Familial defective apolipoprotein B-100: detection and haplotype
analysis of the Arg
3500
Gln mutation in hyperlipidemic Chinese
Yen-Ni Teng
a, Ju-Pin Pan
b, Shiu-Ching Chou
b, Der-Yan Tai
c, Guey-Jen Lee-Chen
a,*
aDepartment of Biology,National Taiwan Normal Uni6ersity,88Ting-Chou Road,Section4,Taipei117,Taiwan,ROC bDepartment of Medicine,Di6ision of Cardiology,Veterans General Hospital-Taipei,National Yang-Ming Uni6ersity,School of Medicine,
Taipei112,Taiwan,ROC
cDepartment of Internal Medicine,Wei Gong Memorial Hospital,Tou Fen,Miaoli351,Taiwan,ROC Received 1 February 1999; received in revised form 12 October 1999; accepted 1 December 1999
Abstract
Familial defective apolipoprotein (apo) B-100 (FDB) is caused by R3500Q mutation of the apo B gene resulting in decreased binding of LDL to the LDL receptor. Two other apo B mutations, R3500W and R3531C, affecting binding are known to date. We screened the apo B gene segment around codon 3500 by heteroduplex analysis and single strand conformation polymorphism (SSCP) analysis in a total of 373 hyperlipidemic individuals. Two single-base mutations were detected and confirmed by DNA sequencing. One mutation, ACA3528ACG change, resulted in degenerate codon with no amino acid substitution. The other
mutation, CGG3500CAG mutation, resulted in an Arg3500Gln substitution (R3500Q). The prevalence of heterozygote in this
selected population was 0.3% (95% confidence interval, 0.01 – 1.5%) for the R3500Q mutation, and 2.4% (95% confidence interval, 1.1 – 4.5%) for the previously described R3500W mutation. The results suggest that the R3500Q mutation is not a significant factor contributing to moderate hypercholesterolemia in Chinese (P=0.027). Family studies of the R3500Q carrier revealed a further two individuals heterozygous for the mutation, both of whom were hypercholesterolemic. Analysis of the R3500Q allele using six diallelic markers and the 3%HVR marker revealed a haplotype which was the same as that reported in a Chinese American but differed from that reported in a Chinese Canadian. Our data support limited multiple recurrent origins for R3500Q in Chinese population. © 2000 Elsevier Science Ireland Ltd. All rights reserved.
Keywords:Familial defective apolipoprotein B-100; R3500Q mutation; Haplotype
www.elsevier.com/locate/atherosclerosis
1. Introduction
Familial defective apolipoprotein (apo) B-100 (FDB) is an autosomal dominant condition resulting in hyper-cholesterolemia [1,2]. It is caused by the substitution of arginine by glutamine at codon 3500 of the apo B gene (apo B R3500Q) [3]. Apo B-100, encoded by the apo B gene, is the main protein of low density lipoprotein (LDL) and is the ligand through which LDL binds to its receptor in the process of receptor-mediated endocy-tosis [4]. The mutation affects the function of apo B-100 by decreasing LDL receptor binding affinity [5].
Recently two other mutations have been identified,
each caused by a CT transition, one at nucleotide
10 707 and the other at nucleotide 10 800. The change at nucleotide 10 707 results in the substitution of arginine by tryptophan at codon 3500 (apo B R3500W) [6], and the change at nucleotide 10800 results in the substitution of arginine by cysteine at codon 3531 (apo B R3531C) [7]. In a general population of whites, R3500Q and R3531C are equally common with a fre-quency of 0.08%, whereas R3500W is mostly likely very rare [8]. R3500W causes hypercholesterolemia and LDL derived from the R3500W heterozygote is dys-functional in that it allows only poor growth of a LDL cholesterol-dependent cell line [6]. R3531C only reduces binding to LDL receptor by about 30% in heterozygote in vitro [7] and R3531C carriers in the general popula-tion are not hypercholesterolemic [8].
* Corresponding author. Tel.: +886-2-29326234; fax: + 886-2-29312904.
E-mail address:[email protected] (G.-J. Lee-Chen).
Haplotype analysis of the affected alleles, using
markers within and/or flanking of the apo B gene,
suggesting that R3500Q mutations in several Western populations were always present on an allele with the same underlying haplotype, designated as haplotype 194 [9 – 13]. On the other hand, three different haplo-types were found in one German [14] and two Chinese subjects [15,16]. Conversely, the same haplotype was associated with R3500W alleles in 10 unrelated Chinese [17,18]. That different haplotypes in subjects of Asian and Scottish descent indicates R3500W mutations arose independently in different ethnic backgrounds [6]. On different chromosomal background, R3531C alleles were also associated with different haplotypes [7].
In a previous study, we reported the detection and haplotype analysis of nine heterozygous R3500W muta-tions in 373 unrelated hyperlipidemic Chinese [18]. However, it is apparent that other functional mutations may be present. We have therefore screened this origi-nal population for the presence of any mutations by heteroduplex analysis and single strand conformation polymorphism analysis.
2. Materials and methods
2.1. Subjects and lipid measurements
Blood samples were obtained after an overnight fast from 373 hyperlipidemic patients attending the lipid clinic of Veterans General Hospital-Taipei and Wei Gong Memorial Hospital. These patients had
choles-terol (TC) concentrations \5.17 mmol/l (not desirable
values according to NCEP guidelines [19]) without ten-don xanthomas. No diagnosis of familial hypercholes-terolemia (FH) was made in these patients. Of these
patients, 258 had triglyceride (TG) values B2.27
mmol/l, and 115 had concentrations above this value.
The mean cholesterol values for Chinese men and women living in Taiwan in 1990 were 5.19 and 5.30
mmol/l, respectively [20]. TC and TG were measured by
enzymatic methods [21,22]; HDL and LDL were
deter-mined by precipitation with phosphotungstic acid/
mag-nesium chloride and heparin/sodium citrate. The lipid
profiles were analysed using reagents supplied by E. Merck (Darmstadt, FRG), according to the CHOD-PAP method.
2.2. DNA preparation and PCR amplification
DNA was isolated from leukocytes, as previously described [18]. A 345-bp fragment flanking nucleotide 10 551 – 10 895 of the apo B gene [23] was amplified by PCR using primers and conditions described by Tybjærg-Hansen et al. [24].
2.3. Mutation screening
PCR products (3 ml) were mixed with an equal
volume of 95% formamide buffer and electrophoresed
at 300 V for 24 h on a low cross-linking 28 cm×16.5
cm×1 mm polyacrylamide gel (20% polyacrylamide
(w/v), 0.2% N,N%-methylenebisacrylamide (w/v),
con-taining 30% (w/v) urea in 0.6× TBE). Gels were
stained with ethidium bromide and photographed un-der ultraviolet light. PCR products were restricted into
141- and 204-bp fragments by EcoRI and subjected to
SSCP analysis using GeneGel Excel gels as recom-mended by the manufacture (Pharmacia Biotech, Upp-sala, Sweden). Aberrant products were sequenced by the dideoxynucleotide chain termination method [25] as previously described [18]. Screening for the identified R3500Q mutation was performed using mutagenic PCR
[26] that creates a MspI restriction site in the normal
allele but not in the mutant allele. The PCR amplified products (nucleotides 10 684 – 10 895) were digested
withMspI and separated on a 10% polyacrylamide gel.
2.4. Haplotype markers
Seven markers were analyzed as described [9] to establish haplotypes at the apo B locus. These markers include a 9-bp insertion/deletion in signal peptide, five biallelic RFLP sites (ApaLI in exon 4;AluI in exon 14; XbaI andMspI sites in exon 26;EcoRI site in exon 29), and a hypervariable repeat region (HVR) downstream
of the apo B gene. Alleles were identified as + or −,
depending on whether or not the 9-bp insertion or particular restriction site was present. The HVR alleles were identified by comparison with known markers and recorded according to the nomenclature of Ludwig et al. [27].
3. Results
3.1. Heteroduplex and SSCP analyses
3.2. Mutation identification
The mutations in A145 and D163 were revealed by DNA sequencing. Individual A145 had a mutation in
codon 3528 (ACAACG), a degeneracy codon change
that did not result in substitution of threonine. By dot
blotting and allele-specific oligonucleotide hybridiza-tion, A145 was shown to be heterozygous for the mutation (data not shown). Individual D163 has
muta-tion in codon 3500 (CGGCAG), which resulted in
substitution of arginine by glutamine (R3500Q). The recurrent R3500Q mutation was confirmed by
muta-genic PCR and MspI digestion (Fig. 2).
3.3. Family studies
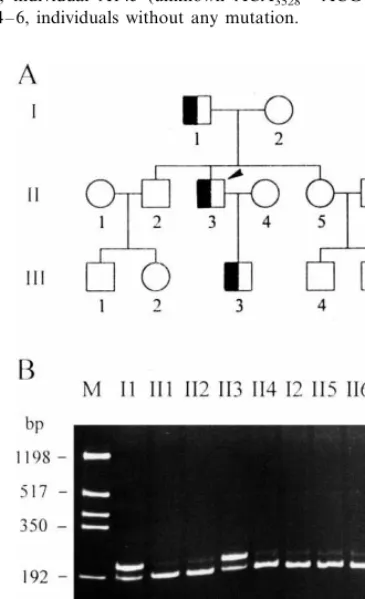
A total of 12 family members of the R3500Q carrier gave consent to be screened for the mutation. As shown in Fig. 2, the proband (II-3) inherited the mutation from his father (I-1) and transmitted the mutation to his son (III-3). None of his siblings (II-2 and II-5) carry the mutation. The clinical and biochemical data for this FDB family are shown in Table 1. All of the FDB subjects in this family were hypercholesterolemic but none of them had evidence for atherosclerotic disease. The abnormal high TG value in the proband is due to his lifestyle as a businessman and inappropriate diet.
3.4. Haplotype analysis on the R3500Q allele
The genotypes at seven polymorphic loci were exam-ined in this FDB family (Table 2). A total of 26 chromosomes yielded three independent haplotypes. The R3500Q allele was associated with the haplotype B: SP+ /ApaLI+ /AluI− /XbaI− /MspI+ /EcoRI+ /30
3%HVR repeats in the family. This haplotype was
desig-nated as 195 according to the nomenclature of Ludwig and McCarthy [9] and was identical to that reported in association with the R3500Q allele in a Chinese Ameri-can [15].
4. Discussion
In this report, a total of 373 unrelated hyperlipidemic individuals were screened by heteroduplex analysis and SSCP analysis for the presence of apo B-100 mutations. The heteroduplex method used in this study appeared able to differentiate among R3500W and R3500Q types. Distinct SSCP band patterns were observed only after performing analysis at both 8 and 15°C (data not shown).
Studies of the prevalence of FDB among hyperc-holesterolemic Caucasian populations range between 1 and 6% [2]. In our study, 10 (one R3500Q and nine R3500W) of the 373 hypercholesterolemic subjects screened were positive. Our data suggest the prevalence of FDB among the Chinese subjects with hypercholes-terolemia is 2.7%. With one R3500Q and nine R3500W detected in our selected population, the results suggest that the R3500Q mutation is not as significant a factor contributing to moderate hypercholesterolemia in Chi-nese as it is in Caucasians.
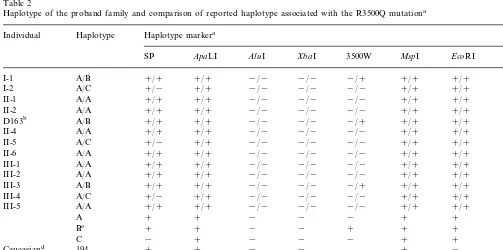
Fig. 1. Heteroduplex analysis of the region spanning codon 3500 of the apo B gene. Lane 1, individual heterozygous for known apo B R3500W; lane 2, individual D163 (unknown R3500Q heterozygote); lane 3, individual A145 (unknown ACA3528ACG heterozygote); lanes 4 – 6, individuals without any mutation.
Table 1
Lipid characteristics of the proband and immediate family
Age (years) R3500Q Serum lipid levels (mmol/l)a
Sex Weight (kg)
aTC, total cholesterol; LDL, LDL cholesterol; HDL, HDL cholesterol; TG, triglyceride. No tendon xanthomas was observed in the proband and immediate family.
bThe proband (individual D163 and II-3 in pedigree).
Table 2
Haplotype of the proband family and comparison of reported haplotype associated with the R3500Q mutationa
Individual Haplotype Haplotype markera
SP ApaLI AluI XbaI 3500W MspI EcoRI 3%HVR
a+and−, presence and absence of 9-bp insertion or restricted site; 3%HVR, number of 15 bp repeat. bThe proband (individual D163 and II-3 in pedigree).
cHaplotype associated with the mutation in this study. dThe common haplotype described in Caucasians [9]. eThe haplotype described in a German patient [14]. fThe haplotype described in a Chinese American [15]. gThe haplotype described in a Chinese Canadian [16].
In the proband and immediate family, clear co-segre-gation of hypercholesterolemia with the R3500Q muta-tion was observed. The cholesterol concentramuta-tions of the R3500Q carriers in the family were moderately
increased (6.12 – 7.08 mmol/l), similar to those reported
(6.43 – 7.29 mmol/l) for Chinese R3500Q carriers
[15,16]. The serum cholesterol concentration of 6.679
het-erozygous for R3500Q ([15,16] and this study) and
6.7791.27 mmol/l in the 15 Chinese R3500W carriers
[17,18] were not significantly different (P=0.727).
Whereas significantly lower cholesterol concentrations were observed in Chinese R3500Q heterozygotes
(6.6790.50 mmol/l, n=6) than in the Caucasian
R3500Q carriers (8.391.9 mmol/l, n=238; PB
0.001) [28]. The milder phenotype of FDB in Chinese is not due to any effect of this mutation, but may be due to differences in dietary fat consumption since Chinese on average have lower cholesterol levels than Caucasians. A traditional low-fat diet modulating the phenotype of Chinese patients with heterozygous fa-milial hypercholesterolemia was also reported [29].
In the kindred, the R3500Q mutation was
unam-biguously assigned to the haplotype SP+ /ApaLI+ /
AluI− /XbaI− /MspI+ /EcoRI+ /30 3%HVR repeats. Comparison of this haplotype with those reported [9,14 – 16] strongly suggests that the R3500Q mutation in the Chinese individual in the study of Bersot et al. [15] and our index case is identical by descent. As the Chinese individual and proband’s father were both natives of China, it is therefore probable that the R3500Q mutation arose in a common ancestor of Chinese origin many years ago.
Haplotype markers are ancient and predate human
racial divergence. Little or no recombination in the 3%
region of the apo B gene was observed [30]. The R3500Q alleles associated with two different haplo-types in the index case of Abdel-Wareth et al. [16] and ours are likely to be recurrent mutation events like that of the factor VIII gene [31]. The hyper-mutable CpG dinucleotide is frequently associated with point mutations of various genes [32].
In summary, we identified an apo B-100 R3500Q heterozygote in hyperlipidemic Chinese by the het-eroduplex method as well as SSCP analysis. Compari-son of nine R3500W carriers detected previously suggests that the R3500Q mutation does not appear to be a significant factor contributing to moderate hypercholesterolemia in Chinese. Together with two cases described, the presence of two different haplo-types associated with this mutation among three Chi-nese individuals supports limited multiple recurrent origins of the R3500Q mutation in the Chinese popu-lation.
Acknowledgements
We thank W.H. Tseng for helpful discussions and
comments. This work was supported by grant
NSC86-2313-B-003-001 from the National Science Council, Executive Yuan, Republic of China.
References
[1] Innerarity TL, Mahley RW, Weisgraber KH, Bersot TP, Krauss RM, Vega GL, Grundy SM, Friedl W, Davignon J, McCarthy BJ. Familial defective apolipoprotein B-100: a mutation of apolipoprotein B that causes hypercholesterolemia. J Lipid Res 1990;31:1337 – 49.
[2] Tybjærg-Hansen A, Humphries SE. Familial defective apolipo-protein B-100: a single mutation that causes hypercholestero-laemia and premature coronary artery disease. Atherosclerosis 1992;96:91 – 107.
[3] Soria LF, Ludwig EH, Clarke HRG, Vega GL, Grundy SM, McCarthy BJ. Association between a specific apolipoprotein B mutation and familial defective apolipoprotein B-100. Proc Natl Acad Sci USA 1989;86:587 – 91.
[4] Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science 1986;232:34 – 47.
[5] Innerarity TL, Weisgraber KH, Arnold KS, Mahley RW, Krauss RM, Vega GL, Grundy SM. Familial defective apolipo-protein B-100: low density lipoapolipo-proteins with abnormal receptor binding. Proc Natl Acad Sci USA 1987;84:6919 – 23.
[6] Gaffney D, Reid JM, Cameron IM, Vass K, Caslake MJ, Shepherd J, Packard CJ. Independent mutations at codon 3500 of the apolipoprotein B gene are associated with hyperlipidemia. Arterioscler Thromb Vasc Biol 1995;15:1025 – 9.
[7] Pullinger CR, Hennessy LK, Chatterton JE, Liu W, Love JA, Mendel CM, Frost PH, Malloy MJ, Schumaker VN, Kane JP. Familial ligand-defective apolipoprotein B: identification of a new mutation that decreases LDL receptor binding affinity. J Clin Invest 1995;95:1225 – 34.
[8] Tybjærg-Hansen A, Steffensen R, Meinertz H, Schnohr P, Nordestgaard BG. Association of mutations in the apolipo-protein B gene with hypercholesterolemia and the risk of is-chemic heart disease. New Engl J Med 1998;338:1577 – 84. [9] Ludwig EH, McCarthy BJ. Haplotype analysis of the human
apolipoprotein B mutation associated with familial defective apolipoprotein B100. Am J Hum Genet 1990;47:712 – 20. [10] Rauh G, Schuster H, Fischer J, Keller C, Wolfram G, Zo¨llner N.
Familial defective apolipoprotein B-100: haplotype analysis of the arginine(3500)glutamine mutation. Atherosclerosis 1991;88:219 – 26.
[11] Leren TP, Rødningen OK, Tonstad S, Røsby O, Urdal P, Ose L. Identification of the apo B-3500 mutation in the Norwegian population. Scand J Clin Lab Invest 1995;55:217 – 21.
[12] Wenham PR, Bloomfield P, Blundell G, Penney MD, Rae PWH, Walker SW. Familial defective apolipoprotein B-100: a study of patients from lipid clinics in Scotland and Wales. Ann Clin Biochem 1996;33:443 – 50.
[13] Rabes JP, Varret M, Saint-Jore B, Erlich D, Jondeau G, Krempf M, Giraudet P, Junien C, Boileau C. Familial ligand-defective apolipoprotein B-100: simultaneous detection of the ARG3500Gln and ARG3531CYS mutations in a French population. Hum Mutat 1997;10:160 – 3.
[14] Rauh G, Schuster H, Schewe CK, Stratmann G, Keller C, Wolfram G, Zo¨llner N. Independent mutation of arginine3500 glutamine associated with familial defective apolipoprotein B-100. J Lipid Res 1993;34:799 – 805.
[15] Bersot TP, Russell SJ, Thatcher SR, Pomernacki NK, Mahley RW, Weisgraber KH, Innerarity TL, Fox CS. A unique haplo-type of the apolipoprotein B-100 allele associated with familiar defective apolipoprotein B-100 in a Chinese man discovered during a study of the prevalence of this disorder. J Lipid Res 1993;34:1149 – 54.
Canadians: identification of a unique haplotype of the apolipo-protein B-100 allele. Atherosclerosis 1997;135:181 – 5.
[17] Choong ML, Koay ESC, Khoo KL, Khaw MC, Sethi SK. Denaturing gradient-gel electrophoresis screening of familial de-fective apolipoprotein B-100 in a mixed Asian cohort: two cases of arginine3500tryptophan mutation associated with a unique haplotype. Clin Chem 1997;43:916 – 23.
[18] Tai DY, Pan JP, Lee-Chen GJ. Identification and haplotype analysis of apolipoprotein B-100 Arg3500Trp mutation in hy-perlipidemic Chinese. Clin Chem 1998;44:1659 – 65.
[19] Report of the National Cholesterol Education Program Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. The Expert Panel, Arch Intern Med 1988;148:36 – 69.
[20] Pan WH, Hung YT, Shaw NS, Huang CJ, Tseng MS, Chen SG. Ecological study on diet, lifestyle, and chronic disease mortality in Taiwan. Report to National Science Council, Executive Yuan, Republic of China, 1990.
[21] Bucolo G, David H. Quantitative determination of serum triglycerides by the use of enzymes. Clin Chem 1973;19:476 – 82. [22] Allain CC, Poon LS, Chan CS, Richmond W, Fu PC. Enzymatic determination of total serum cholesterol. Clin Chem 1974;20:470 – 5.
[23] Ludwig EH, Blackhart BD, Pierotti VC, Caiati L, Fortier C, Knott T, Scott J, Mahley RW, Levy-Wilson B, McCarthy BJ. DNA sequence of the human apolipoprotein B gene. DNA 1987;6:363 – 72.
[24] Tybjærg-Hansen A, Gallagher J, Vincent J, Houlston R, Talmud P, Dunning AM, Seed M, Hamsten A, Humphries SE, Myant NB. Familial defective apolipoprotein B-100: detection in the United Kingdom and Scandinavia, and clinical characteristics of ten cases. Atherosclerosis 1990;80:235 – 42.
[25] Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-termination inhibitors. Proc Natl Acad Sci USA 1977;74:5463 – 7.
[26] Hansen PS, Rudiger N, Tybjærg-Hansen A, Faergeman O, Gregersen N. Detection of the apoB-3500 mutation (glutamine for arginine) by gene amplification and cleavage withMspI. J Lipid Res 1991;32:1229 – 33.
[27] Ludwig EH, Friedl W, McCarthy BJ. High-resolution analysis of a hypervariable region in the human apolipoprotein B gene. Am J Hum Genet 1989;45:458 – 64.
[28] Miserez AR, Keller U. Differences in the phenotypic characteris-tics of subjects with familial defective apolipoprotein B-100 and familial hypercholesterolaemia. Arterioscler Thromb Vasc Biol 1995;15:1719 – 29.
[29] Pimstone SN, Sun XM, du Souich C, Frohlich JJ, Hayden MR, Soutar AK. Phenotypic variation in heterozygous familial hyper-cholesterolemia: a comparison of Chinese patients with the same or similar mutations in the LDL receptor gene in China or Canada. Arterioscler Thromb Vasc Biol 1998;18:309 – 15. [30] Dunning AM, Renges HH, Xu CF, Peacock R, Brasseur R,
Laxer G, Tikkanen MJ, Butler R, Saha N, Hamsten A, Rosseneu M, Talmud P, Humphries SE. Two amino acid substi-tutions in apolipoprotein B are in complete allelic association with the antigen group (x/y) polymorphism: evidence for little recombination in the 3% end of the human gene. Am J Hum Genet 1992;50:208 – 21.
[31] Youssoufian H, Kazazian HH, Phillips DG, Aronis S, Tsiftis G, Brown VA, Antonarakis SE. Recurrent mutations in haemophilia A give evidence for CpG mutation hotspots. Nature 1986;324:380 – 2.
[32] Cooper DN, Youssoufian H. The CpG dinucleotide and human genetic disease (review). Hum Genet 1988;78:151 – 5.