesizer. As long as one muscle somewhere in the body can be voluntarily activated (including the extraocular muscles), the patient should be able to operate a communication device. High-technology solutions to communication problems are not suitable for all patients. Systems must be flexible so that the method of access can be modified as weakness progresses. Respiratory failure is the primary cause of death in ALS. In the absence of underlying intrinsic pulmonary disease, the respiratory failure in ALS is purely mechanical. Because of muscle weakness, the lungs do not inflate fully on inspiration. Most patients with ALS remain asymptomatic until the FVC is less than 50% of predicted. Pulmonary function tests, particularly the FVC, should be monitored every few months, depending on rate of disease progression. The earliest symptoms of respiratory failure are caused by nocturnal hypoventilation and include poor sleep with frequent awakening, nightmares, early morning headaches, and excessive daytime fatigue and sleepiness. Another early sign of respiratory muscle weakness is a weak cough and difficulty in clearing secretions.
The management of respiratory failure in ALS involves prevention of infection when possible and provision of mechanical ventilatory assistance. All patients with ALS should receive a pneumococcal vaccination and a yearly influenza vaccination. If the expiratory muscles are too weak to generate an adequate cough, patients can be helped by either manually assisted coughing or the Cough Assist (J.H. Emerson Co., Cambridge, Mass).24,25 Providing patients
wit supplemental oxygen can relieve symptoms of air hunger and dyspnea but also may suppress respiratory drive, exacerbate alveolar hypoventilation, and ultimately lead to carbon dioxide retention and respiratory arrest.26 Supplemental oxygen is recommended only for
patients with concomitant pulmonary disease or as a comfort measure for those who decline assisted ventilation. Discussion concerning the possibility of respiratory failure should be initiated soon after the diagnosis of ALS so that patients and their families can learn about their choices and, ideally, make a decision about ventilator use in a noncrisis situation. Noninvasive respiratory muscle aids are not a permanent solution to respiratory failure but fo provide many patients with additional time to make a decision concerning tracheostomy. Ultimately, less than 5% of ALS patients choose long-term ventilatory support by tracheostomy.27,28
Noninvasive positive pressure ventilation (NIPPV) can be delivered through a variety of oral or nasal masks and interfaces by use of bilevel positive airway pressure (BiPAP) machines or portable volume-cycled ventilators. BiPAP is the most commonly used form of NIPPV. Several studies suggest that use of NIPPV may prolong survival and slow the decline of FVC.29,30 The American Academy of Neurology’s practice parameter on ALS recommends
that NIPPV be introduced when the FVC falls to 50% of predicted, or earlier if the patient is symptomatic.31 However, others have suggested that earlier introduction of NIPPV may
improve survival and quality of life.32,33 Initially, NIPPV is used only at night. As FVC
continues to decline, ventilator use extends into the day for varying periods and eventually becomes continuous.
PROCEDURES
MANAGEMENT OF SIALORRHEA
Transtympanic neurectomy, salivary duct ligation, and parotid gland irradiation have been used to decrease saliva production but are associated with high failure and complication rates.34 Botulinum toxin injection into salivary glands is a preferred method of management
for patients who do not respond to pharmacologic therapy.8
GASTROCTOMY TUBE
Either a percutaneous endoscopic gastrostomy (PEG) tube or a radiologically inserted gastrostomy tube is recommended when a feeding tube is needed. The American Academy of Neurology’s practice parameter on ALS states that the morbidity and mortality of PEG tube
placement increase when the FVC is below 50% of predicted31 and recommends placement
before that time. However, later studies have shown that PEG tubes can be safely placed with BiPAP assistance in patients with lower FVCs.35,36 In addition, radiologically inserted
gastrostomy tubes may be preferable in patients with low FVCs; less sedation is required, incidence of aspiration is lower, tube placement is more often successful.37 Two studies have
suggested longer survival in patients choosing PEG tube placement38,39 when they are
compared with comparable patients refusing PEG.
SURGERY
TRACHEOSTOMY
This is best performed on a planned basis when patients choose long-term ventilatory support. However, it is more frequently performed in a crisis situation. A cuffless tracheostomy tybe or a tube with a deflated cuff is preferred.
POTENTIAL DISEASE COMPLICATIONS
The potential disease complications are progressive weakness, joint contractures, musculoskeletal pain syndromes, dysphagia, aspiration, dysarthria, depression, progressive respiratory failure, and death.
POTENTIAL TREATMENT COMPLICATIONS
The potential treatment complications include drug reactions (e.g., to riluzole, tricyclic antidepressants) and malfunction or infection of the PEG tube or radiologically inserted gastrostomy tube.
Complications of long-term ventilation by tracheostomy are tracheomalacia, loss of extraocular movements, totally locked-in state, and dementia.
REFERENCES
1. Rosenfeld J, Swash M. What’s in a name? Lumping or splitting ALS, PLS, PMA and other motor neuron diseases. Neurology 2006;66:624-625.
2. Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. ALS Other Motor Neuron Disord 2000;1:293-299.
3. Bensimon G, Lacomblez L, Meininger V, et al. A controlled trial of riluzole in amyotrophic lateral sclerosis. N Engl J Med 1994;330:585-591.
4. Lacomblez L, Bensimon G, Leigh PN, et al. Dose-ranging study of riluzole in amyotophic lateral sclerosis. Lancet 1996;347:1425-1431.
5. Miller R, Mitchell J, Lyon M, Moore D. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev 2002;2:CD001447. 6. de Carvalho M, Costa J, Swash M. Clinical trials in ALS: a review of the role of clinical
and neurophysiological measurements. ALS Other Motor Neuron Disord 2005;6:202-212. 7. Rosenfeld J. ALS combination treatment. Drug cocktails. ALS Other Motor Neuron
Disord 2004;5:115-117.
8. Tan E. Botulinum toxin treatment of sialorrhea; comparing different therapeutic preparations. Eur J Neurol 2006;13:60-64.
9. Brooks B, Thisted R, Appel S, et al. Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: a randomized trial. Neurology 2004;63:1364-1370.
10. Ganzini L, Johnston WS, Hoffman WF. Correlated of suffering in amyotrophic lateral sclerosis. Neurology 1999;52:1434-1440.
11. Lou J, reeves A, Benice T, Sexton G. Fatigue and depression are associated with poor quality of life in ALS. Neurology 2003;60:122-123.
12. Kubler A, Winter S, Ludolph A, et al. Severity of depressive symptoms and quality of life in patients with amyotrophic lateral sclerosis. Neurorehabil Neural Repair 2005;19:182-193.
13. Dal Bello-Haas V, Florence JM, Kloos AD, et al. A randomized controlled trial of resistance exercise in individuals with ALS. Neurology 2007;68:2003-2007.
14. Lindeman E, Leffer P, Spaans F, et al. Strength training in patients with myotonic dystrophy and hereditary motor and sensory neuropathy: a randomized clinical trial. Arch Phys Med Rehabil 1995;76:612-620.
15. Aitkens SG, McCrory MA, Kilmer DD, Bernauer EM. Moderate resistance exercise program: its effect in slowly progressive neuromuscular disease. Arch Phys Med Rehabil 1993;74:711-715.
16. Agre JC, Rodriquez AA, Frankle TM. Strength, endurance, and work capacity after muscle strengthening exercise in postpolio subjects. Arch Phys Med Rehabil 1997;78:681-686.
17. Drory V, Goltsman E, Reznik J, et al. The value of muscle exercise in beneficial to a mouse model of amyotrophic lateral sclerosis. J Neurol Sci 2002;191:133-137.
18. Kirkenzos I, Hernandez D, Bradley W, Moraes C. Regular exercise is beneficial to a mouse model of amyotrophic lateral sclerosis. Ann Neurol 2003;53:804-807.
19. Veldink J, Bar P, Joosten E, et al. Sexual differences in onset of disease and response to exercise in a transgenic model of ALS. Neuromuscul Disord 2003;13:737-743.
20. Kaspar B, Frost L, Christian L, et al. Synergy of insulin-like growth factor-1 and exercise in amyotrophic lateral sclerosis. Ann Neurol 20058;57:649-655.
21. Heffernan C, Jenkinson C, Holmes T, et al. Nutritional management in MND/ALS patients: an evidence based review. ALS Other Motor Neuron Disord 2004;5:72-83. 22. Strand EA, Miller RM, Yorkston KM, Hillel AD. Management of oral-pharyngeal
dysphagia symptoms in amyotrophic lateral sclerosis. Dysphagia 1996;11:129-139. 23. Esposito S, Mitsumoto H, Shanks M. Use of palatal lift and palatal augmentation
protheses to improve dysarthria in patients with amyotrophic lateral sclerosiss: a case series. J Prosthet Dent 2000;83:90-98.
24. Bach J. Respiratory muscle aids for the prevention of morbidity and mortality. Semin Neurol 1995;15:72-83.
25. Winck J, Gonclaves M, Lourenco C, et al. Effects of mechanical insufflation-exsufflation on respiratory parameters for patients with chronic airway secretion encumbrance. Chest 2004;126:774-780.
26. Gay P, Edmonds L. Severe hypercapnia after flow oxygen therapy in patients with neuromuscular disease and diaphragmatic dysfunction. Mayo Clin Proc 1995;70:327-330. 27. Moss A, Casey P, Stocking C, et al. Home ventilation for amyotrophic lateral sclerosis patients: outcomes, costs, and patient, family, and physician attitudes. Neurology 1993;43:438-443.
28. Lechtzin N, Weiner C, Clawson L, et al. Use of noninvasive ventilation in patients with amyotrophic lateral sclerosis. ALS Other Motor Neuron Disord 2204;5:9-15.
29. Kleopa KA, Sher M, Neal B, et al. Bi PAP improves survival and rate of pulmonary function decline in patients with ALS. J Neurol Scri 1999;164:82-88.
30. Aboussouan L, Khan S, Banerjee M, et al. Objective measures of the efficacy of noninvasive positive-pressure ventilation in amyotropic lateral sclerosis. Muscle Nerve 2001;24:403-409.
31. Miller R, Rosenberg J, Gelinas D, et al. A preliminary evaluation of a prospective study of pulmonary function studies and symptoms of hypoventilation in ALS/MND patients. J Neurol Sci 2001;191:75-78.
32. Jackson C, Rosenfeld J, Moore D, et al. A preliminary evaluation of a prospective study of pulmonary function studies and symptoms of hypoventilation in ALS/MND patients. J Neurol SAci 2001;191:75-78.
33. Bourke S, Tomlinson M, Williams T, et al. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial. Lancet Neurol 2006;5:40-147.
34. Yorkston KM, Miller RM, Strand EA. Management of Speech and Swallowing in Degenerative Diseases. Tucson, Communication Skill Builders, 1995:253.
35. Gregory S, Siderowf A, Golaszewski A, et al. Gastrostomy insertion in ALS patients with low vital capacity: respiratory support and survival. Neurology 2002;58:485-487.
36. Boitano L, Jordan T, Benditt J. Noninvasive ventilation allows gastrostomy tube placement in patients with advanced ALS. Neurology 2001;56:413-414.
37. Thornton F, Fotheringham T, Alexander M, et al. Amyotrophic lateral sclerosis: enteral nutrition provision––endoscopic or radiologic gastrostomy? Radiology 2202;224:713-717.
38. Mathus-Vliegen L, Louwerse L, Merkus M, et al. Percutaneous endoscopic gastrostomy in patients with amyotrophic lateral sclerosis and impaired pulmonary function. Gastrointest Endosc 1994;40:463-469.
39. Mazini L, Corra T, Zaccala M, et al. Percutaneous endoscopic gastrostomy and enteral nutrition in amyotrophic lateral sclerosis. J Neurol 1995;242:695-698.
125
PENYAKIT NEURON MOTORIK
Lisa S. Krivickas, MD
DEFINISI
Istilah penyakit motor neuron merujuk pada gangguan neuromuskuler progresif di mana neuron motorik atas atau bawah mengalami degenerasi. Bentuk yang paling umum dari
penyakit motor neuron adalah amyotrophic lateral sclerosis (ALS), yang merupakan fokus
utama pada bab ini. Prinsip-prinsip manajemen untuk bentuk lain dari penyakit motor neuron adalah sama. Untuk memenuhi kriteria diagnostik ALS, seseorang harus memiliki kedua disfungsi pada neuron motorik atas dan bawah. Jika hanya terdapat disfungsi pada neuron motorik bawah, maka penyakit ini disebut atrofi otot progresif; jika hanya disfungsi neuron motorik atas, maka disebut sklerosis lateral primer. Jika hanya terdapat disfungsi bulbar, maka penyakit ini disebut progressive bulbar palsy. Kebanyakan pasien awalnya didiagnosis memiliki atrofi otot progresif, sklerosis lateral primer, atau bulbar palsy progresif, yang pada akhirnya menjadi kesatuan ALS. Mereka yang penyakitnya tidak menjadi ALS memiliki tingkat perkembangan penyakit yang lebih lambat.
Sebagian besar kasus ALS adalah idiopatik. Namun, 5% sampai 10% dari pasien memiliki riwayat kelaian pada keluarga, biasanya diturunkan dengan pola autosomal dominan. Pada sekitar 20% dari kasus-kasus keluarga, mutasi di SOD1 (superoxide dismutase) dapat ditemukan. Bentuk langka lainnya dari penyakit motor neuron yang diturunkan orang
dewasa, yaitu penyakit Kennedy (X-linked resesif) dan atrofi otot tulang belakang dewasa (autosomal resesif), yang keduanya hanya memiliki disfungsi neuron motorik bawah.
ALS secara cepat menghasilkan kelemahan pada otot rangka, yang akhirnya mengarah pada kebutuhan untuk bantuan ventilator atau kematian akibat gagal pernapasan. Timbulnya kelemahan dapat dimulai pada tungkai manapun, otot bulbar, atau otot-otot pernapasan. Otot-otot ekstraokular dan usus dan fungsi kandung kemih umumnya terhindar. Rata-rata kelangsungan hidup, tanpa trakeostomi, adalah 3 tahun dari onset gejala, namun kisaran mungkin kurang dari 1 tahun sampai lebih dari 20 tahun. Satu penjelasan untuk variabilitas ekstrim dalam laju perkembangan penyakit adalah bahwa ALS mungkin kelompok heterogen dari berbagai penyakit daripada sebuah penyakit tunggal.1 Usia rata-rata saat onset adalah
pada pertengahan 50-an, tetapi ALS dapat berkembang pada orang dewasa dari segala usia. Penyebab penyakit ini tidak diketahui, tetapi teori terkemuka tentang patogenesis mengarah pada eksisitotoksisiti glutamat, stres oksidatif, neuroinflamasi, aktivasi sel mikroglial, apoptosis, dan disfungsi mitokondria.
17 LMN signs only ≥ 1 region UMN signs only ≥ 1 region LMN + UMN 1 region Suspected
ALS Possible ALS
Identifie d DNA gene Definite familial ALS LMN + UMN 1 region or UMN ≥ 1 region Probable ALS laboratory supported EMG acute denervation ≥ 2 limbs LMN + UMN 2 regions Probable
ALS Definite ALS
LMN + UMN 3 regions
SKEMA
Kelemahan, atrofi, hiperrefleks, spastisitas Perkembangan dari waktu ke waktu
EMG, NCV, neuroimaging, biopsy
Gambar 125-1. Kriteria El Escorial untuk diagnosis ALS. EMG, electromyography; NCV,
nerve conduction velocity; LMN, lower motor neuron; UMN, upper motor neuron.
GEJALA
Pasien dengan sindrom neuron motorik atas mungkin dijumpai dengan spastisitas, hilangnya ketangkasan, kekakuan, kelemahan akibat spastisitas, dan hilangnya kontrol motorik sadar. Pasien dengan sindrom neuron motorik bawah dapat dijumpai dengan kelemahan, kedutan otot, atrofi otot, dan kram otot. Gejala bulbar termasuk disartria, disfagia, sialorrhea (air liur), aspirasi, dan pseudobulbar afek (tertawa atau menangis tidak sesuai dengan suasana hati). Kegagalan pernafasan dan gejala konstitusional seperti penurunan berat badan dan kelelahan dapat dijumpai. Gejala kognitif meliputi disfungsi perilaku atau eksekutif dan demensia frontotemporal pada sebagian kecil pasien.
PEMERIKSAAN FISIK
Penekanan dari pemeriksaan fisik pasien yang diduga atau didiagnosis penyakit motor neuron adalah pada sistem neurologis, muskuloskeletal, dan kardiorespirasi. Pada pemeriksaan neurologis, salah satu pemeriksaan adalah mencari bukti disfungsi motorik neuron atas dan bawah. Status mental, fungsi non-motor saraf kranial, pemeriksaan sensorik, dan temuan pemeriksaan pada serebelum biasanya normal. Standar baku emas untuk diagnosis penyakit neuron motorik atas adalah adanya refleks patologis-tanda Babinski, tanda Hoffmann, dan
brisk jaw jerk. Pasien dengan ALS mungkin hyperrefleksia atau hiporefleksia, tergantung pada tahap perjalanan penyakit mereka dan apakah mereka memiliki dominasi pada penyakit neuron motorik atas atau bawah. Bukti penyakit neuron motorik bawah termasuk kelemahan otot, atrofi, hipotonia, hiporefleksia, dan kedutan otot. Atrofi sering muncul pertama kali di otot intrinsic tangan. Meskipun kedutan bukan kriteria yang diperlukan untuk diagnosis ALS, seseorang harus mempertanyakan diagnosis ketika tidak ditemukan kedutan. Lidah diperiksa untuk kedutan dan atrofi, dan kekuatan lidah, serta dinilai rentang gerak lidah. Pemeriksaan muskuloskeletal berfokus pada penilaian jangkauan gerak dan evaluasi nyeri sendi atau struktur jaringan lunak. Karena gagal napas berkembang secara progresif, sistem
kardiorespirasi harus dinilai pada setiap kunjungan. Kapasitas vital paksa (FVC) dapat diukur dengan spirometer genggam dalam lingkungan ruangan.
BATASAN FUNGSIONAL
Mayoritas keterbatasan fungsional yang berkembang pada pasien dengan ALS adalah kelemahan otot secara langsung ataupun tidak langsung. Selama penyakit berlangsung, pasien memiliki gangguan mobilitas dan kesulitan dengan kegiatan, bahkan kegiatan yang paling dasar dari kehidupan sehari-hari, seperti makan sendiri. Kelemahan otot bulbar menghasilkan disartria (kesulitan berbicara) dan disfagia (kesulitan menelan); yang pada akhirnya, beberapa pasien menjadi anarthria dan tidak mampu menelan bahkan ludah mereka sendiri. Depresi reaktif, kelelahan umum, dan nyeri muskuloskeletal lebih lanjut membatasi fungsi kehidupan.
STUDI DIAGNOSTIK
Diagnosis ALS didasarkan pada pemeriksaan fisik yang sesuai dan temuan elektrodiagnostik dan penggunaan neuroimaging dan hasil laboratorium klinik untuk mengeksklusi kondisi lain yang mirip ALS. Semua pasien harus menjalani tes elektrodiagnostik. Kriteria El Escorial yang telah direvisi saat ini digunakan untuk mendiagnosis ALS.2 Mereka mengklasifikasikan
tingkat kepastian diagnosis menjadi salah satu dari lima kategori: definite, probable, probable
dengan hasil laboratorium, possible, dan suspected (Gambar 125-1.).
Sistem motorik dibagi menjadi empat wilayah: bulbar, servikal, torakal, dan lumbosakral. Bukti klinis penyakit neuron motorik atas dan bawah dicari di setiap daerah. Tingkat kepastian diagnosis tergantung pada berapa banyak daerah yang menunjukan penyakit neuron motorik atas atau neuron motorik bawah. Temuan elektropsikologi dapat digunakan baik untuk mengkonfirmasi disfungsi neuron motorik bawah di daerah-daerah yang terkena dampak klinis dan untuk mendeteksi disfungsi neuron motorik bawah di daerah klinis tidak terkena dampak klinis.
Studi pencitraan digunakan untuk mengeksklusi kemungkinan lain selain penyakit motor neuron dari diagnosis diferensial. Pencitraan resonansi magnetik adalah modalitas pencitraan utama dalam evaluasi pasien yang diduga ALS. Hampir semua pasien harus memiliki pencitraan resonansi magnetik dari tulang belakang servikal untuk mengeksklusi kompresi spinal, syrinx, atau penyakit sumsum tulang belakang lainnya. Lokasi gejala akan menentukan apakah daerah lain dari sumsum tulang belakang harus dicitrakan. Pada pasien
yang mengalami gejala bulbar, pencitraan resonansi magnetik pada otak harus dilakukan untuk menyingkirkan stroke, tumor, syringobulbia, dan proses patologis lainnya.
Dalam kebanyakan praktek klinik neuromuskuler, sebuah panel rutin tes laboratorium dilakukan untuk semua pasien yang dianggap mengalami ALS. Satu set tes yang disarankan tersebut diberikan pada Tabel 125-1. Alasan di balik banyaknya tes yang dilakukan adalah untuk menilai kesehatan umum pasien dan untuk mengeksklusi kondisi yang dapat diobati. Diferensial diagnosis, dikembangkan setelah anamnesis dan pemeriksaan fisik, dapat menunjukkan tes yang lebih khusus yang harus dilakukan. Tabel 125-2 menunjukkan tes tambahan yang mungkin diperlukan saat ditemukan atrofi otot progresif, sklerosis lateral primer, atau bulbar progresif fenotipe palsy. Ketika ada riwayat keluarga penyakit motor neuron, pengujian SOD1 dapat dipertimbangkan.
Tabel 125-1 Pemeriksaan Laboratorium yang Disarankan
Hematologi
Pemeriksaan darah lengkap Laju sedimentasi
Kimia
Elektrolit, blood urea nitrogen, kreatinin Glukosa Hemoglobin A Kalsium Fosfor Magnesium Kreatin kinase Tes fungsi hati Kadar timbal serum Tes logam berat urin Vitamin B12 Folat Endokrin Tiroksin Thyroid-stimulating hormone Imunologi Imunoelektroforesis serum Tes protein Bence Jones urin
Antinuclear antibody
Faktor reumatoid
Lyme titer
Tes VDRL
Opsional
Tes HIV (jika terdapat faktor risiko)
Anti-Hu antibody (jika curiga keganasan)
Tes DNA untuk mutasi SOD1 (dengan riwayat keluarga)
Tabel 125-2 Pemeriksaan Laboratorium Khusus
Fenotipe Tes Diagnosis Tereksklusi
Atrofi muskular progresif
Tes DNA: CAG repeat on X
chromosome
Penyakit Kennedy
Tes DNA: mutasi gen SMN Atrofi muskular spinal
Hexosaminidase A Defisiensi hexosaminidase A
(heterozygous Tay-Sachs disease)
Voltage-gated calcium channel antibody test
Sindrom miastenik Lambert-Eaton
Pemeriksaan cairan serebrospinal Poliradikulopati, infeksi atau
neoplastik
Panel antibodi GM1 Neuropati motorik multifokal
Sklerosis lateral primer
Asam lemak rantai sangat panjang Adrenoleukodistrofi
Antibodi Human T-lymphotropic
virus 1 (HTLV-1)
Mielopati HTLV-1 (paraparesis spastik tropis)
Hormon paratiroid Mielopati hiperparatiroid
Pemeriksaan serebrospinal Sklerosis multipel
Palsi bulbar progresif
Antibodi reseptor asetilkolin Miastenia gravis
Tes DNA: CAG repeat on X
chromosome
Penyakit Kennedy
Pemeriksaan serebrospinal Sklerosis multipel
Diferensial Diagnosis
Diferensial diagnosis bergantung pada apakah di neuron motorik bawah, neuron motorik atas, bulbar, atau gabungan neuron motor atas dan
Degenerasi kombinasi subakut Paraparesis spastik herediter Mielopati
bawah. Siringomielia
Neuron Motroik Bawah Saja Bulbar
Atrofi muskular progresif Palsi bulbar progresif
Atrofi muskular spinal Miastenia gravis
Penyakit Kennedy Sklerosis multipel
Polimielitis, sindrom post-poliomielitis Tumor foramen magnum
Amiotrofi monomelik benigna Glioma batang otak
Defisiensi hexosaminidase A Stroke
Poliradikulopati Siringobulbia
Neuropati motorik multifokal dengan blok konduksi
Kanker kepala dan leher
Polineuropati demielinasi inflamasi kronis Polimiositis
Neuropati motorik atau neuronopati Distrofi muskular okulofaringeal
Sindrom Lambert-Eaton Penyakit Kennedy
Pleksopati
Fasikulasi benigna Motor Neuron Atas dan Bawah
ALS
Neuron Motorik Atas Saja Mielopati servikal dengan radikulopati
Sklerosis lateral primer Siringomielia
Sklerosis multipel Tumor tulang belakang atau malformasi
arteriovenosa
Adrenoleukodistrofi Penyakit Lyme
PENATALAKSANAAN INISIAL
FARMAKOLOGI
Walaupun belum ditemukan pengobatan ALS, telah dilakukan berbagai penelitian untuk mengidentifikasi obat yang dapat memperlambat progresivitas penyakit ALS. Pemberian terapi farmakologi memberikan dampak psikologi yang jauh lebih nyata dari pada memperlambat progresivitas penyakit. Riluzole (Rilutek) adalah satu-satunya obat yang disetujui oleh FDA untuk terapi ALS. Meskipun demikian banyak ahli-ahli neuromuskular yang merekomendasikan untuk mengkombinasikannya dengan antioksidan, vitamin, dan suplemen lainnya.
Penggunaan Riluzole (agen antiglutamat) untuk terapi ALS telah disetujui pada 1995 setelah adanya bukti penurunan progresivitas penyakit pada dua uji klinis.3,4 Cochrane melaporkan
meta-analisis dari uji klinis Riluzole menunjukkan manfaat kurang lebih 2 bulan.5 Sayangnya,
tidak ditemukan perbedaan manfaat fungsional dari mereka yang mengkonsumsi riluzole dan placebo. Efek samping yang paling sering ditemukan adalah lelah, mual, dan peningkatan enzim hepar. Riluzole dikontraindikasikan pada pasien dengan peningkatan aktivitas enzim hepar lima kali di atas normal.
Karena stres oksidatif akibat produksi radikal bebas berlebih pada motor neuron adalah salah satu faktor patogen pemicu pada ALS, para dokter menyarankan untuk mengkonsumsi
antioksidan seperti vitamin E, vitamin C, dan coenzyme Q10. Belum ada uji buta ganda
dengan placebo yang membuktikan keberhasilan terapi ini. Dosis aman harian yang direkomendasikan adalah 1000 sampai 3000 mg untuk vitamin C, 400 IU untuk vitamin E,
dan sampai 1200 mg untuk coenzyme Q10.
Uji klinis negatif ALS terbaru menguji sejumlah faktor pertumbuhan, keratin, glutamat antagonis, agen antiinflamasi (celecoxib), calcium channel blockers, dan asam amino.6 Kini,
sedang diuji mengenai neurotrophic factor, antioksidan, glutamate antagonis, coenzyme Q10, dan tamoxifen. Di masa yang akan datang, pendekatan koktail mungkin merupakan strategi terapi yang paling ideal untuk menurunkan progresivitas penyakit.7
Berbagai terapi farmakologi bermanfaat untuk mengatasi spastisitas, sialorrhea,
pseudobulbar affect, depresi, dan ansietas.
Spastisitas perlu diterapi hanya bila hal menimbulkan gangguan fungsi. Terapi nonfarmakologi meliputi mengajari pasien mengenai latihan peregangan dan positioning technique untuk mengurangi tonus otot. Baclofen (Lioresal) adalah terapi farmakologi yang paling efektif, diikuti dengan tizanidine (Zanaflex). Diazepa, (Valium) perlu dihindari karena dapat menyebabkan supresi pada system respirasi, dan dantrolene (Dantrium) juga tidak direkomendasikan karena dapat menyebabkan kelemahan otot berlebih. Secara keseluruhan, terapi farmakologi untuk spastisitas kurang berhasil pada pasien ALS dibandingkan dengan pasien multiple sclerosis atau spinal cord injury karena komponen motor neuron bawah pada ALS membuat penderitanya sangat rentan terhadap kelemahan otot yang sangat progresif. Pasien dengan disfungsi bulbar mengalami sialorrhea karena adanya kesulitan menelan dan mengatur sekresi oral dari produksi normal mereka. Beragam obat antikolinergik mungkin digunakan untuk mengeringkan mulut. Antidepresan trisiklik sering menjadi obat pertama yang dipilih namun tidak sebanding dengan efek sampingnya (mulut kering, somnolen, retensi urin). Ketika antidepresan trisiklik tidak dapat ditolerir, scopolamine patch
(Transderm Scop) atau glycopyrrolate (Robinul) mungkin dapat digunakan sebagai alternatifnya. Bila terapi tersebut belum adekuat, pasien dapat menggunakan injeksi toksin botulinum pada kelenjar ludah.8
Pseudobulbar affect, yang sering disebut inkontinensi emosi merujuk pada ketidakmampuan pasien untuk menggambarkan emosi yang sedang dirasakannya secara tepat. Pasien tertawa saat sedih atau menangis saat bahagia. Mereka juga memberikan respon yang berlebihan saat memiliki perasaan yang sesuai dengan situasi saat itu. Amitriptyline (Elavil), kombinasi dextromethorphan (Robitussin, Drixoral) dan quinidine, atau fluvoxamine (Luvox) dapat membantu mengurangi intensitas dari reaksi yang tidak sesuai atau berlebihan tersebut. Kombinasi dextromethorphan dan quinidine terbukti efektif pada uji klinik acak bersampel besar9 namun kombinasi ini belum disetujui oleh FDA
Depresi yang reaktif dan ansietas merupakan reaksi normal untuk diagnosis ALS.10 Pasien
dan keluarganya dapat diberikan konseling individu dan berpartisipasi dalam kelompok pendukung ALS. Ansietas dapat diterapi dengan benzodiazepine selama pasien tidak menunjukkan penurunan kapasitas vital yang signifikan. Hipoventilasi yang tidak terdeteksi dapat menyebabkan dan memicu perasaan cemas. Depresi harus diterapi secara farmakologi karena dapat memberikan dampak negatif pada kualitas hidup bila tidak diobati.11,12Selective serotonin reuptake inhibitors (SSRIs) adalah pilihan pertama yang baik karena efek sampingnya sangatlah sedikit. Meskipun demikian, antidepresan triskilik mungkin lebih disukai jika diperlukan juga terapi untuk gejala lain seperti sialorrhea dan pesudobulbar affect.
REHABILITASI
Kelemahan otot rangka adalah gangguan utama pada ALS dan penyebab sebagian besar masalah klinis. Pada tahap awal ALS, pasien sering bertanya tentang peran latihan dalam mencegah perburukan pada kelemahan otot. Kemudian untuk penyakitnya sendiri, strategi rehabilitasi harus digunakan untuk mempertahankan fungsi dan untuk mengkompensasi kelemahan otot. Tiga bentuk latihan pelatihan yang relevan untuk penderita ALS: latihan fleksibilitas, penguatan, dan aerobik. Latihan fleksibilitas membantu mencegah perkembangan kontraktur yang menyakitkan, secara nonfarmakologi membantu mengurangi kelenturan dan menghilangkan kejang otot yang berlebihan. Umumnya, para dokter enggan
untuk merekomendasikan latihan penguatan karena mereka takut bahwa pemakaian berlebih pada kelemahan otot akan mempercepat terjadinya disabilitas. Filosofi ini mendorong perkembangan kelemahan otot akibat tidak digunakan dan dekondisi otot yang dapat memperparah kelemahan yang disebabkan oleh ALS tersebut. Literatur yang mendukung mengenai perkembangan kelemahan otot akibat pemakaian berlebih pada pasien neuromuskular tidaklah dapat dipercaya, dan kelemahan akibat pemakaian berlebihan tidak dapat dibuktikan dalam studi kontrol prospektif manapun. Studi mengenai latihan penguatan pada ALS menunjukkan adanya perlambatan penurunan fungsi fisik dan tidak ada efek sampingnya.13 Studi pada pasien dengan penyakit neuromuskuler yang lebih lambat
progresivitasnya dan pasien sindrom post-poliomyelitis menggambarkan bahwa otot-otot yang hanya sedikit terpengaruh oleh proses penyakit dapat diperkuat dengan program penguatan beresistensi sedang.14-16 Latihan penguatan eksentrik beresistensi tinggi harus
dihindari karena dapat merusak otot.
Saya menyarankan pasien ALS yang berminat untuk memulai atau melanjutkan program penguatan untuk memaksimalkan kekuatan otot-otot yang masih normal atau yang mengalami kelemahan ringan guna menunda terjadinya kerusakan fungsi otot tersebut. Latihan beban harus dilakukan dengan beban yang dapat diangkat 20 kali, dan individu harus diinstruksikan untuk melakukan submaximal set dengan pengulangan kurang lebih 10-15 kali; hal ini untuk memastikan bahwa latihan tersebut dilakukan dalam level resistensi rendah sampai sedang. Jika rejimen latihan terus menerus menyebabkan nyeri otot atau kelelahan yang berlangsung lebih dari setengah jam setelah latihan, itu berarti latihan terlalu berat. Latihan aerobik membantu mempertahankan kesehatan kardiorespirasi. Studi mengenai aktivitas aerobik moderat pada pasien dengan ALS menunjukkan efek positif jangka pendek pada disabilitas, pasien yang melakukan latihan akan lebih lama berada pada level penyakit yang lebih ringan.17 Selain itu, studi dengan model tikus transgenik pada ALS telah
menunjukkan bahwa latihan aerobik memperpanjang kelangsungan hidup.18-20 Mengingat
kurangnya kontraindikasi yang jelas, latihan aerobik hanya dianjurkan untuk pasien dengan ALS selama dapat dilakukan dengan aman tanpa risiko jatuh atau cedera. Selain dari manfaat fisik, latihan penguatan dan aerobik memberikan efek positif pada suasana hati, kesejahteraan psikologis, nafsu makan, dan tidur.
Seiring dengan progresivitas ALS, fokus rehabilitasi bergeser dari latihan ke pemeliharaan mobilitas dan fungsi independen selama mungkin. Intervensi termasuk alat bantu seperti tongkat, alat bantu berjalan, penyangga kaki, bidai tangan, kursi roda, dan skuter; peralatan rumah seperti alat bantu untuk berpakaian, peralatan yang telah disesuaikan, palang
pegangan, raised toilet seats, bangku mandi, dan lift; modifikasi rumah (pintu yang landai, lebar); dan mobil dengan adaptasi, seperti hand control, environmental control unit, dan
voice-activated software. Intervensi rehabilitasi ini paling baik disediakan oleh tim multidisiplin yang mencakup psikiater, terapis fisik, terapis okupasi, dan ortotis.
Pasien sering mengalami sindrom nyeri muskuloskeletal seperti adhesive capsulitis (kaku bahu), nyeri punggung bawah, dan nyeri leher akibat kelemahan otot dan ketidakmampuan untuk mengubah posisi tubuhnya sendiri. Langkah-langkah untuk mencegah bahu kaku
meliputi latihan range of motion dan sebisa mungkin penggunaan penyangga tangan daripada
dibiarkan menjuntai di sisi tubuh. Nyeri pinggang bawah dapat ditimbulkan karena posisi duduk yang tidak nyaman. Langkah-langkah pencegahan termasuk di dalamnya yaitu
penyangga lumbal pada kursi roda, bantal empuk pada dudukan kursi yang kokoh, frequent
weight shift, dan reclining back or tilt-in-space wheelchair. Neck pain associated with head drop adalah salah satu masalah nyeri muskuloskeletal yang paling sulit untuk diatasi. Penyangga kepala pada kursi roda atau kursi baring lebih baik dan nyaman daripada penyangga leher. Asetaminofen, obat anti-inflamasi non steroid, Lidoderm patch, dan jika
perlu, opioid harus digunakan untuk meringankan nyeri muskuloskeletal. Transcutaneous
electrical nerve stimulation (TENS) juga dapat digunakan untuk mengontrol rasa sakit. Kekhawatiran utama dalam penggunaan obat opiate adalah distres pernapasan dan sembelit. Distres pernapasan mungkin muncul pada tahap akhir penyakit, namun pada kenyataannya, morfin adalah cara yang baik untuk mengurangi kekurangan oksigen dalam tahap terminal. Fungsi menelan yang adekuat diperlukan untuk mempertahankan status nutrisi pasien dengan ALS (kecuali yang menggunakan selang makanan). Jika status gizi tidak benar-benar diperhatikan, pasien cenderung menggunakan otot sebagai bahan bakar dan dengan demikian pasien dapat kehilangan massa dan kekuatan otot lebih cepat dari biasanya.21 Disfungsi
menelan juga dapat memicu pneumonia aspirasi atau kegagalan pernafasan. Tanda dan gejala awal disfagia adalah drooling, suara yang basah, batuk saat atau setelah minum air, regurgitasi nasal, dan membutuhkan waktu yang lama untuk menghabiskan makanan. Pasien harus dirujuk ke patologi wicara saat tanda-tanda pertama disfagia muncul. Pasien dengan kesulitan menelan ringan dapat diajarkan teknik mengurangi risiko aspirasi dan tersedak.22
Rekomendasi mengenai cara modifikasi konsistensi makanan yang disajikan dapat diberikan. Perkembangan pneumonia aspirasi, kehilangan lebih dari 10% berat badan, dan waktu yang lama untuk makan sedemikian rupa sehingga gangguan kualitas hidup adalah indikasi unuk pemberian makanan melalui selang.
Disartria awal atau ringan mungkin dapat diatasi oleh para terapis wicara yang mengajarkan pasien dengan strategi adaptif, seperti overarticulation dan memperlambat kecepatan berbicara. Pada pasien yang berbicara dengan hipernasal yang disebabkan oleh kelemahan palatum dan terutama disfungsi neuron motorik bawah, pengangkatan atau augmentasi prostesis palatum dapat meningkatkan kejelasan dalam berbicara.23 Seiring dengan
perburukan disartria, pasien memerlukan bentuk-bentuk alternatif komunikasi. Menulis adalah alternatif yang baik untuk pasien yang memiliki fungsi tangan yang masih baik. Bagi mereka yang tidak bisa menulis, intervensi berteknologi rendah berupa huruf dan word boards. Solusi teknologi yang lebih maju adalah perangkat komunikasi augmentatif dengan penghasil suara. Selama satu otot di tubuh dapat diaktifkan secara volunter (termasuk otot-otot ekstraokular), pasien tentu mampu mengoperasikan perangkat komunikasi. Solusi berteknologi tinggi belum tentu cocok untuk semua pasien dengan masalah komunikasi. Sistem tersebut haruslah fleksibel sehingga cara pengaksesan dapat dimodifikasi seiring dengan progresivitas kelemahan pada pasien.
Gagal nafas adalah penyebab utama kematian pada pasien ALS. Saat tidak ada penyakit intrinsik paru yang mendasarinya, gagal nafas pada ALS murni adalah masalah mekanis. Karena kelemahan otot, paru-paru tidak mengembang sepenuhnya pada fase inspirasi. Kebanyakan pasien dengan ALS tetap asimtomatik sampai FVC kurang dari 50% dari yang diprediksikan. Tes fungsi paru-paru, khususnya FVC, harus dipantau setiap beberapa bulan, tergantung pada tingkat perkembangan penyakit. Gejala awal gagal napas disebabkan oleh hipoventilasi nokturnal dan termasuk kurangnya tidur karena sering terbangun, mimpi buruk, sakit kepala pada pagi hari, dan kelelahan serta kantuk yang berlebihan pada siang hari. Tanda awal lainnya dari kelemahan otot pernapasan adalah batuk ringan dan kesulitan dalam pembersihan sekresi.
Pengelolaan kegagalan pernafasan di ALS melibatkan pencegahan infeksi bila mungkin dan pemberian bantuan ventilasi mekanis. Semua pasien dengan ALS harus menerima vaksinasi pneumokokus dan vaksinasi influenza tahunan. Jika otot-otot ekspiratori terlalu lemah untuk menghasilkan batuk yang memadai, pasien dapat dibantu baik bantuan batuk manual maupun dengan Cough Assist (J.H. Emerson Co., Cambridge, Mass).24,25 Memberikan pasien bantuan
tambahan oksigen dapat meringankan gejala hipoksia dan dyspnea tetapi juga dapat menekan hantaran pernafasan, memperburuk hipoventilasi alveolar, dan pada akhirnya menyebabkan retensi karbondiksida dan henti nafas. 26
Oksigen tambahan hanya direkomendasikan untuk pasien dengan penyakit paru konkomitan atau sebagai tolak ukur kenyamanan bagi mereka yang menolak ventilasi bantuan. Diskusi
mengenai kemungkinan kegagalan pernapasan harus dimulai segera setelah diagnosis ALS sehingga pasien dan keluarga mereka dapat mempelajari pilihan mereka dan membuat keputusan secara ideal mengenai penggunaan ventilator dalam situasi yang tidak kritis. Bantuan otot pernapasan noninvasive bukan merupakan solusi permanen untuk kegagalan pernapasan tetapi memang memberikan pasien tambahan wantu untuk membuat keputusan mengenai trakeostomi. Pada akhirnya, kurang dari 5% ALS pasien memilih trakeostomi sebagai pendukung ventilasi jangka panjang.27,28
Ventilasi tekanan positif non-invasif (NIPPV) dapat diberikan melalui berbagai macam
sungkup mulut dan/atau hidung dengan menggunakan mesin bilevel positive airway pressure
(BiPAP) atau portable volume-cycled ventilators. BiPAP adalah bentuk penggunaan paling
umum dari NIPPV. Beberapa penelitian menunjukkan bahwa penggunaan NIPPV dapat
memperpanjang kelangsungan hidup dan memperlambat penurunan FVC. 29,30
Parameter praktik American Academy of Neurology pada ALS merekomendasikan
penggunaan NIPPV ketika FVC turun sampai 50% dari perkiraan atau lebih awal pada pasien simptomatik.31 Meskipun demikian, beberapa pihak lain menyatakan penggunaan NIPPV
yang lebih awal dapat meningkatkan kelangsungan dan kualitas hidup.32,33 Pada awalnya,
NIPPV digunakan hanya pada malam hari. Seiring FVC terus menurun, penggunaan ventilator memanjang sampai satu hari dalam beragam kondisi dan akhirnya menjadi terus-menerus.
PROSEDUR
MANAJEMEN SIALORRHEA
Transtympanic neurectomy, salivary duct ligation, dan parotid gland irradiation telah digunakan untuk mengurangi produksi saliva namun memiliki tingkat kegagalan dan komplikasi yang tinggi. Injeksi toksin botulinum pada kelenjar ludah juga sering digunakan pada pasien yang tidak memberikan respon pada terapi farmakologi.
SELANG GASTROSTOMI
Percutaneous endoscopic gastrostomy (PEG) atau pemasangan selang gastrostomi dengan bantuan radiologi direkomendasikan bila pasien membutuhkan selang makanan. Parameter praktik American Academy of Neurology pada ALS menyatakan bahwa morbiditas dan mortalitas pemasangan selang PEG meningkat ketika FVC di bawah 50% dari prediksi dan direkomendasikan pemasangan sebelum hal tersebut terjadi. Namun, studi studi selanjutnya menunjukkan bahwa selang PEG aman dipasang dengan bantuan BiPAP pada pasien dengan FVC rendah. Selain itu, pemasangan selang gastrostomi dengan bantuan radiologi lebih
dipilih pada pasien dengan FVC rendah; lebih sedikit sedasi yang dibutuhkan, insiden aspirasi lebih rendah, keberhasilan pemasangan selang lebih tinggi. Dua studi lainnya menunjukkan angka kelangsungan hidup yang lebih baik pada pasien yang memilih pemasangan selang PEG dibanding dengan pasien yang menolak pemasangan selang PEG.
PEMBEDAHAN TRAKEOSTOMI
Paling baik dilakukan secara terencana ketika pasien memilih menggunakan bantuan ventilator jangka panjang. Namun, tindakan ini lebih sering dilakukan saat situasi kritis.
Cuffless tracheostomy tube atau tube withdeflated cuff lebih sering digunakan.
KOMPLIKASI POTENSIAL PENYAKIT
Komplikasi potensial penyakit adalah kelemahan progresif, kontraktur sendi, musculoskeletal pain syndrome, disfagia, aspirasi, disartria, depresi, gagal nafas progresif, dan kematian.
KOMPLIKASI POTENSIAL TERAPI
Komplikasi potensial terapi meliputi reaksi farmakologi (seperti dengan riluzole, antidepresan trisiklik) dan malfungsi atau infeksi pada selang PEG atau selang gastrostomi. Komplikasi ventilasi jangka panjang dari trakeostomi adalah trakeomalasia, hilangnya pergerakan ekstraokular, locked-in state, dan demensia.
REFERENSI
1. Rosenfeld J, Swash M. What’s in a name? Lumping or splitting ALS, PLS, PMA and other motor neuron diseases. Neurology 2006;66:624-625.
2. Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. ALS Other Motor Neuron Disord 2000;1:293-299.
3. Bensimon G, Lacomblez L, Meininger V, et al. A controlled trial of riluzole in amyotrophic lateral sclerosis. N Engl J Med 1994;330:585-591.
4. Lacomblez L, Bensimon G, Leigh PN, et al. Dose-ranging study of riluzole in amyotophic lateral sclerosis. Lancet 1996;347:1425-1431.