Laporan Tugas Akhir
PERANCANGAN DAN ANALISA SIMULASI DINAMIKA
MOLEKUL ENSEMBLE MIKROKANONIKAL DAN KANONIKAL
DENGAN POTENSIAL LENNARD JONES
Diajukan untuk memenuhi syarat kelulusantahap sarjana di departemen Teknik Fisika Institut Teknologi Bandung
Oleh:
AREE WITOELAR
13396018Pembimbing:
Hermawan K. Dipojono, Dr., Ir. Nugraha, Dr., Ir.
DEPARTEMEN TEKNIK FISIKA FAKULTAS TEKNOLOGI INDUSTRI
INSTITUT TEKNOLOGI BANDUNG 2002
PERANCANGAN DAN ANALISA SIMULASI DINAMIKA
MOLEKUL ENSEMBLE MIKROKANONIKAL DAN KANONIKAL
DENGAN POTENSIAL LENNARD JONES
Oleh:
AREE WITOELAR
13396018Disahkan oleh:
Pembimbing I Pembimbing II
Hermawan K. Dipojono, Dr., Ir. Nugraha, Dr., Ir.
DEPARTEMEN TEKNIK FISIKA FAKULTAS TEKNOLOGI INDUSTRI
INSTITUT TEKNOLOGI BANDUNG 2002
1.1 ABSTRAK
Perancangan material baru dengan sifat-sifat tertentu sangat berguna untuk berbagai bidang. Namun perancangan material dengan eksperimen membutuhkan biaya mahal. Salah satu solusi adalah dengan melakukan eksperimen komputer dengan teknik
dinamika molekul.
Dinamika Molekul adalah teknik simulasi komputer yang mengamati pergerakan molekul-molekul yang saling berinteraksi. Dari molekul-molekul ini, dapat diperoleh sifat material yang diamati. Dinamika molekul banyak digunakan dalam berbagai bidang ilmu untuk mempelajari material.
Berdasarkan itu, maka dirancang suatu simulasi dinamika molekul untuk sistem terisolasi (ensemble mikrokanonikal) dan sistem dengan pengendalian temperatur
(ensemble kanonikal). Untuk menguji kebenaran simulasi, maka digunakan model
potensial Lennard-Jones dan dilakukan analisa hasil simulasi dengan berbagai parameter. Perangkat lunak simulasi dikembangkan dengan bahasa pemrograman C++ dengan berorientasi obyek.
Data yang diperoleh menunjukkan bahwa simulasi dinamika molekul untuk ensemble mikrokanonikal dan kanonikal memenuhi spesifikasi ketelitian yang dibutuhkan. Simulasi dinamika molekul dapat digunakan atau dikembangkan dalam penelitian selanjutnya.
DAFTAR ISI
DAFTAR ISI ... i
DAFTAR GAMBAR... iv
DAFTAR NOTASI ... vi
BAB 1 PENDAHULUAN...1
1.1 Latar Belakang Masalah ...1
1.2 Perumusan Masalah...2
1.3 Tujuan Penelitian...2
1.4 Pembatasan Masalah ...2
1.5 Sistematika Pembahasan ...3
BAB 2 DASAR TEORI...5
2.1 Dinamika Molekul...5
2.1.1 Ensemble ...6
2.1.1.1 Ensemble Mikrokanonikal (E, V, N)...6
2.1.1.2 Ensemble Kanonikal (T, V, N)...7
2.1.1.3 Ensemble Isobarik-Isotermal (P, T, N)...7
2.1.2 Langkah-Langkah Simulasi Dinamika Molekul...7
2.1.3 Syarat Batas Periodik ...8
2.2 Mekanika Klasik...10 2.3 Mekanika Statistik ...11 2.3.1 Energi Total ...11 2.3.2 Temperatur ...12 2.3.3 Distribusi Kecepatan...12 2.3.4 Tekanan ...13 2.3.5 Entalpi...14
2.4 Model Interaksi Antar Molekul ...14
2.4.1 Potensial Lennard-Jones...15
2.4.2 Gaya...16
2.4.3 Pemotongan Potensial ...17
2.5.1 Velocity Scaling ...18
2.5.2 Termostat Nose-Hoover ...18
2.5.3 Termostat Berendsen ...20
2.6 Pengendalian Tekanan...21
2.7 Pengembangan Persamaan Gerak Diskrit...21
2.7.1 Algoritma Verlet...22
2.7.2 Algoritma Leap-Frog...23
2.7.3 Algoritma Verlet dengan Pengendalian Temperatur ...24
2.7.4 Pendiskritan ζ untuk Termostat Nose-Hoover dan Berendsen...25
BAB 3 PERANCANGAN SIMULASI DINAMIKA MOLEKUL...26
3.1 Garis Besar ...26
3.1.1 Prosedur Simulasi...26
3.1.2 Parameter Simulasi...29
3.1.2.1 Kotak Simulasi dan Syarat Batas Periodik...29
3.1.2.2 Pemotongan Potensial ...30
3.1.2.3 Posisi Awal...30
3.1.3 Paralelisasi...31
3.1.3.1 Dinamika molekul paralel dengan dekomposisi atom...32
3.1.3.2 Dinamika molekul paralel dengan dekomposisi geometrik ...32
3.2 Perangkat Lunak...33 3.2.1 Kandidat Kelas ...33 3.2.2 Atribut Kelas ...33 3.2.2.1 Simulation...34 3.2.2.2 Molekul...34 3.2.2.3 Parameter...35 3.2.2.4 Property ...36 3.2.2.5 Initialize...37 3.2.2.6 Verlet ...37 3.2.2.7 LennardJones...37 3.2.3 Operasi Kelas...37 3.2.3.1 Simulation...37
3.2.3.2 Molecule ...38 3.2.3.3 Parameter...38 3.2.3.4 Property ...38 3.2.3.5 Initialize...38 3.2.3.6 Verlet ...40 3.2.3.7 LennardJones...40
3.2.4 Hubungan Antar Kelas ...41
3.2.5 Interface (Antar Muka)...42
BAB 4 ANALISA ...43
4.1 Simulasi Sistem Mikrokanonikal ...43
4.1.1 Uji Kesalahan Energi Simulasi...43
4.1.2 Momentum ...46
4.1.3 Uji Simulasi Terhadap Temperatur Awal...48
4.2 Simulasi Ensemble Kanonikal...50
4.2.1 Termostat Nose Hoover...50
4.2.1.1 Pengujian Terhadap Parameter Nose Hoover Q...50
4.2.1.2 Uji Hamiltonian ...52
4.2.1.3 Error Temperatur pada Perubahan Tset...53
4.2.1.4 Penurunan Matematis ...54
4.2.2 Termostat Berendsen ...57
4.2.2.1 Pengujian Terhadap Parameter Berendsen Q...57
4.2.2.2 Error Temperatur pada Perubahan Tset...58
4.2.2.3 Penurunan Matematis ...59
4.3 Tekanan terhadap Temperatur ...61
4.4 Entalpi...63
4.5 Posisi...64
BAB 5 KESIMPULAN DAN SARAN...66
5.1 Kesimpulan...66
5.2 Saran ...66
DAFTAR PUSTAKA...68
DAFTAR GAMBAR
Gambar 2.1 Sistem yang diamati dalam simulasi dinamika molekul...5
Gambar 2.2 Langkah-langkah simulasi dinamika molekul. [2] ...8
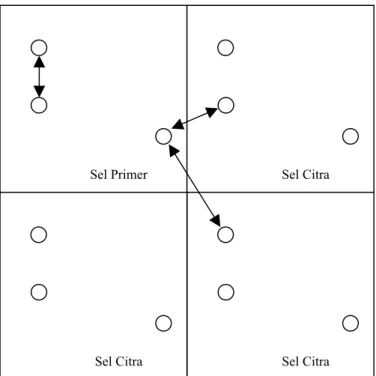
Gambar 2.3 Pembagian materi menjadi sel primer dan sel citra...9
Gambar 2.4 Distribusi kecepatan molekul pada berbagai temperatur [2] ...13
Gambar 2.5 Potensial Lennard-Jones ...16
Gambar 2.6 Pemasangan reservoir kalor pada sistem ...19
Gambar 2.7 Pemasangan reservoir kalor dan reservoir tekanan pada sistem...21
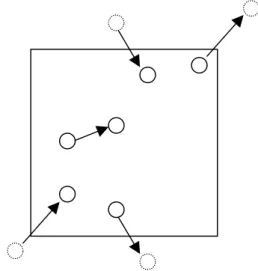
Gambar 3.1 Penghasilan trajektori molekul. [1]...26
Gambar 3.2 Prosedur simulasi dinamika molekul...28
Gambar 3.3 Syarat batas periodik untuk molekul yang keluar dari kotak simulasi. ...29
Gambar 3.4 Syarat batas periodik untuk jarak antar molekul ...30
Gambar 3.5 Penggunaan linked-list untuk molekul ...35
Gambar 3.6 Penambahan molekul dalam linked-list [11] ...39
Gambar 3.7 Hubungan antar kelas ...42
Gambar 3.8 Contoh tampilan Simulasi Dinamika Molekul ...42
Gambar 4.1 Grafik perbandingan energi kinetik dan energi potensial...44
Gambar 4.2 Grafik error energi total pada ∆t= 0,005 ...44
Gambar 4.3 Grafik error energi total pada ∆t= 0,01 ...45
Gambar 4.4 Grafik error energi total pada ∆t= 0,02 ...45
Gambar 4.5 Grafik error energi total pada ∆t= 0,05 ...46
Gambar 4.6 Grafik momentum total pada sumbu x, y dan z...47
Gambar 4.7 Grafik temperatur terhadap waktu...48
Gambar 4.8 Error energi total besar pada temperatur tinggi ...49
Gambar 4.9 Lompatan energi potensial...49
Gambar 4.10 Grafik temperatur dengan termostat Nose-Hoover Q = 1 ...50
Gambar 4.11 Grafik temperatur dengan termostat Nose-Hoover Q = 5 ...51
Gambar 4.12 Grafik temperatur dengan termostat Nose-Hoover Q = 15 ...51
Gambar 4.14 Temperatur pada perubahan Tset dengan termostat Nose-Hoover...53
Gambar 4.15 Perubahan ζ dan ζ& terhadap waktu...54
Gambar 4.16 Grafik temperatur dengan termostat Berendsen Q = 0,1 ...57
Gambar 4.17 Grafik temperatur dengan termostat Berendsen Q = 1 ...58
Gambar 4.18 Grafik temperatur dengan termostat Berendsen Q = 10 ...58
Gambar 4.19 Temperatur pada perubahan Tset dengan termostat Berendsen ...59
Gambar 4.20 Perbandingan tekanan dan temperatur sistem...61
Gambar 4.21 Perbandingan komponen Pm dan Pf dari tekanan sistem ...62
Gambar 4.22 Perbandingan entalpi dan tekanan sistem...63
DAFTAR NOTASI
A Luas i a Percepatan molekul i E Energi total F Gaya H Hamiltonian H Entalpi K Energi kinetik B k Konstanta Boltzmann P Tekanan p MomentumL Rusuk kotak simulasi
m Massa
N Jumlah molekul
Q Parameter pengendalian temperatur c
R Radius cutoff i
R Posisi molekul i
ij
R Jarak antar molekul
T Temperatur set T Temperatur set U Energi potensial V Volume i v Kecepatan molekul i t ∆ Time step
ε Parameter kekuatan interaksi σ Parameter panjang interaksi ζ Variabel pengendali temperatur
β ≡ . .∆t
2 1ζ β
BAB 2
PENDAHULUAN
2.1 LATAR BELAKANG MASALAH
Teori-teori baru mengenai material pada skala atomik mempermudah peneliti untuk memprediksi perilaku material pada skala makroskopik dan memberikan kemampuan untuk merancang material-material baru dengan sifat-sifat tertentu yang diinginkan. Namun analisa dan rancangan material dahulu hanya dapat dilakukan dengan eksperimen berkali-kali yang memerlukan biaya yang sangat mahal dan waktu yang lama. Selain itu, ada berbagai kondisi yang sulit atau tidak bisa diimplementasikan, antara lain eksperimen pada suhu yang sangat tinggi atau tekanan yang sangat besar.
Dengan adanya kemajuan dalam ilmu komputer dan kemampuan komputasi yang jauh lebih kuat daripada dahulu, terbuka kemungkinan baru yaitu eksperimen komputer. Eksperimen komputer adalah jembatan antara teori dan eksperimen yang telah diterima sebagai salah satu metoda penelitian dan pengembangan material. Suatu eksperimen material secara fisik dapat didahului oleh eksperimen komputer untuk menentukan kondisi yang dibutuhkan atau memprediksi hasilnya. Eksperimen komputer dapat berfungsi untuk mengkomplemen eksperimen [1].
Keuntungan eksperimen komputer adalah harga yang relatif lebih murah dan mudah meskipun untuk sistem yang kompleks. Selain itu, kemampuan eksperimen komputer meningkat sejalan dengan kemajuan komputer.
Dinamika Molekul adalah teknik simulasi komputer yang mengamati pergerakan molekul-molekul yang saling berinteraksi. Teknik ini mensimulasikan molekul-molekul yang saling menarik dan mendorong dan menabrak satu sama lain. Simulasi dinamika molekul memberikan informasi statik dan dinamik pada skala atomik, seperti posisi dan kecepatan. Informasi ini lalu dapat diolah menjadi informasi pada skala makroskopis seperti tekanan, temperatur dan lain-lain.
Berbeda dengan beberapa teknik simulasi lainnya(lihat di [2]), dinamika molekul bersifat deterministik. Jika keadaan suatu materi diketahui pada waktu tertentu, maka keadaan materi tersebut pada waktu berbeda dapat ditentukan dengan sempurna.
2.2 PERUMUSAN MASALAH
Terdapat dua masalah utama saat merancang simulasi dinamika molekul. Pertama adalah pemodelan sistem yang terdiri dari model interaksi antar molekul dan interaksi antara sistem dengan lingkungan. Kedua adalah pemilihan algoritma dan metoda numerik.
Pemodelan sistem akan menentukan kebenaran hasil simulasi dari segi fisis. Namun hasil yang lebih akurat memerlukan model yang lebih kompleks dan sulit dipecahkan dan membutuhkan waktu komputasi lebih lama. Pemodelan dan metoda numerik yang semakin akurat memerlukan komputasi yang lebih mahal. Karena salah satu hambatan terbesar adalah keterbatasan kemampuan komputasi, maka suatu kompromi dibutuhkan antara ketelitian simulasi dengan kecepatan simulasi.
2.3 TUJUAN PENELITIAN Penelitian ini memiliki tujuan:
1. Memahami prinsip dinamika molekul
2. Mengembangkan simulasi dinamika molekul dengan algoritma yang tepat dan efisien
3. Melakukan uji coba simulasi dinamika molekul dengan berbagai parameter dan kondisi yang berbeda
2.4 PEMBATASAN MASALAH
Penelitian dengan simulasi dinamika molekul di laporan ini dibatasi: 1. Materi yang diamati berada dalam fasa padat sepanjang simulasi dengan
2. Simulasi dilakukan dibatasi pada energi total sistem konstan dan temperatur sistem konstan.
3. Model potensial intermolekuler yang digunakan adalah potensial Lennard-Jones [2].
4. Algoritma persamaan gerak yang digunakan adalah algoritma Verlet [2]. 5. Model interaksi sistem dengan lingkungan dibatasi pada metoda Velocity
Scaling [1], thermostat Nose-Hoover [8] dan termostat Berendsen [9].
2.5 SISTEMATIKA PEMBAHASAN
Penelitian dilakukan dengan langkah-langkah sebagai berikut:
1. Studi literatur mengenai termodinamika statistik dan simulasi dinamika molekul.
2. Studi literatur mengenai teknik dan bahasa pemrograman C++. 3. Perancangan perangkat lunak simulasi dinamika molekul. 4. Menjalankan simulasi dengan berbagai parameter.
5. Analisa hasil simulasi. 6. Penulisan laporan.
Laporan tugas akhir ini disusun dalam lima bab. Bab I Pendahuluan
Bab ini menjelaskan latar belakang, perumusan masalah, tujuan penelitian, pembatasan masalah penelitian, dan sistematika pembahasan.
Bab II Dasar teori
Bab ini menjelaskan landasan teori yang digunakan dalam penelitian ini, yaitu dasar dinamika molekul, model interaksi antar molekul, model interaksi sistem dengan lingkungan dan dasar mekanika statistik yang digunakan untuk mengolah informasi.
Bab III Perancangan
Bab ini membahas implementasi teori yang dijelaskan pada bab II ke dalam perangkat lunak. Perancangan perangkat lunak menggunakan bahasa pemrograman C++ dengan pemrograman berorientasi obyek.
Bab ini memberikan hasil uji coba simulasi dinamika molekul untuk melihat kesesuaian dengan spesifikasi yang diinginkan dan analisa hasil yang diperoleh.
Bab V Kesimpulan dan Saran
Bab ini memberikan kesimpulan penelitian dan saran-saran untuk penelitian selanjutnya.
BAB 3
DASAR TEORI
3.1 DINAMIKA MOLEKUL
Dinamika molekul mengamati molekul-molekul dalam suatu sistem
tertutup, di mana jumlah materi (molekul) dalam sistem tidak berubah. Energi
dapat keluar atau masuk sistem, tergantung dari jenis simulasi yang dilakukan. Suatu sistem adalah suatu kuantitas materi atau volume yang dipilih untuk diamati, sedangkan materi dan volume di luar sistem disebut lingkungan [3]. Pemisah antara sistem dan lingkungan disebut batas, yang secara teoritis tidak memiliki massa ataupun volume tersendiri. Sistem terbagi atas sistem tertutup, jika materi tidak dapat menembus batas, dan sistem terbuka, jika materi dapat menembus batas.
SISTEM
MOLEKUL BATAS
LINGKUNGAN
Gambar 3.1 Sistem yang diamati dalam simulasi dinamika molekul
Materi pada skala makroskopis terdiri dari molekul-molekul berjumlah sangat besar (bilangan Avogadro berorde 1023). Karena keterbatasan komputasi, maka simulasi dinamika molekul hanya dapat melakukan perhitungan untuk ratusan atau ribuan molekul. Semakin banyak molekul dalam simulasi, semakin realistis hasil yang diperoleh, tetapi biaya komputasi semakin mahal.
Tujuan pertama simulasi dinamika molekul adalah menghasilkan trajektori molekul-molekul sepanjang suatu jangka waktu terhingga. Pada setiap waktu, molekul-molekul dalam simulasi memiliki suatu posisi dan momentum tertentu untuk masing-masing sumbu. Untuk N molekul dalam ruang 3 dimensi, terdapat ruang posisi berdimensi 3N dan ruang momentum berdimensi 3N, sehingga terbentuk ruang fasa berdimensi 6N. Suatu konfigurasi posisi dan momentum molekul-molekul dapat diartikan sebagai koordinat dalam ruang fasa tersebut.
Menurut Boltzmann [2], dengan jangka waktu yang cukup, suatu sistem akan pernah memiliki semua konfigurasi yang mungkin terjadi. Dengan kata lain, sistem tersebut akan pernah berada pada setiap koordinat dalam ruang fasa.
Ruang fasa ini merupakan informasi keadaan mikroskopis sistem. Informasi ini lalu digunakan untuk memperoleh keadaan makroskopis sistem dan memprediksi ruang fasa pada waktu selanjutnya. Ruang fasa yang berbeda dapat menghasilkan keadaan makroskopis yang sama. Dengan kata lain, suatu keadaan makroskopis belum pasti berasal dari satu keadaan mikroskopis yang unik.
3.1.1 Ensemble
Suatu ensemble adalah koleksi dari keadaan sistem yang mungkin yang memiliki keadaan mikroskopis berbeda tetapi memiliki keadaan makroskopis sama [4]. Contohnya adalah sistem dengan konfigurasi posisi atau momentum yang berbeda namun memiliki temperatur yang sama.
Beberapa ensemble yang sering digunakan dalam dinamika molekul adalah ensemble mikrokanonikal, ensemble kanonikal dan ensemble isobarik-isotermal.
3.1.1.1 Ensemble Mikrokanonikal (E, V, N)
Ensemble mikrokanonikal adalah ensemble yang memiliki karakteristik jumlah molekul N dan volume yang tidak berubah serta energi total yang konstan. Ensemble ini diperoleh dari sistem yang terisolasi sehingga tidak ada interaksi sistem dengan lingkungan. Dengan demikian energi tidak dapat keluar dan masuk ke sistem dan energi total memiliki harga konstan. Ensemble ini biasa dinamakan
ensemble (E, V, N). Ensemble mikrokanonikal adalah ensemble yang paling sederhana untuk simulasi dinamika molekul, namun kurang praktis mensimulasi keadaan eksperimen dalam laboratorium. Ini disebabkan energi total sistem sulit dipertahankan konstan dalam eksperimen.
3.1.1.2 Ensemble Kanonikal (T, V, N)
Ensemble kanonikal adalah ensemble dengan keadaan makroskopis suhu yang tetap. Selain itu jumlah molekul N dan volume tidak berubah, maka dinamakan ensemble (T, V, N). Dalam laboratorium, temperatur sistem lebih mudah dikendalikan daripada energi total sistem, maka eksperimen sering dilakukan pada temperatur konstan. Ensemble kanonikal mendekati keadaan eksperimen pada temperatur konstan.
3.1.1.3 Ensemble Isobarik-Isotermal (P, T, N)
Dinamika molekul juga dapat dilakukan dengan mempertahankan tekanan dan temperatur sistem pada harga yang konstan. Tekanan dan temperatur adalah sifat makroskopis yang mudah dikendalikan dalam eksperimen. Dalam ensemble isobarik-isotermal, volume sistem dapat berubah atau menjadi suatu variabel. Jumlah molekul tidak berubah, maka ensemble ini juga dinamakan ensemble (P, T, N).
Pengembangan Model Simulasi Dinamika Molekul Pemodelan Interaksi Antar Molekul Pemodelan Interaksi Sistem-Lingkungan Pengembangan Persamaan Gerak Generasi
Trajektori Analisa Trajektori
Inisialisasi Ekuilibrasi Produksi
Pemodelan Dinamika Molekul
Gambar 3.2 Langkah-langkah simulasi dinamika molekul. [2] Dinamika molekul dilakukan dengan langkah-langkah berikut: 1. Pengembangan Model
Pengembangan model harus dilakukan sebagai persiapan simulasi dinamika molekul. Model dapat diperoleh dari teori atau eksperimen. Model interaksi antar molekul dibutuhkan jika molekul berinteraksi satu sama lain. Model interaksi antara sistem dan lingkungan dibutuhkan jika sistem tidak terisolasi. Interaksi antara sistem dan lingkungan dipergunakan untuk pengendalian temperatur sistem dan pengendalian tekananc sistem.
2. Simulasi Dinamika Molekul
Tahap simulasi dilakukan dengan penghasilan trajektori molekul-molekul dalam simulasi, lalu analisa yang dapat dilakukan bersamaan atau setelah simulasi. Generasi trajektori dibagi menjadi 3 tahap, yang akan dibahas lebih mendalam pada bab 4.1.
3.1.3 Syarat Batas Periodik
Molekul-molekul dekat batas sistem atau permukaan akan menimbulkan masalah karena memiliki molekul tetangga yang lebih sedikit daripada molekul yang berada di tengah sistem. Pada materi berskala makroskopis, molekul dekat
permukaan jauh lebih sedikit daripada molekul total sistem sehingga efek permukaan sangat kecil. Simulasi dinamika molekul sangat terpengaruh oleh efek permukaan. Ini menyebabkan informasi yang didapatkan adalah sifat materi dekat permukaan, padahal yang lebih penting diamati adalah sifat materi itu sendiri. Untuk menghindari ini, maka interaksi molekul dengan batas dihilangkan dengan menggunakan syarat batas periodik. [5]
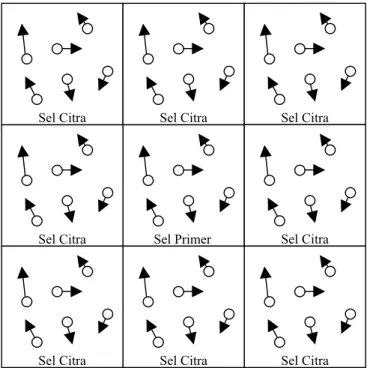
Material yang diamati akan dibagi menjadi sel-sel yang identik satu sama lain. Sel yang diamati disebut sebagai sel primer atau sel komputasi, sedangkan sel lain yang tidak diamati disebut sebagai sel citra. Di dalam sel citra terdapat citra-citra dari molekul-molekul dalam sel utama. Sel citra memiliki semua informasi (misal jumlah, posisi relatif dan kecepatan molekul) sama dengan sel primer. Sel citra adalah hasil replika dari sel primer secara periodik ke segala arah.
Sel Primer Sel Citra
Sel Citra Sel Citra Sel Citra
Sel Citra
Sel Citra Sel Citra Sel Citra
Gambar 3.3 Pembagian materi menjadi sel primer dan sel citra.
Ada dua implikasi dari syarat batas periodik. Pertama, jika sebuah molekul meninggalkan sel primer, molekul tersebut akan digantikan oleh citranya yang masuk ke dalam sel primer secara bersamaan. Posisi molekul yang keluar dari sel primer tersebut diganti dengan posisi baru yaitu posisi citranya yang masuk ke dalam sel primer. Kondisi ini akan menyebabkan jumlah atom N yang berada dalam sel primer akan konstan.
Implikasi kedua syarat batas periodik adalah konvensi citra minimum
(minimum-image convention). Molekul-molekul dalam sel utama dapat
berinteraksi baik dengan molekul-molekul lain dalam sel utama maupun dengan citra-citranya dalam sel-sel citra. Interaksi antara dua molekul hanya diperhitungkan satu kali, dengan memilih interaksi molekul pertama dengan molekul kedua yang terdekat, baik dalam sel utama atau citranya dalam sel citra [6].
3.2 MEKANIKA KLASIK
Dalam dinamika molekul digunakan ketiga Hukum Newton:
1. Suatu partikel akan tetap diam atau bergerak dengan kecepatan tetap kecuali jika menerima gaya-gaya eksternal dengan resultan tidak sama dengan nol.
2. Jika partikel dengan massa m menerima gaya Fr, maka partikel itu akan mengalami percepatan sebesar
m F a
r
r= (3.1)
3. Jika partikel i memberikan gaya pada partikel j sebesar Frij, maka partikel j
memberikan gaya pada partikel i sebesar −Frij. ji
ij F
Fr =−r (3.2)
Hukum Newton ini memberikan konsekuensi hukum kekekalan
momentum. Dalam suatu sistem terisolasi, momentum masing-masing molekul
dapat berubah-ubah akibat interaksi satu sama lain, namun momentum total tidak akan berubah. Momentum total sistem dapat diamati untuk memeriksa kebenaran simulasi ensemble mikrokanonikal.
0 ) ( ) (
∑
=∑
= i i i i i mv dt d p dt d (3.3) di mana m adalah massa molekul dan p adalah momentum molekul.3.3 MEKANIKA STATISTIK
Mekanika statistik atau termodinamika statistik dibutuhkan untuk mengkonversikan informasi pada skala atomik menjadi informasi pada skala makroskopik [5]. Konfigurasi posisi dan momentum molekul-molekul menentukan sifat-sifat yang dimiliki materi tersebut. Sifat-sifat itu antara lain adalah energi, temperatur, tekanan dan entalpi. Menurut mekanika statistik, kuantitas fisis diperoleh sebagai rata-rata konfigurasi tersebut terhadap waktu.
3.3.1 Energi Total
Energi total suatu sistem tersusun dari energi potensial sistem dan energi kinetik sistem. Energi potensial adalah jumlah dari semua energi potensial molekul-molekul dalam sistem.
) ( N i i R U U =
∑
(3.4)RN adalah set posisi titik pusat massa atom atau molekul, RN = {R1, R2, R3, … ,
RN}.
Energi kinetik sistem adalah jumlah dari energi kinetik setiap molekul.
∑
= i i i v K K ( ) (3.5) dengan i i i i i m p v m K 2 ) ( 2 1 2 2 = = .Untuk sistem terisolasi di mana tidak ada energi yang menembus batas, sistem bersifat konservatif atau energi sistem konstan. Konservasi energi ini adalah salah satu cara untuk memperiksa kebenaran simulasi ensemble mikrokanonikal.
3.3.2 Temperatur
Menurut termodinamika statistik, temperatur tidak lain adalah suatu skala dari energi kinetik molekul-molekul penyusunnya. Untuk tiga dimensi, hubungan antara energi kinetik dengan temperatur dinyatakan oleh
T Nk K B 2 3 = (3.6) Atau, B Nk K T 3 2 = (3.7)
di mana K adalah energi kinetik total sistem, N adalah jumlah molekul sistem, kB
adalah konstanta Boltzmann dan T adalah temperatur.
3.3.3 Distribusi Kecepatan
Molekul-molekul dalam materi dapat memiliki kecepatan yang berbeda-beda, sehingga terbentuk suatu distribusi kecepatan. Secara statistik dapat diperoleh bahwa molekul-molekul akan paling banyak berada pada suatu kecepatan tertentu, dan semakin berkurang jumlah molekulnya dengan semakin jauh kecepatan dari kecepatan tersebut.
Salah satu penyebabnya adalah karena molekul-molekul dalam materi akan saling bertabrakan dan berinteraksi. Interaksi ini menyebabkan adanya pemerataan energi kinetik, karena molekul yang bergerak lebih cepat memberikan tambahan momentum pada molekul yang bergerak lebih lambat dan sebaliknya. Distribusi kecepatan yang terjadi berbentuk distribusi normal, dan dinamakan Distribusi Maxwell-Boltzmann [7]. Distribusi Maxwell-Boltzmann bergantung dari temperatur, dirumuskan:
dv kT mv C dv v f − = 2 exp 1 ) ( 2 (3.8)
Gambar 3.4 Distribusi kecepatan molekul pada berbagai temperatur [2]
3.3.4 Tekanan
Tekanan didefinisikan sebagai gaya yang bekerja tegak lurus pada suatu satuan luas.
A F
P x
x = (3.9)
Dengan menggunakan hukum Newton kedua,
dt mv d A P x x ) ( 1 = (3.10)
Maka tekanan adalah suatu fluks momentum atau momentum yang menembus suatu satuan luas dalam suatu satuan waktu [2]. Menurut termodinamika statistik, ini terdiri dari dua bagian yaitu:
1. Pm, adalah fluks momentum akibat molekul yang menembus suatu
permukaan luas selama dt.
K V N Pm 3 2 = (3.11)
2. Pf, adalah fluks momentum akibat gaya yang bekerja antara dua molekul
yang berada pada sisi yang berbeda dari permukaan luas.
∑∑
⋅ = i j ij ij f F R V P r r 3 1 (3.12) Maka tekanan total menurut termodinamika statistik adalah∑∑
⋅ + = i j ij ij R F V K V N P r r 3 1 3 2 (3.13) 3.3.5 EntalpiEntalpi didefinisikan sebagai gabungan dari energi total sistem dengan energi aliran [3]:
PV E
H = + (3.14)
3.4 MODEL INTERAKSI ANTAR MOLEKUL
Model interaksi antar molekul yang diperlukan adalah hukum gaya antar molekul, yang ekivalen dengan fungsi energi potensial antar molekul. Pemilihan fungsi energi potensial harus dilakukan sebelum simulasi apa pun dapat dikerjakan.
Pemilihan model interaksi antar molekul sangat menentukan kebenaran simulasi dari sudut pandang fisika. Karena berada dalam skala atomik, interaksi secara prinsip harus diturunkan secara kuantum, di mana berlaku prinsip ketidakpastian Heisenberg. Namun kita dapat melakukan pendekatan mekanika klasik di mana atom atau molekul dianggap sebagai suatu titik massa.
Model interaksi itu harus memenuhi dua buah kriteria. Pertama, molekul-molekul harus mampu menahan tekanan pasangan molekul-molekul yang saling berinteraksi. Ini dapat diartikan bahwa ada gaya tolak-menolak antar molekul. Kedua, molekul-molekul itu harus saling mengikat, atau ada gaya tarik-menarik antara molekul. Jika molekul-molekul terlalu dekat, gaya resultan adalah gaya tolak-menolak. Sebaliknya, jika terlalu jauh, maka gaya resultan adalah gaya tarik-menarik. Pada suatu jarak tertentu, kedua gaya tersebut saling meniadakan sehingga gaya resultannya sama dengan nol.
Untuk N jumlah atom dalam suatu simulasi maka fungsi energi potensial adalah U(RN) di mana RN adalah set posisi titik pusat massa atom atau molekul,
RN = {R
1, R2, R3, … , RN}. Model energi potensial yang sederhana dan umum
digunakan adalah pair-wise, di mana potensial adalah jumlah dari interaksi antara dua molekul yang diisolasikan.
∑∑
= i j ij R U U ( ); i < j (3.15) 3.4.1 Potensial Lennard-JonesSalah satu model energi potensial antara dua molekul yang dikembangkan adalah Potensial Lennard-Jones. Model ini dianggap paling sederhana, namun memiliki ketelitian yang baik untuk simulasi. Model Potensial ini dirumuskan:
− = m ij n ij ij R R k R U( ) ε σ σ (3.16)
n dan m adalah bilangan bulat positif yang dipilih di mana n > m.
) /(n m m m n m n n k − − = dan n>m
Dan i dan j adalah indeks dari molekul, Rij ≡ Ri −Rj atau jarak antara molekul i
dan j. Sedangkan σ adalah parameter jarak, dan ε adalah parameter yang menyatakan kekuatan interaksi. Pilihan yang umum untuk m dan n adalah m=6
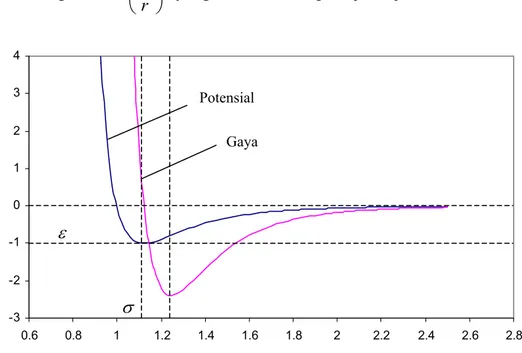
− = 6 12 4 ) ( ij ij ij R R R U ε σ σ (3.17) 3.4.2 Gaya
Gaya adalah negatif dari gradien potensial. Untuk potensial Lennard-Jones, besar gaya adalah:
− = − = 7 13 2 24 ) ( ) ( ij ij ij ij R R R U dr d R F σ σ σ ε (3.18) Menurut konvensi, gaya positif adalah gaya tolak-menolak, dan gaya
negatif adalah gaya tarik-menarik. Model ini menggambarkan adanya gaya tolak-menolak dengan suku
13 r σ
yang mendominasi pada jarak dekat dan gaya
tarik-menarik dengan suku
7 r σ
yang mendominasi pada jarak jauh.
-3 -2 -1 0 1 2 3 4 0.6 0.8 1 1.2 1.4 1.6 1.8 2 2.2 2.4 2.6 2.8 σ Potensial Gaya ε
Gambar 3.5 Potensial Lennard-Jones
Dapat diturunkan untuk masing-masing sumbu: )
(Rij U
− = ∂ ∂ − = − = ∂ ∂ − = − = ∂ ∂ − = 6 12 6 12 6 12 2 24 ) ( 2 24 ) ( 2 24 ) ( ij ij ij z ij z ij ij ij y ij y ij ij ij x ij x R R R r R U z F R R R r R U y F R R R r R U x F σ σ ε σ σ ε σ σ ε (3.19) 3.4.3 Pemotongan Potensial
Penghitungan gaya-gaya yang terjadi antar molekul adalah proses yang paling lama dalam simulasi dinamika molekul. Dalam prakteknya, sering kali potensial diberikan jarak cutoff Rc dan interaksi antar atom yang berjarak lebih
besar dari Rc diabaikan. Cutoff ini dipasang pada jarak di mana interaksi seperti
potensial dan gaya sudah kecil dan dapat diabaikan.
> ≤ − = c ij c ij ij ij ij R R R R R R R U 0 4 ) ( 6 12 σ σ ε (3.20) 3.5 PENGENDALIAN TEMPERATUR
Metoda dinamika molekul dengan ensemble mikrokanonikal tidak selalu adalah cara terbaik untuk mendapatkan rata-rata statistik tertentu. Eksperimen laboratorium lebih sering dilakukan pada temperatur konstan (ensemble kanonikal T,V,N) daripada energi konstan (ensemble kanonikal E,V,N), karena temperatur lebih mudah dikendalikan pada skala makroskopis.
Namun, untuk ensemble kanonikal diperlukan adanya fungsi tambahan untuk menjaga temperatur. Fungsi tambahan ini merupakan interaksi sistem dengan lingkungan di sekitarnya.
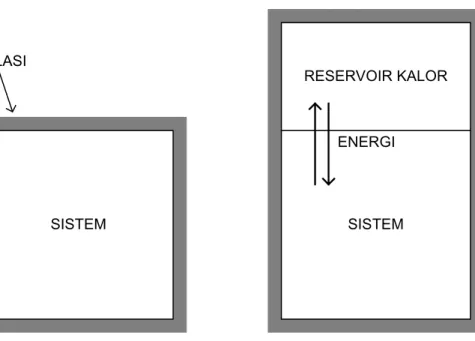
Beberapa model interaksi sistem-lingkungan yang dikembangkan melibatkan adanya heat bath atau reservoir kalor yang berinteraksi dengan sistem. Reservoir adalah sesuatu di luar sistem yang memiliki temperatur tertentu yang
tidak akan berubah meskipun menerima dari atau memberikan kalor pada sistem. Kalor berpindah dari temperatur tinggi ke temperatur rendah, maka jika reservoir lebih panas daripada sistem, kalor berpindah dari reservoir menuju sistem, dan sebaliknya.
3.5.1 Velocity Scaling
Pengendalian temperatur dapat dilakukan dengan menyesuaikan harga kecepatan masing-masing molekul terus menerus atau pada periode tertentu.
i set i v T T v′ = (3.21)
Dengan metoda ini dapat dipastikan bahwa temperatur sistem sesaat akan tepat berada pada temperatur set point. Namun teknik ini tidak menjelaskan bagaimana interaksi sistem-lingkungan bekerja. Kelemahan lain teknik ini adalah sifat-sifat materi tidak dapat diturunkan dengan benar, karena pengambilan informasi harus dilakukan pada saat sistem bebas dan tanpa paksaan.
Velocity scaling berguna dalam kecepatan awal atau jika simulasi suatu saat perlu menaikkan atau menurunkan temperatur. Cara lain adalah dengan secara periodik, namun tidak setiap saat, melakukan velocity scaling sehingga sistem lambat laun akan berkisar pada temperatur yang diinginkan. Teknik ini disebut quench method [1].
3.5.2 Termostat Nose-Hoover
Shuichi Nose [8] melakukan pengendalian temperatur dengan memasangkan reservoir kalor pada sistem. Reservoir kalor dapat memberi atau menerima energi kalor dari sistem. Perpindahan energi dari lingkungan ke sistem bersifat non-lokal, atau terjadi di semua lokasi dalam sistem secara bersamaan. Molekul-molekul dalam sistem lalu akan dipercepat jika menerima kalor atau diperlambat jika memberi kalor sehingga distribusi kecepatannya mendekati bentuk distribusi kecepatan Maxwell-Boltzmann pada temperatur yang diinginkan.
SISTEM SISTEM RESERVOIR KALOR
ENERGI ISOLASI
Gambar 3.6 Pemasangan reservoir kalor pada sistem
Dengan termostat Nose, molekul-molekul dalam sistem menerima gaya tambahan yang merupakan fungsi dari kecepatannya. Gaya ini dapat diumpamakan sebagai gaya gesek. Nose membuat suatu variabel baru s yang mengatur pertukaran kalor sistem dengan lingkungan. Hoover menyederhanakan ini dengan mendefinisikan variabel dinamis ζ :
s s&
≡
ζ (3.22)
Pengendalian temperatur ini lalu dinamakan termostat Nose-Hoover dengan persamaan gerak baru dan penentuan harga ζ sebagai berikut:
) ( ) ( }) ({ 1 ) ( t r t R R U M t r i i i & && =− ∂ ∂ −ζ (3.23)
(
)
B set i i i T Nk t r m t Q ( )=∑
( )2 −3 & & ζ (3.24)Dengan definisi temperatur dalam persamaan (2.7), persamaan dapat dituliskan
(
kBT kBTs)
Qζ&=3 − (3.25)
di mana Q adalah parameter termostat yang menunjukkan kekuatan pengendalian temperatur dan Ts adalah temperatur set-point.
Termostat Nose-Hoover mengendalikan temperatur sistem dengan mengatur (perubahan ζ& ζ terhadap waktu). Jika temperatur sistem melebihi Tset,
akan menjadi positif dan
ζ& ζ bertambah. Karena ζ berkisar di sekitar nol, maka setelah beberapa waktu ζ akan menjadi positif. Jika ζ positif maka umpan balik negatif bekerja dengan memperlambat molekul-molekul, sehingga temperatur sistem turun. Jika temperatur sistem lebih rendah daripada Tset, akan menjadi
negatif dan
ζ&
ζ berkurang. Setelah beberapa waktu, ζ menjadi bernilai negatif dan molekul-molekul dipercepat, sehingga temperatur sistem naik.
Termostat Nose-Hoover mengumpamakan perpindahan kalor memiliki ‘inersia termal’, dan seluruh sistem dengan reservoir kalor mengkonservasi nilai Hamiltonian H. Hamiltonian tidak memiliki nilai fisis namun dapat dipergunakan untuk memerika kebenaran simulasi. Hamiltonian ini dirumuskan
H M sR U
( )
{ }
R Qs gkBT s i i i ln 2 1 ) ( 2 1 2 + + 2 + =∑
& & (3.26) 3.5.3 Termostat BerendsenMetoda pengendalian temperatur lain yang memiliki banyak kesamaan dengan termostat Nose-Hoover dirumuskan oleh Berendsen et al. [9]. Termostat Berendsen menggunakan persamaan gerak yang sama dengan termostat Nose-Hoover, ) ( ) ( }) ({ 1 ) ( t r t R R U M t r i i i & && −ζ ∂ ∂ − =
Perbedaannya adalah termostat Berendsen bukan mengatur harga melainkan harga ζ& ζ dengan persamaan: ) ( 2 1 s B B B T k T k T Qk − = ζ (3.27)
di mana Q di sini adalah parameter termostat yang berupa konstanta waktu.
Metoda ini mengarahkan temperatur sistem lebih langsung daripada metoda Nose-Hoover, sehingga kurva temperatur akan lebih tajam.
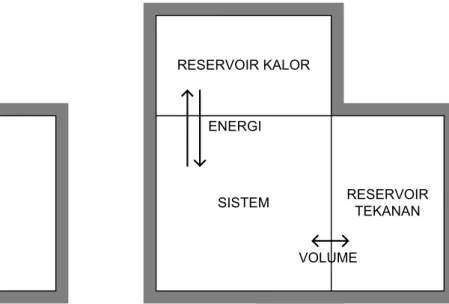
3.6 PENGENDALIAN TEKANAN
Tekanan merupakan fungsi dari temperatur dan volume sistem. Pengendalian tekanan dilakukan untuk simulasi ensemble isobarik-isotermal (P, V, N) dengan mengatur perubahan volume sistem sehingga tekanan tidak berubah. Sistem dijadikan piston yang dapat bergerak sehingga volume membesar atau mengecil. Sebuah reservoir tekanan lalu ditambahkan di luar sistem yang menggerakkan piston jika terdapat perbedaan tekanan.
SISTEM SISTEM RESERVOIR KALOR ENERGI ISOLASI RESERVOIR TEKANAN VOLUME
Gambar 3.7 Pemasangan reservoir kalor dan reservoir tekanan pada sistem
3.7 PENGEMBANGAN PERSAMAAN GERAK DISKRIT
Dinamika molekul mengamati posisi dan derivasinya, antara lain kecepatan dan percepatan. Maka persamaan gerak dapat dituliskan:
) ( ) ( ) ( ) ( t a dt t dv t v dt t dr i i i i = = (3.28)
Persamaan gerak ini harus dijadikan diskrit agar dapat dipecahkan secara numerik. Pendiskritan ini dilakukan dengan metoda beda hingga (finite-difference). Waktu yang diskrit memiliki langkah waktu (time step) yang merupakan selisih antara dua waktu berturutan.
t
∆
Metoda beda hingga dilakukan dengan melakukan ekspansi Taylor. Suatu metoda beda hingga yang mengaproksimasi solusi sebuah persamaan diferensial,
akan selalu menimbulkan truncation error (error pemotongan). Truncation error dihitung dari suku bukan nol pertama yang dihilangkan dari suatu ekspansi.
Metoda yang dipilih harus memberikan error kecil namun tidak terlalu kompleks sehingga memerlukan waktu komputasi yang lama. Beberapa metoda integrasi persamaan gerak antara lain adalah algoritma Verlet, algoritma Leap-Frog dan algoritma Velocity Form [9].
3.7.1 Algoritma Verlet
Algoritma Verlet sering digunakan karena algoritmanya yang sederhana namun memiliki ketelitian yang baik. Caranya adalah dengan menggunakan ekspansi Taylor untuk t+∆t dan t−∆tsebagai berikut:
) ) (( ) ( ) ( 6 1 ) ( ) ( 2 1 ) ( ) ( ) ( 3 4 3 3 2 2 2 t O t dt t r d t dt t r d t dt t dr t r t t r i i i i i +∆ = + ∆ + ∆ + ∆ + ∆ (3.29) ) ) (( ) ( ) ( 6 1 ) ( ) ( 2 1 ) ( ) ( ) ( 3 4 3 3 2 2 2 t O t dt t r d t dt t r d t dt t dr t r t t r i i i i i −∆ = − ∆ + ∆ − ∆ + ∆ (3.30)
Penjumlahan persamaan (2.28) dan (2.29) menghasilkan: ) ) (( ) ( ) ( ) ( 2 ) ( ) ( 2 4 2 2 t O t dt t r d t r t t r t t r i i i i +∆ + −∆ = + ∆ + ∆ ) ) (( ) ( ) ( ) ( ) ( 2 ) ( 2 4 2 2 t O t dt t r d t t r t r t t r i i i i +∆ = + −∆ + ∆ + ∆ atau ) ) (( ) ).( ( ) ( ) ( 2 ) (t t r t r t t a t t 2 O t 4 ri +∆ = i + i −∆ + i ∆ + ∆ (3.31) ) ) (( ) .( ) ( ) ( ) ( 2 ) ( t 2 O t 4 m t F t t r t r t t r i i i i i +∆ = + −∆ + ∆ + ∆ (3.32)
Maka diperoleh posisi molekul pada t+dt dengan truncation error berorde (∆t)4. Sedangkan kecepatan pada t diperoleh dengan mengurangkan persamaan (2.29) dengan persamaan (2.30), menjadi
) ) (( ) ( 2 ) ( ) ( t O t 3 dt t dr t t r t t r i i i +∆ − −∆ = ∆ + ∆ (3.33)
yang dapat dituliskan
t t O t t r t t r dt t dr t v i i i i ∆ ∆ + ∆ − − ∆ + = = 2 ) ) (( ) ( ) ( ) ( ) ( 3 (3.34)
Kelemahan dari algoritma Verlet adalah penanganan kecepatan yang kurang praktis, karena harus memprediksi posisi berikut sebelum dapat menghitung kecepatan sesaat. Selain itu, posisi sama sekali tidak ditentukan oleh kecepatan pada saat t, maka algoritma ini tidak mudah mempergunakan velocity scaling untuk simulasi pada T konstan.
3.7.2 Algoritma Leap-Frog
Algoritma Leap-Frog adalah metoda di mana kecepatan melompati percepatan, dan posisi melompati kecepatan. Menggunakan ekspansi Taylor sampai orde kedua untuk t+21∆t dan t− ∆t
2 1 : ) ) (( ) ( ) ( 2 1 ) ( ) ( ) ( 2 3 2 1 2 2 2 1 2 1 t O t dt t r d t dt t dr t r t t r i i i i + ∆ = + ∆ + ∆ + ∆ (3.35) ) ) (( ) ( ) ( 2 1 ) ( ) ( ) ( 2 3 2 1 2 2 2 1 2 1 t O t dt t r d t dt t dr t r t t r i i i i − ∆ = − ∆ + ∆ + ∆ (3.36)
Dengan cara yang sama dengan penghitungan kecepatan Verlet yaitu pengurangan (2.35) dengan (2.36) diperoleh
) ) (( ) ( ) ( 2 ) ( ) ( 3 2 1 2 1 2 1 t O t dt t dr t t r t t r i i i + ∆ − − ∆ = ∆ + ∆ ) ) (( ) ( ) ( ) ( 3 2 1 2 1 t O t dt t dr t t r t t r i i i + ∆ = − ∆ + ∆ + ∆ (3.37)
Dengan mentranslasi seluruh persamaan sebesar 21∆t, diperoleh: ) ) (( ) ( ) ( ) ( 21 t O t 3 dt t t dr t r t t r i i i ∆ + ∆ ∆ + + = ∆ + atau ) ) (( ) ( ) ( ) ( 3 2 1 t t O t t v t r t t ri +∆ = i + i + ∆ ∆ + ∆ (3.38)
Dengan cara yang sama diperoleh persamaan untuk kecepatan:
) ) (( ) ( ) ( 2 ) ( ) ( 3 2 1 2 1 2 1 t O t dt t dv t t v t t v i i i + ∆ − − ∆ = ∆ + ∆ ) ) (( ) ( ) ( ) ( 3 2 1 2 1 t O t dt t dv t t v t t v i i i + ∆ = − ∆ + ∆ + ∆ (3.39)
atau ) ) (( . ) ( ) ( ) ( 3 2 1 2 1 t O t m t F t t v t t v i i i i + ∆ = − ∆ + ∆ + ∆ (3.40)
3.7.3 Algoritma Verlet dengan Pengendalian Temperatur
Algoritma Verlet dapat digunakan dengan pengendalian temperatur dengan modifikasi dengan menggabungkan persamaan (2.23), persamaan (2.32) dan persamaan (2.34): ) ( ) ( }) ({ 1 ) ( t r t R R U M t r i i i & && =− ∂ ∂ −ζ ) ) (( ) ).( ( ) ( ) ( 2 ) (t t r t r t t a t t 2 O t 4 ri +∆ = i + i −∆ + i ∆ + ∆ t t O t t r t t r dt t dr t v i i i i ∆ ∆ + ∆ − − ∆ + = = 2 ) ) (( ) ( ) ( ) ( ) ( 3
maka persamaan gerak menjadi
) ) (( ) .( ) ( ) ( }) ({ 1 ) ( ) ( 2 ) ( t r t t 2 O t 4 R R U M t t r t r t t r i i i i i ∆ + ∆ − ∂ ∂ − ∆ − + = ∆ + ζ & (3.41) ) ) (( ) ( . 2 ) ) (( ) ( ) ( ) ( }) ({ 1 ) ( ) ( 2 ) ( 4 2 3 t O t t t O t t r t t r t R R U M t t r t r t t r i i i i i i ∆ + ∆ ∆ ∆ + ∆ − − ∆ + − ∂ ∂ − ∆ − + = ∆ + ζ ) ) (( ) .( }) ({ 1 ) .( 2 ) ( ) ( ) ( ) ( ) ( 2 ) ( 3 2 2 t O t R R U M t t t t r t t r t t t r t r t t r i i i i i i ∆ + ∆ ∂ ∂ − ∆ ∆ ∆ − − ∆ + − ∆ − + = ∆ + ζ Didefinisikan t t t ≡ ( )∆ 2 1 ) ( ζ β (3.42)
sehingga persamaan (2.41) menjadi
) ) (( ) .( }) ({ 1 )) ( ) ( ( ) ( ) ( ) ( 2 ) ( 3 2 t O t R R U M t t r t t r t t t r t r t t r i i i i i i ∆ + ∆ ∂ ∂ − ∆ − − ∆ + − ∆ − + = ∆ + β (3.43)
) ) (( ) .( }) ({ 1 ) 1 )( ( ) ( 2 ) 1 )( ( t 2 O t 3 R R U M t t r t r t t r i i i i ∂ ∆ + ∆ ∂ − − ∆ − + = + ∆ + β β ∆ + ∆ ∂ ∂ − − ∆ − + + = ∆ + 2 ( ) ( )(1 ) 1 ({ }).( ) (( ) ) 1 1 ) ( t 2 O t 3 R R U M t t r t r t t r i i i i β β (3.44)
3.7.4 Pendiskritan ζ untuk Termostat Nose-Hoover dan Berendsen
Dengan melakukan ekspansi Taylor untuk ζ pada t+∆t dan t , maka diperoleh persamaan-persamaan: t ∆ − ) ) (( ) ).( ( 2 1 ). ( ) ( ) (t+∆t =ζ t +ζ t ∆t+ ζ t ∆t 2 +O ∆t 3 ζ & && (3.45) ) ) (( ) ).( ( 2 1 ). ( ) ( ) (t−∆t =ζ t −ζ t ∆t− ζ t ∆t 2 +O ∆t 3 ζ & && (3.46)
Persamaan (2.41) dikurangi persamaan (2.42) menjadi ) ) (( ). ( 2 ) ( ) (t+∆t −ζ t−∆t = ζ t ∆t+O ∆t 3 ζ & (3.47)
Dengan definisi ς&:
(
)
B s i i ir t Nk T m t Q ( )=∑
( )2 −3 & & ζ maka diperoleh(
( ))
3 (( ) ) 2 ) ( ) ( mr t 2 Nk T O t 3 Q t t t t t B s i i i + ∆ − ∆ + ∆ − = ∆ + ζ∑
& ζ (3.48)BAB 4
PERANCANGAN SIMULASI DINAMIKA
MOLEKUL
4.1 GARIS BESAR
4.1.1 Prosedur Simulasi
Simulasi dinamika molekul terdiri atas tiga tahap: inisialisasi, ekuilibrium, dan produksi. Dalam penelitian ini bagian ekuilibrium tidak dilakukan secara spesifik.
1. Tahap inisialisasi terdiri dari penentuan sistem unit, algoritma dan parameter simulasi dan inisialisasi molekul-molekul. Inisialisasi molekul melibatkan penentuan posisi awal dan kecepatan awal molekul-molekul. 2. Tahap ekuilibrium diperlukan agar keadaan awal simulasi tidak dominan
mempengaruhi analisa dari simulasi. Dalam penelitian ini bagian ekuilibrium sudah termasuk ke dalam tahap produksi.
3. Tahap produksi adalah tahap utama dalam simulasi dinamika molekul, saat memperoleh informasi dalam simulasi. Algoritma finite-difference
digunakan berulang kali sambil mengumpulkan trajektori ruang fasa. Tahap produksi harus dilakukan dengan jangka waktu yang cukup untuk mengurangi ketidakpastian statistik sampai memenuhi spesifikasi.
Inisialisasi Parameter dan
Molekul
Maju Satu Iterasi
Hitung Interaksi Antar Molekul Kalkulasi Posisi dan Kecepatan Peroleh Sifat Materi Tidak Iterasi Selesai? Ya Ulangi Simulasi Tidak Ya Pengendalian Temperatur
4.1.2 Parameter Simulasi
4.1.2.1 Kotak Simulasi dan Syarat Batas Periodik
Simulasi dinamika dilakukan dalam kubus dengan dimensi L x L x L, karena bentuk ini paling memudahkan syarat batas periodik. Maka syarat batas periodik diimplementasikan sebagai berikut:
1. Jika sebuah molekul melebihi batas kotak simulasi, maka xi =xi +αL
dengan memilih α = …,-1,0,1,… sehingga molekul masih berada dalam kotak simulasi.
Gambar 4.3 Syarat batas periodik untuk molekul yang keluar dari kotak simulasi.
2. Jarak suatu molekul ke molekul kedua ditentukan sebagai jarak terdekat di dalam sel utama atau dengan citranya. Maka jarak dua molekul pada suatu sumbu adalah Rijx =Rijx +αL dengan memilih α = …,-1,0,1,… untuk mendapatkan Rijxterkecil.
Sel Citra Sel Citra Sel Citra Sel Primer
Gambar 4.4 Syarat batas periodik untuk jarak antar molekul
4.1.2.2 Pemotongan Potensial
Penghitungan interaksi antar molekul memakan waktu paling lama dalam simulasi dinamika molekul. Agar komputasi tidak mahal, maka interaksi molekul hanya dihitung dengan tetangga terdekat saja. Teknik ini banyak digunakan dalam dinamika molekul materi dalam fasa padat. Pemotongan potensial dilakukan dengan memasang jarak cutoff di antara tetangga pertama dan tetangga kedua molekul.
Jarak cutoff dipasang di antara jarak molekul ke tetangga pertama dan tetangga keduanya. Jarak cutoff dipasang di antara tetangga pertama yang berjarak
L dan tetangga kedua yang berjarak L 2, yaitu sebesar 1,2L.
4.1.2.3 Posisi Awal
Penempatan molekul-molekul secara acak dapat menyebabkan adanya molekul-molekul yang saling menumpuk atau berjarak terlalu dekat sehingga terjadi gaya tolak-menolak yang terlalu besar. Untuk mencegah hal ini, maka
molekul-molekul disusun dalam bentuk kubus sebagai posisi awal. Struktur ini sesuai dengan struktur materi dalam fasa padat.
Panjang rusuk kubus adalah jarak di mana molekul-molekul berada pada potensial terendah. Untuk model potensial Lennard-Jones, maka jarak tersebut kalau hanya memperhitungkan tetangga terdekatnya adalah
0 ) ( ) ( 2 24 ) ( 12 6 = − = ∂ ∂ − = ij ij ij x ij x R R R r R U x F ε σ σ (4.1) 6 12 ( ) ) ( 2 0 ij ij R R σ σ − = 6 12 ( ) ) ( 2 ij ij R R σ σ = 2 ) ( 6 = σ ij R 6 2 σ = ij R (4.2) 4.1.3 Paralelisasi
Simulasi dinamika molekul memerlukan kemampuan komputasi yang besar untuk jumlah molekul banyak dan algoritma yang kompleks. Salah satu cara meningkatkan kemampuan komputasi adalah dengan menggunakan paralelisasi komputer [10].
Paralelisasi simulasi dinamika molekul dengan P prosesor terdiri dari mengambil sistem N partikel yang berinteraksi dan mendekomposisi menjadi P
sistem yang lebih kecil yang dapat diproses secara paralel. Dengan P proses bekerja simultan maka kecepatan komputasi secara ideal akan berlipat P kali dari kecepatan satu prosesor. Dalam praktek, komunikasi antar komputer dan beban yang berbeda pada masing-masing prosesor akan menambah waktu simulasi.
Dua teknik utama adalah dekomposisi molekular dan dekomposisi geometrik.
4.1.3.1 Dinamika molekul paralel dengan dekomposisi atom
Teknik ini adalah cara paling mudah untuk membagi-bagi suatu sistem. Untuk P prosesor, masing-masing prosesor menangani N/P molekul. Alokasi molekul pada satu prosesor tidak berdasar pada kondisi tertentu, seperti lokasi yang dekat. Molekul itu akan ditangani oleh prosesor tersebut sepanjang simulasi.
Untuk mengetahui suatu molekul berinteraksi dengan molekul lain yang mana, setiap prosesor memiliki daftar tetangga dari molekul yang ditanganinya. Daftar tetangga adalah daftar molekul-molekul lain yang berada dalam bola dengan radius Rc dan berpusat pada molekul. Daftar tetangga perlu disusun
kembali setiap periode tertentu.
Teknik ini mempunyai kelemahan yaitu komunikasi antar prosesor untuk mempertukarkan informasi molekulnya dapat memakan waktu banyak.
4.1.3.2 Dinamika molekul paralel dengan dekomposisi geometrik
Pada teknik ini, ruang dipartisi menjadi banyak kotak yang disusun sehingga seluruh ruang kotak simulasi terpenuhi. Kotak partisi lalu didistribusikan pada banyak prosesor sehingga kotak partisi yang berdekatan secara fisik akan berada pada prosesor yang berdekatan. Pertukaran data jarak jauh akan diminimisasikan.
Kelemahan teknik ini adalah jika molekul-molekul berpindah kotak partisi sehingga beban yang ditangani prosesor menjadi tidak seimbang. Prosesor dengan beban lebih kecil akan selesai lebih dahulu dan harus menunggu prosesor dengan beban terbesar selesai. Alternatif lain adalah dengan menyesuaikan distribusi ruang agar distribusi beban prosesor merata, namun ini menambah kompleksitas simulasi.
Dekomposisi geometrik baik untuk digunakan dalam simulasi molekul dengan perpindahan kecil sehingga molekul-molekul kurang lebih berada tetap dalam kotak partisinya.
4.2 PERANGKAT LUNAK
Simulasi dinamika molekul dirancang dengan bahasa pemrograman C++ berorientasi objek dengan Borland C++ Builder Version 4.0. Simulasi dibentuk dengan berbagai kelas yang masing-masing memiliki tugas tertentu dalam simulasi. Penggunaan kelas dalam program ini mempermudah modifikasi program andaikata perlu mempergunakan model atau algoritma lain.
4.2.1 Kandidat Kelas
Nama Kelas Type Tanggung Jawab
Simulation Process Mengatur
langkah-langkah simulasi dinamika molekul
Molekul Actor -
Parameter Process Menginisialisasi parameter simulasi
Property Process Memperoleh sifat materi
dari simulasi
Initialize Process Mengatur jalan inisialisasi
simulasi dinamika molekul
Verlet Process Menjalankan tahap
produksi dengan algoritma Verlet
LennardJones Process Menghitung interaksi
antar molekul dengan potensial LennardJones
4.2.2.1 Simulation
Atribut Data Type Keterangan
t integer Nomor time step simulasi
4.2.2.2 Molekul
Atribut Data Type Keterangan
index integer Nomor indeks molekul
dalam kotak simulasi
m double Massa molekul
R array[3] of double Posisi molekul pada sumbu x, y dan z pada t
Rnext array[3] of double Posisi molekul pada sumbu x, y dan z pada
t
t+∆
Rprev array[3] of double Posisi molekul pada sumbu x, y dan z pada
t
t−∆
v array[3] of double Kecepatan molekul pada sumbu x, y dan z pada t
F array[3] of double Gaya total yang diterima molekul pada sumbu x, y dan z
next Molecule* Pointer ke molekul
berikut dalam kotak simulasi
Atribut kelas molekul bersifat public dan akan dimanipulasi oleh kelas-kelas lain. Algoritma Verlet membutuhkan posisi pada waktu t−∆t, t, dan t+∆t, kecepatan dan percepatan, namun percepatan diggantikan dengan gaya yang dialami molekul agar lebih fleksibel untuk simulasi ensemble mikrokanonikal dan ensemble kanonikal.
Molekul-molekul dalam simulasi disusun dalam bentuk linked list (daftar terkait) di mana suatu molekul memiliki pointer (penunjuk) untuk molekul berikutnya. Pointer ini diberi nama next.
Gambar 4.5 Penggunaan linked-list untuk molekul
4.2.2.3 Parameter
Atribut Data Type Keterangan
N integer Jumlah molekul dalam
simulasi
Naxis integer Jumlah molekul
sepanjang rusuk kubus
SIG double Parameter panjang dalam
interaksi
EPS double Parameter kekuatan
interaksi
L0 double Panjang rusuk satu kubus
molekul
L double Panjang kubus simulasi
RC double Jarak cutoff
DT double Besar satu time step
T0 double Temperatur awal sistem
Kb double Konstanta Boltzmann
Q double Parameter kekuatan untuk pengendalian temperatur
Tset double Temperatur sistem yang
diinginkan
4.2.2.4 Property
Atribut Data Type Keterangan
U double Energi potensial sistem
K double Energi kinetik sistem
E double Energi total sistem
E0 double Energi total awal sistem
error double ) 0 ( ) 0 ( ) ( E E t E error= −
momentum array[3] of double Momentum total sistem
pada masing-masing sumbu
zeta double ζ
zetadot double ζ&
beta double t ∆ ≡ ζ β 2 1 s double s
sdot double s&
T double Temperatur sistem
P double Tekanan
Pf double Komponen tekanan yang
bergantung pada gaya
Pm double Komponen tekanan yang
bergantung pada momentum
H double Entalpi
Ham0 double Hamiltonian awal sistem HamError double ) 0 ( ) 0 ( ) ( H H t H HamError = − 4.2.2.5 Initialize
Kelas Initialize tidak memiliki atribut.
4.2.2.6 Verlet
Atribut Data Type Keterangan
zetaprev double ζ(t−∆t)
zetanext double ζ(t+∆t)
sprev double s(t−∆t)
snext double s(t+∆t)
4.2.2.7 LennardJones
Atribut Data Type Keterangan
Raxis array[3] of double Jarak dua molekul pada sumbu x, y dan z
Rij double Jarak dua molekul
force array[3] of double Gaya yang terjadi antar molekul i dan j atau Fij
4.2.3 Operasi Kelas 4.2.3.1 Simulation
Operasi Return Type Keterangan
Simulation() - Konstruktor kelas
dinamika molekul
Reset() void Menghapus hasil simulasi
NextStep() void Memindahkan
variabel-variabel t+∆t menjadi t
4.2.3.2 Molecule
Operasi Return Type Keterangan
Molecule() - Konstruktor kelas
4.2.3.3 Parameter
Operasi Return Type Keterangan
Parameter() - Konstruktor kelas
Set() void Memasang harga
parameter sesuai dengan harga input
4.2.3.4 Property
Operasi Return Type Keterangan
Property() - Konstruktor kelas
Run() void Menghitung sifat-sifat
materi dari simulasi
Kinetic() void Menghitung energi
kinetik sistem
Energy() void Menghitung energi total
sistem dan besar error
4.2.3.5 Initialize
Operasi Return Type Keterangan
Run() void Menjalankan inisialisasi simulasi dinamika
molekul
CreateMolecule() void Menambahkan
molekul-molekul baru ke dalam simulasi
InitialPosition() void Memasang
molekul-molekul pada posisi awal
InitialVelocity() void Memberikan kecepatan
awal pada molekul-molekul head NULL head NULL Molekul N head Molekul N Molekul N-1 NULL head ... Molekul 1 Molekul N NULL
4.2.3.6 Verlet
Operasi Return Type Keterangan
Verlet() - Konstruktor kelas
Run() void Mengatur jalan algoritma
Verlet pada setiap iterasi
RunOne() void Mengatur jalan algoritma
Verlet untuk iterasi pertama
PredictPosition() void Memprediksi posisi
molekul-molekul pada iterasi berikut
PredictPositionOne() void Memprediksi posisi
molekul-molekul setelah iterasi pertama
Velocity() void Menghitung kecepatan
molekul-molekul PredictZeta() void Memprediksi )ζ(t+∆t
PredictS() void Memprediksi s(t+∆t)
VelocityScale() void Melakukan velocity
scaling jika diperlukan
4.2.3.7 LennardJones
Operasi Return Type Keterangan
LennardJones() - Konstruktor kelas
Run() void Mengatur interaksi antar
molekul
ClearValues() void Mengosongkan harga
potenial dan gaya yang dialami molekul
p2: Molecule*) molekul pada Force(p1: Molecule*, p2:
Molecule*)
void Menghitung gaya yang
terjadi antar dua molekul Potential(p1: Molecule*,
p2: Molecule*)
void Menghitung potensial
yang terjadi antar dua molekul
PressureByForce(p1:
Molecule*, p2: Molecule*)
void Menghitung komponen
tekanan yang berupa fungsi gaya antar dua molekul
4.2.4 Hubungan Antar Kelas
+Simulation() +Run() : void +Reset() : void -NextStep() : void +t : int Simulation +LennardJones() +Run() : void -ClearValues() : void
-Distance(in p1 : Molecule*, in p2 : Molecule*) : void -Force(in p1 : Molecule*, in p2 : Molecule*) : void -Potential(in p1 : Molecule*, in p2 : Molecule*) : void -PressureByForce(in p1 : Molecule*, in p2 : Molecule*) : void -RAxis[3] : double -Rij : double -force : double LennardJones +Molecule() +index : int +m : double +R[3] : double +Rnext[3] : double +Rprev[3] : double +v[3] : double +F[3] : double +next : Molecule* Molecule +Parameter() +Set() : void +N : int +NAxis : int +SIG : double +EPS : double +L0 : double +L : double +RC : double +DT : double +T0 : double +Kb : double +TMAX : int +Q : double +Tset : double Parameter +Property() +Run() : void -Kinetic() : void -Energy() : void +U : double +K : double +E : double +E0 : double +error : double +momentum[3] : double +zeta : double +zetadot : double +beta : double +s : double +sdot : double +T : double +P : double +Pf : double +Pm : double +H : double +Ham : double +Ham0 : double +HamError : double Property +Verlet() +Run() : void +RunOne() : void -NextStep() : void -PredictPosition() : void -PredictPositionOne() : void -Velocity() : void -PredictZeta() : void -PredictS() : void -VelocityScale() : void -zetaprev : double -zetanext : double -snext : double -sprev : double Verlet +Initialize() +Run() : void -CreateMolecules() : void -InitialPosition() : void -InitialVelocity() : void Initialize * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * *
Gambar 4.7 Hubungan antar kelas
4.2.5 Interface (Antar Muka)
Tampilan perangkat lunak simulasi dinamika molekul dalam penelitian ini:
BAB 5
ANALISA
5.1 SIMULASI SISTEM MIKROKANONIKAL 5.1.1 Uji Kesalahan Energi Simulasi
Energi diamati untuk melihat kebenaran simulasi, karena dalam sistem terisolasi atau ensemble mikrokanonikal, energi tidak berubah sepanjang simulasi. Namun karena simulasi dinamika molekul menggunakan persamaan diskrit atau metode beda hingga, maka tidak terhindarkan timbulnya error.
Perubahan energi potensial akan diimbangi oleh perubahan energi kinetik sehingga energi total tidak berubah.
Gambar 5.1 Grafik perbandingan energi kinetik dan energi potensial
Kesalahan energi total dihitung dengan ) 0 ( ) 0 ( ) ( E E t E error= − (5.1)
Hasil perhitungan error dalam beberapa simulasi 512 molekul dengan berbagai besar time step ∆t:
1. ∆t= 0,005, 100000 time step
Gambar 5.2 Grafik error energi total pada ∆t= 0,005
Gambar 5.3 Grafik error energi total pada ∆t= 0,01 3. ∆t= 0,02, 25000 time step
Gambar 5.4 Grafik error energi total pada ∆t= 0,02 4. ∆t= 0,05, 10000 time step
Gambar 5.5 Grafik error energi total pada ∆t= 0,05
Time step (∆t) Orde Error
0,005 10-9
0,01 10-8
0,02 10-8
0,05 10-7
Tabel 5.1 Orde error terhadap besar time step ∆t
Dapat terlihat bahwa simulasi memiliki error lebih besar dengan yang lebih besar. Namun simulasi dengan
t
∆
t
∆ lebih kecil memerlukan lebih banyak time step untuk menjalankan simulasi dengan waktu yang sama. Dari analisa ini kita pilih ∆t=0,02 untuk simulasi lain dalam penelitian agar dapat mensimulasikan waktu lebih lama namun error masih berorde 10-8.
5.1.2 Momentum
Dalam simulasi ensemble mikrokanonikal, perubahan kecepatan dan momentum molekul hanya terjadi akibat gaya antar molekul. Karena Fij = -Fji,
maka perubahan momentum molekul i akan diimbangi molekul j, sehingga momentum total tidak berubah.
Gambar 5.6 Grafik momentum total pada sumbu x, y dan z
Terlihat momentum total pada ketiga sumbu hanya mengalami perubahan kecil (berorde 10-10) dan dapat diabaikan. Simulasi berjalan dengan baik dari
![Gambar 3.2 Langkah-langkah simulasi dinamika molekul. [2]](https://thumb-ap.123doks.com/thumbv2/123dok/2272902.2727354/18.892.170.808.121.440/gambar-langkah-langkah-simulasi-dinamika-molekul.webp)
![Gambar 3.4 Distribusi kecepatan molekul pada berbagai temperatur [2]](https://thumb-ap.123doks.com/thumbv2/123dok/2272902.2727354/23.892.190.784.146.690/gambar-distribusi-kecepatan-molekul-berbagai-temperatur.webp)
![Gambar 4.1 Penghasilan trajektori molekul. [1]](https://thumb-ap.123doks.com/thumbv2/123dok/2272902.2727354/36.892.240.683.563.993/gambar-penghasilan-trajektori-molekul.webp)
![Gambar 4.6 Penambahan molekul dalam linked-list [11]](https://thumb-ap.123doks.com/thumbv2/123dok/2272902.2727354/49.892.160.773.134.526/gambar-penambahan-molekul-dalam-linked-list.webp)
