REFERAT
DISTROFI KORNEA
Disusun oleh :
Abrilia Octafijayanti ; Nim 1102007001 Fifthya Syabrina ; Nim 1102006104
Pembimbing : Dr. Bambang Rianto, Sp.M.
Dr. Yanuar, Sp. M.
KEPANITERAAN BAGIAN ILMU PENYAKIT MATA RSUD SUBANG
UNIVERSITAS YARSI JAKARTA
KATA PENGANTAR
Assalamualaikum. Wr. Wb.
Alhamdulillahirabbil’alamin segala puji bagi Allah SWT atas segala rahmat dan hidayah-Nya, shalawat serta salam atas nabi besar Muhammad SAW. Terimakasih yang sebesar besarnya kepada Dr. Yanuar atas kesediaan, waktu dan kesempatan yang diberikan sebagai pembimbing referat ini, kepada teman sesama kepaniteraan Ilmu Penyakit Mata dan para perawat yang selalu mendukung, memberi saran, motivasi, bimbingan dan kerjasama yang baik sehingga dapat terselesaikannya referat ini
Referat ini disusun untuk memenuhi tugas kepaniteraan bagian Ilmu Penyakit Mata di RSUD Subang yang merupakan salah satu dari prasyarat ujian. Referat ini membahas dan menganalisa berbagai hal mengenai “Distrofi Kornea”. Bahasan referat ini diambil dari berbagai macam sumber.
Penyusun sadar bahwa dalam penyusunan referat ini masih banyak sekali kekurangan. Oleh karena itu kritik dan saran yang membangun diharapkan, demi perbaikan laporan kasus ini.
Akhir kata dengan mengucapkan Alhamdulillah, semoga Allah SWT selalu meridhoi kita semua dan semoga referat ini bermanfaat bagi semua pihak yang terkait.
Wassalamualaikum. Wr. Wb.
Jakarta, 1 Agustus 2011
DAFTAR ISI
KATA PENGANTAR...i
BAB I. PENDAHULUAN...1
BAB II. ANATOMI KORNEA...2
BAB III. DISTROFI KORNEA...4
2.1. DISTROFI KORNEA SUPERFISIAL...4
2.1.1. EPIDEMIOLOGI...4
2.1.2. ETIOLOGI...4
2.1.3. MANIFESTASI KLINIS, ETIOLOGI, MANAJEMEN...5
2.1.3.1. DISTROFI MEESMANN...5
2.1.3.2. DISTROFI KORNEA REIS-BUCKLERS...6
2.1.3.3. DISTROFI THIEL-BEHNKE...7
2.1.3.4. DISTROFI KORNEA GELATINOUS DROP-LIKE..8
2.1.3.5. DISTROFI EPITEL KORNEA LISCH...9
2.1.3.6. DISTROFI EROSI EPITEL REKUREN...9
2.1.3.7. DISTROFI SUBEPITEL KORNEA MUSINOSA...10
2.1.4. DIAGNOSA BANDING...10
2.1.5. TERAPI...10
2.2. DISTROFI KORNEA SUPERFISIAL...11
2.2.1. MANIFESTASI KLINIS, ETIOLOGI, MANAJEMEN...11
2.2.1.1. DISTROFI KORNEA MAKULAR...11
2.2.1.2. DISTROFI KORNEA GRANULAR...12
2.2.1.3. DISTROFI KORNEA LATTICE...14
2.2.1.4. DISTROFI KORNEA SCHYNDER...15
2.2.1.5. DISTROFI KORNEA FLECK...16
2.2.1.6. DISTROFI STROMA KORNEA KONGENITAL...17
2.2.1.7. DISTROFI KORNEA AMORPHOUS POSTERIOR18 2.2.2. DIAGNOSIS BANDING...18
2.3. DISTROFI KORNEA POSTERIOR...19
2.3.1. EPIDEMIOLOGI...19
2.3.2. ETIOLOGI...19
2.3.3. MANIFESTASI KLINIS, ETIOLOGI, MANAJEMEN...20
2.3.3.1. DISTROFI KORNEA FUCHS...20
2.3.3.2.DISTROFI KORNEA POLIMORPORUS POSTERIOR...21
2.3.3.3. DISTROFI ENDOTEL KORNEA HEREDITER KONGENITAL...22
2.3.3.4. DISTROFI ENDOTEL KORNEA X-LINKED...23
2.3.4. DIAGNOSIS BANDING...23
2.3.5. KRITERIA DIAGNOSIS...24
2.3.6. KONSELING GENETIK...24
BAB I
PENDAHULUAN
Distrofi kornea mencakup sekelompok heterogen bilateral ditentukan secara genetis non-inflamasi penyakit kornea yang dibatasi untuk kornea. Secara klinis, distrofi kornea dapat dibagi menjadi tiga kelompok berdasarkan lokasi anatomi tunggal atau dominan dari kelainan. Beberapa mempengaruhi terutama epitel kornea dan membran basement atau lapisan Bowman dan stroma kornea superfisial (dystrophies kornea anterior), stroma kornea (dystrophies kornea stroma), atau membran Descemet dan endotelium kornea (dystrophies kornea posterior). Kebanyakan dystrophies kornea tidak memiliki manifestasi sistemik dan hadir dengan variabel kekeruhan kornea berbentuk dalam kornea jernih atau berawan dan mereka mempengaruhi ketajaman visual untuk derajat yang berbeda.
Dystrophies kornea mungkin memiliki autosomal dominan yang sederhana, resesif autosom atau X-linked resesif. Dystrophies kornea yang berbeda yang disebabkan oleh mutasi pada gen CHST6, KRT3, KRT12, PIP5K3, SLC4A11, TACSTD2, TGFBI, dan UBIAD1. Gen untuk dystrophies kornea lainnya telah dipetakan untuk lokus kromosom tertentu, tetapi belum diidentifikasi. Sebagai manifestasi klinis bervariasi secara luas dengan entitas yang berbeda, dystrophies kornea harus dicurigai saat transparansi kornea hilang atau kekeruhan kornea terjadi secara spontan, terutama di kedua kornea, dan terutama dengan adanya riwayat keluarga yang positif atau keturunan dari orang tua kerabat.
Diagnosis diferensial utama meliputi berbagai penyebab gammopathy monoklonal, lesitin-kolesterol-acyltransferase defisiensi, penyakit Fabry, cystinosis, defisiensi transaminase tirosin, penyakit penyimpanan lisosomal sistemik (mucopolysaccharidoses, lipidoses, mucolipidoses), dan beberapa penyakit kulit (terkait-X ichthyosis, keratosis follicularis spinolosa decalvans).
Pengelolaan dystrophies kornea bervariasi dengan penyakit tertentu. Beberapa ditangani secara medis atau dengan metode yang cukai atau mengikis jaringan kornea yang abnormal, seperti keratoplasty endotel dalam lamelar (DLEK) dan keratectomy phototherapeutic (PTK). Dystrophies kurang melemahkan atau asimtomatik lainnya tidak menjamin pengobatan.
BAB II
ANATOMI KORNEA
Kornea (Latin cornum = seperti tanduk) adalah selaput bening mata, bagian selaput mata yang tembus cahaya, merupakan lapis jaringan yang menutup bola mata sebelah depan. Kornea berbentuk elips dengan diameter 12 mm secara horizontal dan 11 mm secara vertical. Ketebalannya berkisar 1 mm pada daerah limbus dan semakin berkurang ke tengah hingga 0,52 mm. Kornea merupakan permukaan refraksi yang paling penting pada mata, dengan kekuatan 43 dioptri, dan banyak teknik bedah refraksi yang bergantung pada perubahan kelengkungan permukaan kornea. Kornea tersusun atas 5 lapisan yaitu epitel, membrane Bowman, stroma, membrane Descemet, dan endotel.
1. Epitel
Terdiri atas 5 lapis sel epitel skuamosa tidak bertanduk yang saling tumpang tindih, berbentuk kolumnar pada daerah basal dan semakin mendatar membran permukaan dengan tebal keseluruhan 50 pm. Membrane basal sering terjadi mitosis sel, dan sel muda ini terdorong ke depan menjadi lapis sel sayap dan semakin maju ke depan menjadi sel gepeng. Sel basal berikatan erat dengan sel basal di sampingya dan sel membrane di depannya melalui desmosom dan membran okluden. Ikatan ini menghambat pengaliran air, elektrolit, dan glukosa dan merupakan barrier.
2. Membran Bowman
Terletak di bawah membrane basal epitel komea, merupakan jaringan kolagen yang tersusun tidak teratur seperti stroma dan berasal dari bagian depan stroma. Lapisan ini tidak mempunyai daya regenerasi dan bila rusak akan membentuk jaringan ikat. 3. Stroma
Terdiri atas lamela yang merupakan susunan kolagen yang sejajar satu dengan lainnya, pada permukaan terlihat anyaman yang teratur sementara di bagian perifer serat kolagen ini bercabang. Pembentukan kembali serat kolagen memakan waktu lama, kadang-kadang sampai 15 bulan. Di antara lamella terdapat sel keratosit yang bertanggung jawab untuk produksi serat kolagen dan substansi dasar yakni mukopolisakarida dan glikosaminoglikan.
4. Membran Descement
Merupakan membran aselular dan merupakan batas belakang stroma komea. Lapisan ini bersifat sangat elastic dan tahan terhadap trauma dan infeksi.
5. Endotel
Berasal dari mesotelium, merupakan 1 lapis sel berbentuk heksagonal dengan besar 20-40 pm. Endotel melekat pada membrane descement melalui hemidesmosom dan zonula okluden. Tidak seperti epitel, endotel tidak dapat beregenerasi. Jika terjadi kerusakan sel endotel, sel lain yang tersisa akan menjadi datar untuk menutupi area endotel yang rusak namun hal ini sangat menurunkan fungsi sel endotel.
Kornea dipersarafi oleh banyak saraf sensoris terutama berasal dari saraf siliar longus dan saraf nasosiliar. Saraf ke V saraf siliar longus berjalan suprakoroid, masuk ke dalam stroma kornea, menembus membrane Bowman dan melepaskan selubung Schwannya. Seluruh lapis epitel dipersarafi sampai pada kedua lapis terdepan tanpa ada akhir saraf. Bulbul Krauseuntuk sensasi dingin ditemukan di daerah limbus. Daya regenerasi saraf sesudah dipotong didaerah limbus terjadi dalam waktu 3 bulan. Kejernihan dari kornea dipengaruhi oleh susunan dan keadaan hidrasi dari serat kolagen dalam stroma. Jika air masuk ke dalam serat kolagen maka akan terjadi pemisah antar sel sehingga mengurangi kejernihan kornea. Lapisan epitel dan endotel mencegah masuknya cairan ke dalam stroma dengan bekerja sebagai penghalang. Sebagai tambahan, endotel dapat mengeluarkan kelebihan air di jaringan kornea melalui proses transport aktif (pompa endotel). Kornea mendapatkan asupan nutrisi dan oksigen dari air mata, humor akuosus, dan kapiler-kapiler daerah limbus [1].
BAB III
DISTROFI KORNEA
Distrofi kornea adalah suatu kondisi bilateral simetrik dan diturunkan, yang sedikit berhubungan atau tidak ada hubungannya dengan lingkungan atau faktor sistemik. Distrofi dimulai pada awal kehidupan tetapi bisa tidak menimbulkan gejala klinis dikemudian hari. Berkembang secara progresif lambat. Distrofi kornea dapat diklasifikasikan menurut genetik, keparahan, gambaran karakteristik biokemis atau lokasi anatomis. Skema anatomik yang mengklasifikasikan distrofi tergantung pada level kornea yang terkena yaitu anterior distrofi, stromal distrofi, posterior distrofi, dan ektatik distrofi [2].
Banyak manisfestasi kornea dari penyakit sistemik mempengaruhi kejernihan kornea diakibatkan oleh penumpukan abnormal substansi metabolik di epitel, stroma atau endotel. Substansi abnormal secara tipikal menumpuk pada lisosom atau struktur intrasitoplasmik seperti lisosom sebagai penyebab defek enzim tunggal. Kebanyakan kelainan ini adalah autosomal resesif. Yang termasuk kelainan metabolik ini adalah kelainan metabolism karbohidrat, lemak, asam amino, protein, sintesa imunoglobulin, metabolisme nukleotida dan mineral [2].
2.1. Distrofi Kornea Superfisial
Kelompok distrofi kornea ini terdiri dari Meesmann dystrophy (MECD), Reis-Bücklers corneal dystrophy (RBCD), Thiel-Behnke dystrophy (TBCD), gelatinous drop-like corneal dystrophy (GDCD), Lisch epithelial corneal dystrophy (LECD), epithelial recurrent erosion dystrophy (ERED) and subepithelial mucinous corneal dystrophy (SMCD).
2.1.1. Epidemiologi
Prevalensi distrofi kornea superfisial tidak dapat diketahui dengan pasti, tetapi kelainan ini jarang terjadi dan khususnya ditemukan pada populasi yang responsif dengan mutasi gen [2].
2.1.2. Etiologi
Seperti pada distrofi kornea lainnya, distrofi kornea superfisial ditentukan secara genetik dan selalu diwariskan. Fenotip unik pada distrrofi kornea ini sebagian besar disebabkan oleh perbedaan mutasi gen yang spesifik. Dihasilkan dari mutasi gen yang
berbeda dan perbedaan fenotip mungkin dihasilkan dari perbedaan mutasi di dalam gen yang sama. Adanya mutasi dalam 4 gen (KRT3, KRT12, TGFB1, dan TACSTD2) merupakan penyebab dari kelainan genetik yang ada di kornea superfisial [2].
2.1.3. Manifestasi klinis, Histopatologi, Etiologi, Manajemen.
2.1.3.1. Meesmann dystrophy (MECD, Stocker-Holt dystrophy)
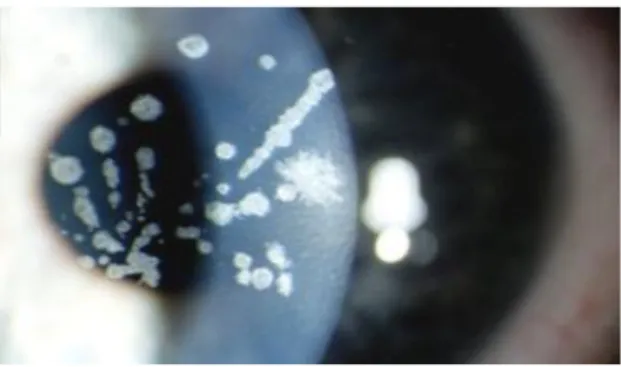
Karakteristik distrofi Meesmann ditandai adanya gelembung kecil yang jelas terlihat, pungtata dengan gambaran keruh berbentuk bulat sampai oval yang terbentuk di epitel sentral kornea dan ke tingkat yang lebih rendah arah perifer kornea pada kedua mata selama masa kanak-kanak [3].
Gambar 1 : titik titik keruh yang multipel pada epitel kornea
Kekeruhan ini disebabkan oleh kista intraepitelial yang terlihat retroiluminasi sebagai tetesan embun yang transparan dan sangat sulit untuk dilihat kecuali dengan menggunakan biomikroskop slit lamp [3].
MECD disebabkan oleh mutasi pada salah satu pasangan gen (KRT3 dan KRT12) yang mengkodekan 2 unit sitokeratin di epitel kornea. Mutasi tersebut mempunyai batasan gambaran keratin. Sebagai contoh, pada sitokeratin 12 meliputi gambaran berupa inisiasi dan terminasi helix. Secara dominan mutasi ini memberikan efek pada molekul keratin lain sehingga merusak fungsi sitoskeletal. Epitel sel yang normal akan berpindah tempat ke permukaan kornea karena desakan kista dan sel yang cacat. Distrofi kornea Stocker-Holt merupakan varian dari MECD yang disebabkan oleh perubahan ap. Arg19Leu asam amino di sitokeratin 12 [5].
MECD berlanjut seumur hidup. Terapi dengan cara pembuangan sel epitel kornea abnormal tetapi tidak bersifat kuratif. Meskipun dengan membuang sel epitel yang rusak, kelainan ini akan berulang pada epitel sel yang regenerasi [11].
2.1.3.2. Reis-Bücklers corneal dystrophy (RBCD, corneal dystrophy of Bowman layer type I, geographic corneal dystrophy, superficial granular corneal dystrophy (GCD), atypical GCD, GCD type III, anterior limiting membrane dystrophy type I)
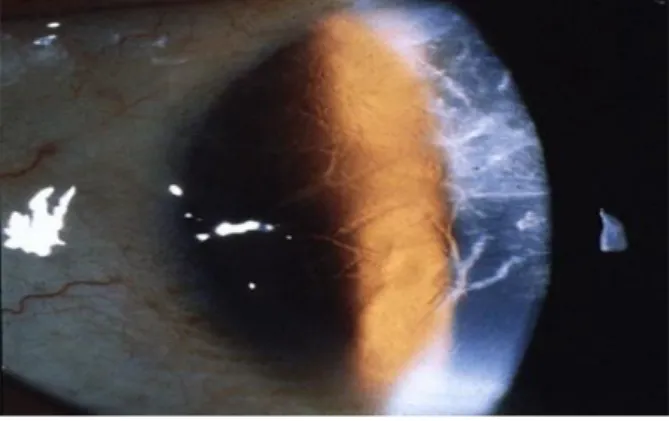
RBCD adalah kekeruhan retikuler yang simetris di kornea sentral superfisial pada kedua mata antara umur 4-5 tahun. Kelainan kornea ini pertama kali dideskripsikan oleh Reis dan dilanjutkan oleh Bucklers [7].
Gambar 3 : kekeruhan retikuler di kornea superfisial
Kekeruhan ini terlihat pola berbentuk cincin yang ireguler pada titik yang berlainan dan pada garis epitel kornea yang meninggi. RBCD tetap asimptomatik sampai adanya presipitat epitel yang erosi pada mata hiperemis, nyeri, dan fotofobia. Ketajaman penglihatan nantinya berkurang selama 2-3 dekade kehidupan karena kabut yang progresif di superfisial dan permukaan kornea yang ireguler [7].
RBCD menjadi simptomatis dan mempunyai frekuensi rekurensi yang tinggi timbulnya erosi pada pasien dengan varian lain yaitu GCD. Kornea stroma superfisial pada RBCD berisi mutasi gen yang mengubah faktor pertumbuhan beta sehingga menginduksi protein yang tidak dapat dibedakan dari pasien yang mempunyai varian GCD oleh karena itu dinamakan GCD tipe III [7].
Pada semua kasus RBCD disebabkan oleh mutasi spesifik gen TGFBI (p. Arg124 Leu). analisis haplotipe pada keluarga yang berbeda merupakan bukti awal adanya mutasi yang multipel. Mutasi lain pada gen TGFBI dilaporkan adanya dugaan mempunyai RBCD atau variasi yang berbeda (p. Gly623Asp) berdasarkan kekeruhan secara klinis di anterior sampai pertengahan stroma kornea, tetapi tanpa keterangan histopatologis. Pada beberapa kasus yang mungkin mempunyai TCBD, tidak mungkin dapat dibedakan dengan RBCD tanpa menggunakan mikroskop transmisi elektron untuk melihat defek pada kornea [7].
2.1.3.3. Thiel-Behnke dystrophy (TBCD, corneal dystrophy of Bowman layer type II, honeycomb corneal dystrophy, anterior limiting membrane dystrophy type II, curly fibers corneal dystrophy, Waardenburg-Jonker corneal dystrophy)
Distrofi kornea pada jenis ini, kekeruhan sub-epitel kornea membentuk pola sarang lebah di kornea superfisial. Sebuah zona yang jelas ada di limbus corneoscleral. Erosi kornea dimulai pada dekade pertama dan kedua dan menyebabkan ketidaknyamanan okular dan nyeri. Erosi terjadi secara berulang dan penglihatan secara bertahap menjadi terganggu [4].
TBCD merupakan kelainan dominan autosomal yang diwariskan di kromosom 5 (5q31) yang trerkait dengan mutasi Arg555Gln pada TGFBI. Heterogenitas genetik tampaknya ada dan lain lokus untuk TBD juga telah diidentifikasi pada kromosom 10 (10q23-Q24) [4].
Seperti pada banyak kelainan kornea yang lain, kelainan TBCD diwariskan dan bersifat rekuren apabila dilakukan terapi dengan metode graft, yakni mengikis kornea yang abnormal [4].
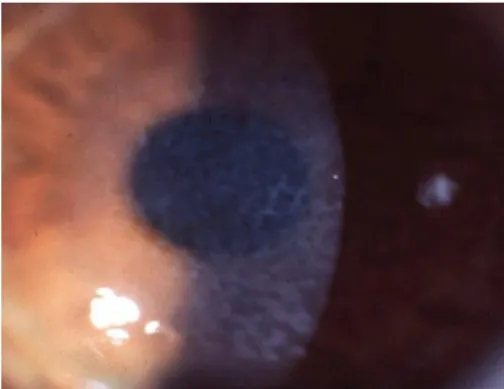
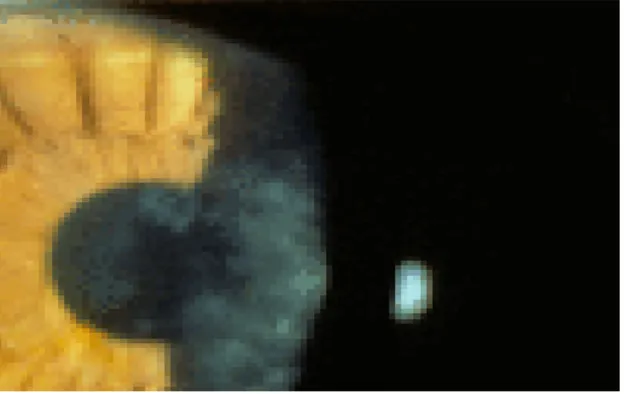
2.1.3.4. Gelatinous drop-like corneal dystrophy (GDCD, subepithelial amyloidosis, primary familial amyloidosis)
Adanya tonjolan nodul berbentuk agar agar murbei warna putih susu di bawah epitel kornea selama dekade pertama kehidupan GDCD [8]
Gambar 4. Kekeruhan pada kornea dengan nodul putih multipel. Terlihat pembuluh darah di kornea yang keruh
Gambar 5. Adanya titik amiloid yang rekuren di subepitel kornea setelah transplantasi jaringan kornea.
Keluhan lain adanya fotofobia, berair, sensasi ada benda asing di kornea dan penurunan tajam penglihatan yang progresif. Pada GDCD nodul multiple amiloid berada di jaringan subepitel di kedua kornea. Amiloid terdiri dari lactoferin tetapi pada kelainan ini tidak berhubungan dengan gen lactoferin [8].
Lebih dari 20 mutasi pada TACSTD2 (khususnya M1S1, TROP2, GA733-1) gen yang mengkode tumor menyebabkan GDCD. Mutasi p. Gln118X terdeteksi paling sering. Beberapa individu yang terkena ditemukan tidak memiliki mutasi pada TACSTD2
menunjukkan adanya heterogenitas genetik dalam penyakit resesif autosomal. Dalam GDCD, respons terhadap LPK dan PK serta keratekromi superfisial tidak memuaskan [9].
2.1.3.5. Lisch epithelial corneal dystrophy (LECD, band-shaped and whorled microcystic dystrophy of the corneal epithelium)
LECD ditandai dengan kekeruhan halus dan microkista dalam epitel kornea berbentuk pita. Penglihatan kabur tanpa rasa sakit kadang-kadang dimulai setelah enam puluh tahun kehidupan [11].
Mutasi gen untuk LECD terjadi di lengan pendek kromosom X (Xp22.3). Seperti yang diharapkan karena modus warisan itu tidak terkait dengan KRT3 dan KRT12 gen. Kekeruhan epitel secara perlahan progresif dan dapat menyebabkan kerusakan penglihatan. Debridement selular epitel kornea telah dicoba sebagai terapi, tetapi setelah pengobatan ini LECD tetap terjadi rekurensi [11].
2.1.3.6. Epithelial recurrent erosion dystrophy (ERED, recurrent hereditary corneal erosions, Dystrophia Helsinglandica, Dystrophia Smolandiensis)
Jenis distrofi kornea ditandai dengan erosi epitel yang berulang dari masa kanak-kanak dan tidak berhubungan dengan penyakit lain. Erosi mulai secara spontan atau dipicu oleh trauma ringan, debu atau asap. Kondisi ini dapat menjadi jelas pada usia 6 bulan, tetapi sebagai aturan hanya dimulai pada usia 4 sampai 6 tahun. Erosi dapat disertai dengan kabut dan kekeruhan di subepitel, karena fibrosis atau keloid seperti nodul yang berkembang. Kelainan morfologi khas belum diidentifikasi di ERED. Etiologinya tidak sepenuhnya dipahami. Gen untuk ERED masih harus dicari ke lokus kromosom tertentu [12].
ERED dapat diterapi secara medis dengan upaya terhadap penyembuhan epitel yng cacat dan melindungi epitel yang normal. Sebuah antibiotik topikal, siklopegik amat penting. Sebuah salep pelumas berguna pada malam hari. Hipertonik salin dan perban lensa kontak terapi mungkin juga memiliki peran. Dalam ERED intensitas dan frekuensi dari erosi epitel berulang cenderung berkurang dengan waktu dan biasanya berhenti pada akhir dekade keempat [13].
2.1.3.7. Subepithelial mucinous corneal dystrophy (SMCD)
Intensitas rekurensi erosi kornea yang tinggi dalam dekade pertama kehidupan mencirikan SMCD dan di publikasikan kondisi ini hanya pada orang tua yang ditemukan dengan kekeruhan subepitel dan kabut pada kornea. Mutasi gen untuk SMCD masih harus dicari dalam lokus kromosom tertentu [10].
Gambar 6. kekeruhan yang tidak teratur pada kornea superfisial
Awalnya ketika terdapat rekurensi epitel kornea, pengobatan dapat dilakukan seperti pada ERED. Seperti distrofi ini hanya dilaporkan dalam satu keluarga saja, terapi yang ideal masih belum jelas. SMCD akhirnya berkembang dari waktu ke waktu dan menyebabkan kekeruhan kornea [10].
2.1.4. Diagnosis Banding
MECD dan LECD memiliki kesamaan klinis dan perlu dibedakan satu sama lain dan ini mudah dilakukan dengan perbedaan mutasi gen yang diwariskan. TBCD memiliki kesamaan dengan RBCD, tapi mungkin memiliki perjalanan klinis yang tidak terlalu berat. Tanpa pemeriksaan jaringan atau analisis genetika molekuler TBCD umumnya misdiagnosed sebagai RBCD. ERED perlu dibedakan dari kondisi lain yang disertai dengan erosi epitel berulang, terutama bila mereka adalah manifestasi awal kehidupan, seperti membran [2].
2.1.5. Terapi
Pasien dengan kekeruhan kornea superfisial cocok diterapi dengan keratektomi superfisial, keratoplasty lamelar atau keratektomi phototherapeutic. Distrofi kornea superfisial tidak perlu dilakukan keratoplasty penetrating [2].
2.2. Stroma Kornea Dystrophies
Kelompok ini mencakup macular corneal dystrophy (MCD), granular corneal
dystrophy (GCD) type I, the lattice corneal dystrophies (LCD), Schnyder corneal dystrophy (SCD), fleck corneal dystrophy (FCD), congenital stromal corneal dystrophy (CSCD) and posterior amorphous corneal dystrophy (PACD)[2].
2.2.1. Deskripsi temuan klinis, histopatologi, etiologi, manajemen
2.2.1.2. Macular distrofi kornea (MCD, distrofi kornea Groenouw tipe II, Fehr distrofi kornea)
Gambaran samar berbentuk kabut biasanya muncul pertama kali dalam stroma kedua kornea selama masa remaja, tetapi kekeruhan dapat menjadi jelas pada masa pertumbuhan atau bahkan dalam dekade keenam [14].
Gambar 7. Kekeruhan yang samar dalam kornea.
Kekeruhan kornea bilateral secara bertahap meluas ke seluruh stroma kornea sentral dan perifer. Stroma kornea lebih tipis dari normal. Kebanyakan pasien dengan MCD tidak memiliki keratan sulfat dalam serum (MCD tipe I dan IA), tetapi beberapa tingkat keratan sulfat antigen dalam serum normal (tipe II MCD). Immunophenotypes ini tidak dapat dibedakan satu sama lain secara klinis dan tidak memiliki signifikansi klinis [14].
MCD telah diidentifikasi terjadi di seluruh dunia, tetapi jarang di sebagian besar populasi. Hal ini paling umum di India, Arab Saudi, Islandia dan bagian Amerika Serikat. Adanya mutasi gen CHST6 bertanggung jawab untuk kebanyakan kasus MCD [15].
Pada kelainan ini, penglihatan dapat dikembalikan dengan transplantasi kornea, tapi penyakit ini dapat kambuh setelah bertahun-tahun walaupun sudah di transplantasi. Karena kondisi ini mempengaruhi stroma kornea secara keseluruhan, membran Descemet dan endotel kornea maka tindakan keratoplasti tidak mencakup semua jaringan patologis [15].
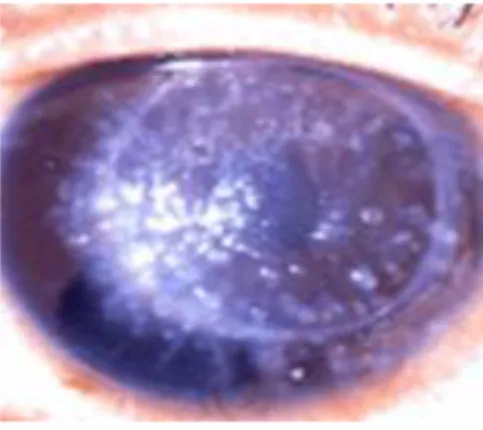
2.2.1.3.Granular corneal dystrophy (GCD) type I (classic GCD, corneal dystrophy Groenouw type I)
Berbentuk bintik-bintik kecil putih multipel yang tidak beraturan yang menyerupai serpihan roti atau salju. Gambaran tersebut terlihat jelas di daerah membran Bowman dalam stroma kornea sentral superfisial [6].
Gambar 8. Kekeruhan kornea berbentuk banyak serpihan bintik kecil yang ireguler
Gambar 9. Variasi ukuran kekeruhan dalam stroma kornea berbentuk serpihan yang menyatu sehingga terlihat seperti bintang dan memanjang.
Muncul pada dekade pertama kehidupan dan terlihat jelas pada umur 3 tahun. Awalnya terlihat seperti titik kekurahan dan dengan seiring waktu mereka secara bertahap membesar dan menjadi lebih banyak. Pada anak-anak terlihat permukaan kornea halus, namun pada orang dewasa sering tidak merata. Ketajaman penglihatan kurang lebih normal. Pada akhir dekade kedua, banyak kekeruhan terdapat di kornea sentral dan superfisial, namun jarang di stroma bagian dalam. Terdapat perbedaan klinis yang terlihat pada kekeruhan kornea antara 2 tipe GCD yaitu GCD tipe 1 dan GCD tipe 2 [16].
GCD telah dipelajari secara ekstensif di Denmark oleh Moller. GCD1 paling umum di Eropa, tetapi GCD2 lebih umum di Jepang, Korea dan Amerika Serikat. Kekeruhan kornea pada GCD mudah dilihat setelah kornea dipotong [6].
Gambar 10. Setengah potongan bedah kornea berbentuk kekeruhan putih pada kornea yang ireguler dan banyak bergabung satu sama lain.
Sebuah studi menunjukkan bahwa individu keluarga heterozigot dan homozigot untuk gen TGFBI fenotip identik, tetapi mutasi genetik telah dilakukan dalam kasus ini. Banyak mutasi TGFB1 ditemukan di fenotipe histopatologi dan klinis yang berbeda tetapi GCD1 hasil mutasi Arg555Trp, sementara GCD2 adalah efek dari mutasi Arg124His pada gen TGFBI [6].
Dalam kebanyakan kasus GCD, ketajaman visual tetap baik sampai akhir dalam perjalanan penyakit. Setelah keratoplasti, biasanya tidak terjadi kekambuhan selama kurang lebih 30 bulan, tetapi kekeruhan dapat kambuh dalam setahun [16]
2.2.1.4.Lattice corneal dystrophies (LCD) type I (Biber-Haab-Dimmer dystrophy)
Sebuah jaringan filamen bercabang kekeruhan kornea interdigitating halus dalam bentuk kelainan bawaan pada dua genetika yang berbeda [19].
Gambar 11. Sebuah garis jaringan tebal kekeruhan kornea pada pasien dengan varian LCD1 (LCD tipe III) karena mutasi homozigot Leu527Arg pada gen TGFBI
Satu tanpa manifestasi sistemik yang disebabkan oleh mutasi spesifik dalam gen TGFBI (LCD tipe I dan variannya) (LCD1), yang lain dihasilkan dari mutasi pada gen GSN (LCD tipe II) (LCD2) dan memiliki manifestasi sistemik. LCD1 biasanya menjadi jelas pada kedua mata pada akhir dekade pertama kehidupan, tapi kadang-kadang dimulai pada usia pertengahan dan jarang pada masa bayi. Berbentuk garis buram dan lainnya menumpuk terutama dalam stroma kornea sentral, sedangkan kornea perifer relatif transparan [19].
Kelainan kornea disertai dengan neuropati perifer dan kranial yang progresif, dysarthria, kulit kering dan gatalpada kulit. Karakteristik ekspresi wajah "seperti topeng", bibir menonjol dengan gerakan terganggu dan blepharochalasis juga terlihat [19].
Transplantasi kornea mungkin diperlukan dalam LCD 1 pada usia 20 tahun, tetapi biasanya tidak diindikasikan sampai setelah dekade keempat. Hasil dari PK adalah sangat baik, tetapi deposit amiloid dapat terjadi pada 2-14 tahun kemudian. Lesi kornea pada LCD2 jarang surat perintah keratoplasty menembus, tetapi ketika melakukan cacat epitel neurotropik persisten dapat berkembang [19].
2.2.1.4. Schnyder corneal dystrophy (SCD, Schnyder crystalline corneal dystrophy, crystalline stromal dystrophy, Schnyder crystalline dystrophy sine crystals, hereditary crystalline stromal dystrophy of Schnyder)
SCD biasanya menjadi jelas pada awal kehidupan dengan adanya kabut kristal di kornea atau stroma kornea. Seiring waktu, sebuah stroma kornea awal biasa-biasa saja memperoleh kekeruhan putih kecil dan menyebar kabut. SCD disebabkan oleh salah satu mutasi gen UBIAD1 [20].
Gambar 12. Kekeruhan kristal yang jelas di kornea pusat
Gambar 13. Kornea sentral berisi kristal dan cincin buram yang menonjol jelas di kornea perifer.
Ketajaman penglihatan umumnya baik dalam SCD dan secara umum setelah masa kanak-kanak. Tetapi kekeruhan kornea dapat berubah dari waktu ke waktu dan membentuk kekeruhan kornea sentral berbentuk cakram padat. Penglihatan scotopic sangat baik dan
berlanjut sampai usia pertengahan, namun mereka yang terkena dampak perlu keratoplasty penetrating sebelum dekade ketujuh [20].
2.2.1.5.Fleck corneal dystrophy (FCD, Francois-Neetens speckled corneal dystrophy)
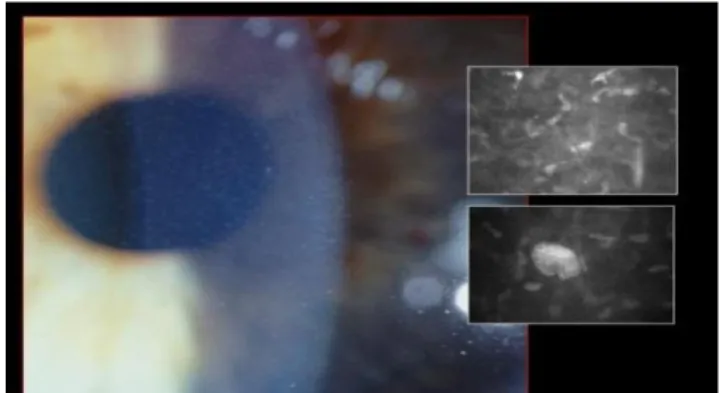
Karakteristik FCD adalah asimtomatis, kekeruhan simetris yang non progresif di seluruh stroma kornea. Salah satu jenis kekeruhannya berbentuk oval banyak dan kecil, bulat, seperti lingkaran atau setengah lingkaran dengan batas yang berbeda ("spot") di kornea pusat dan perifer [21].
Gambar 14. Penampilan kornea dengan biomicroscopy slit lamp (gambar kiri) dan mikroskop confocal (gambar kanan)
Gambar 15. Kornea berisi bintik kecil yang keruh. Penampilan kornea dengan biomicroscopy slit lamp (gambar kiri) dan mikroskop confocal (gambar kanan)
Kekeruhan lain menyerupai salju atau awan dan terdiri dari warna abu-abu kecil tanpa batas yang jelas dan terjadi terutama di tiga kornea sentral. Mereka berada di anterior dan perifer stroma, terkadang lebih padat terdapat di stroma bagian dalam dekat dengan membran Descement. FCD mempengaruhi pria dan wanita adalah sama dan telah diamati sepanjang hidup dan bahkan pada anak-anak usia 2 tahun. Epitel kornea, lapisan Bowman, dan membran Descemet normal. Sensasi kornea biasanya normal. FCD disebabkan oleh mutasi dari gen PIP5K3 [21].
FCD adalah non-progresif, tidak mempengaruhi penglihatan dan biasanya tanpa gejala dan tidak memerlukan pengobatan, tetapi fotofobia ringan telah dilaporkan. Pada satu pasien yang menjalani keratoplas penetrating, tidak ada bukti klinis rekurensi FCD pada jaringan transplantasi setelah 10 tahun follow up [21].
2.2.1.6.Congenital stromal corneal dystrophy (CSCD, congenital hereditary stromal dystrophy, Witschel dystrophy)
CSCD adalah gangguan non-progresif yang ditandai dengan mengaburkan stabil atau berbulu banyak stroma kornea buram. Serpih dan tempat menjadi lebih banyak dengan usia dan akhirnya mencegah evaluasi klinis endothelium kornea [25].
Gambar 16. Bawaan distrofi stroma. Kornea buram dalam stroma anterior dengan celah-lampu biomicroscopy.
Erosi kornea, vaskularisasi kornea dan fotofobia tidak hadir. Beberapa individu yang terkena strabismus atau primer sudut terbuka glaukoma. CSCD sangat jarang, hanya empat keluarga telah dilaporkan. CSCD satu keluarga besar dikenal memiliki keturunan di Jerman
dan Perancis. Individu dipengaruhi oleh CSCD telah dipelajari secara ekstensif dalam keluarga besar Perancis dan Norwegia [25].
2.2.1.7.Posterior amorphous corneal dystrophy (PACD, posterior amorphous stromal dystrophy)
PACD adalah gangguan kornea tidak teratur ditandai secara klinis oleh lembar-seperti "amorf" kekeruhan di dalam stroma kornea, terutama posterior, dan membran Descemet. Sesuai dengan gagasan ini bahwa ini adalah gangguan perkembangan, kelainan telah diamati pada masa bayi dan masa kanak-kanak, dan kontras dengan dystrophies kornea tradisional, non-kornea manifestasi telah dilaporkan, termasuk kelainan dari adhesi iris (iridocorneal, corectopia, dan pseudopolycoria ) [24].
Stroma kornea transparan mungkin ikut campur antara kekeruhan, yang kadang-kadang indent membran Descemet dan endotelium kornea, yang mungkin memiliki kelainan fokal. Bentuk PACD Centroperipheral dan perangkat diakui. Jenis centroperipheral meluas ke limbus corneoscleral dan disertai dengan penipisan kornea dan kelengkungan kornea adalah datar. Ketajaman visual Gangguan biasanya minimal [24].
PACD cenderung lambat progresif atau nonprogressive. Ketajaman visual biasanya minimal terganggu, tetapi dapat cukup parah untuk menjamin keratoplasty menembus [24].
2.2.2. Diagnosis Banding
Anggota keluarga yang terkena dengan LCD1 dapat mengembangkan fenotipe klinis menyerupai RBCDS. Fitur klinis Kementerian telah dilaporkan dalam keluarga dengan LCD1, tapi studi ini tidak menganalisis gen KRT3 dan KRT12 [79]. MCD harus dibedakan pada mucopolysaccharidoses sistemik (MASC) (seperti IH jenis mucopolysaccharidosis dan IS) dan mucolipidosis. Berbeda dengan deposito sistemik materi MASC abnormal antara serat-serat kolagen dalam stroma kornea di MCD. GCD harus dibedakan dari gammopathy monoklonal karena lesi histopatologi bisa sangat mirip. SCD harus dibedakan dari lemak lain dan keratopathy lethithin khusus: penyakit kolesterol acyltransferase (penyakit LCAT, penyakit Norum) dan penyakit ikan mata disebabkan oleh mutasi yang berbeda pada gen LCAT [2].
2.2.3. Manajemen Pengobatan
Karena stroma kornea dystrophies ekstensif atau sepenuhnya memperpanjang melalui stroma kornea seluruh, sebuah keratoplasty menembus atau keratoplasty lamelar mungkin pada akhirnya diperlukan bila visi menjadi gangguan signifikan. Sebagai tindakan sementara ablasi kornea dangkal dapat praktis, terutama jika jaringan donor tidak tersedia [2].
2.3. Distrofi Kornea Posterior
Kelompok dystrophies kornea posterior termasuk Fuchs corneal dystrophy (FECD),
posterior polymorphous corneal dystrophy (PPCD), congenital hereditary endothelial corneal dystrophy (CHED) and X-linked endothelial corneal dystrophy (XECD). Penyakit ini
ditandai oleh kelainan dari endotelium kornea dan selaput Descemet. Dalam sebagian besar dari mereka, merusak transportasi fluida aktif dengan endotelium kornea menyebabkan edema stroma kornea berlebihan dan merusak kejernihan kornea dan mengurangi ketajaman visual [2].
2.3.1. Epidemiologi
Prevalensi FECD berbeda di berbagai belahan dunia. Hal ini umum dan distrofi kornea yang paling umum di Amerika Serikat, mempengaruhi sekitar 4% dari populasi lebih dari 40 tahun. Hal serupa juga terjadi di negara-negara maju lainnya. FECD jauh lebih umum dan lebih parah pada wanita dibandingkan pada pria. FECD biasa di Arab Saudi dan Cina di Singapura dan FECD sangat langka di Jepang [2]
2.3.2. Etiologi
Sementara beberapa yang berbeda dystrophies kornea posterior ada hubungan antara beberapa dari mereka telah disarankan berdasarkan penelitian genetika molekuler. Heterogenitas fenotipik dan heterogenitas alelik ada. Misalnya, Gly455Lys mutasi pada gen COL8A2, yang mengkode rantai α2 kolagen tipe VIII, telah terdeteksi di kedua FECD dan PPCD. Demikian pula, mutasi pada gen yang menyebabkan tipe 3 TCF8 PPCD, telah ditemukan di salah satu dari 74 pasien Cina dengan FECD dan mutasi pada gen SLC4A11 telah terdeteksi dalam 4 individu dengan akhir-onset FECD [2].
2.3.3. Deskripsi temuan klinis, histopatologi, etiologi, manajemen
2.3.3.1.Fuchs corneal dystrophy (FECD, Fuchs endothelial corneal dystrophy, endo-epithelial corneal dystrophy, late hereditary endothelial dystrophy)
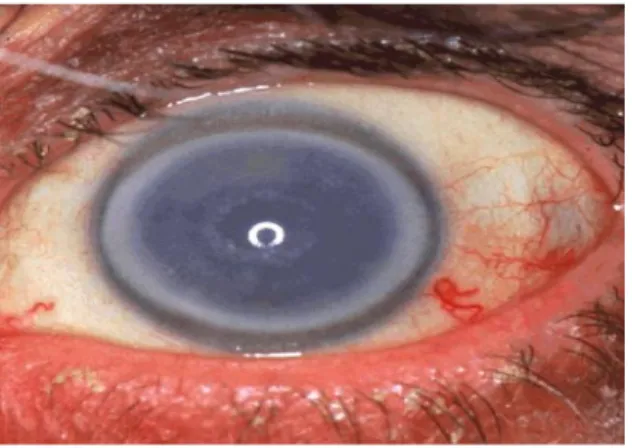
FECD merupakan kelainan endotel kornea yang progresif, menyebabkan edema kornea dan kehilangan penglihatan. Stadium awal penyakit FECD biasanya dimulai sejak dekade ke 5 sampai 7, dan ditandai dengan akumulasi dari pertumbuhan jaringan lokal yang disebut gutata, dan penebalan membran Descemet. Pada akhirnya akan terjadi kehilangan densitas dan fungsi sel endotel sebagai pompa kornea yang menyebabkan edema kornea. Walaupun gutata kornea bukan merupakan tanda khas dari FECD, perkembangan edema stroma semakin menegaskan kelainan ini. Edema kornea diikuti oleh erosi kornea berulang, dan kerusakan parah pada ketajaman visual, bahkan kebutaan pada lanjut usia [18].
Gambar 17. Sebuah kornea buram yang nyata disebabkan oleh edema yang luas karena hilangnya sel endotel normal yang menjaga hidrofilik stroma kornea.
Awalnya, edema stroma menghasilkan kabut abu-abu biru di anterior ke membran Descemet. Seluruh kornea stroma mengental, sementara membrana Descement mengerut. Ketajaman visual terus memburuk, kelainan kornea mulai terpusat, tetapi menyebar ke limbus corneoscleral. FECD umumnya terkait dengan katarak [18].
Penyebab FECD tidak diketahui, tetapi tampaknya menjadi gangguan heterogen herediter disebabkan oleh interaksi kompleks dari faktor genetik dan lingkungan. FECD
diekspresikan lebih sering pada wanita. Pada kasus yang jarang FECD telah dikaitkan dengan mutasi pada gen COL8A2 [23].
Klinis FECD biasanya mencakup 10-20 tahun dan ditandai dengan edema progresif dari jaringan stroma, dan fibrosis subepitel (keratopathy bulosa). Katarak umum pada individu dengan ekstraksi katarak dan mempercepat dekompensasi kornea FECD. Mikroba keratitis dan neovaskularisasi kornea adalah FECD komplikasi yang sangat jarang [18].
Kebanyakan pasien dengan keratoplasty penetrating FECD akhirnya membutuhkan salah satu prosedur baru untuk memperbaiki permukaan posterior kornea, seperti di keratoplasty endotel pipih (DLEK), keratoplasty Descemet pengupasan endotel (DSEK), atau Descemet pengupasan endotel keratoplasty otomatis (DSAEK) [22].
2.3.3.2.Posterior polymorphous corneal dystrophy (PPCD, posterior polymorphous dystrophy)
Bentuk umum dari distrofi kornea dapat hadir pada saat lahir (dengan kekeruhan dari kornea) atau lambat selama hidup dan ditandai oleh lesi mempengaruhi endotelium. Kebanyakan orang tidak mengembangkan gejala-gejala (asimtomatik). Efek pada kornea mungkin progresif lambat. Kedua mata biasanya terkena, tetapi satu mata mungkin lebih sangat terpengaruh dari yang lain (asimetris). Dalam kasus yang parah, individu dengan distrofi polymorphous posterior dapat mengembangkan pembengkakan (edema) dari stroma, kepekaan yang abnormal terhadap cahaya (fotofobia), penurunan penglihatan, dan perasaan (sensasi) dari bahan asing di mata. Dalam kasus yang jarang terjadi, tekanan meningkat dengan mata (tekanan intraokuler) dapat terjadi [24].
PPCD biasannya kebanyakan kasus tidak memerlukan pengobatan. Namun, mereka biasanya dilakukan keratoplasty penetrating atau memakai salah satu prosedur terapipada FECD. PPCD bisa kambuh setelah keratoplasty penetrating [24].
2.3.3.3.Congenital hereditary endothelial corneal dystrophy: congenital hereditary endothelial dystrophy type 1 (CHED1, autosomal dominant CHED), congenital hereditary endothelial dystrophy type 2 (CHED2, Maumenee corneal dystrophy, autosomal recessive CHED, infantile hereditary endothelial dystrophy)
CHED merupakan kelainan bilateralyang melibatkan degenerasi dari lapisan endotel kornea. CHED memiliki 2 bentuk,autosomal dominan (AD-CHED : CHED1) dan autosomal resesif (AR-CHED : CHED2).CHED ditandai dengan penampakan kornea yang seperti kaca dan nampak lebih tebal.CHED1 biasanya timbul pada 2 tahun pertama setelah lahir, dengan gejala fotofobia dan keluar air mata, namun tidak didapatkan adanya nistagmus. Pada CHED2, individu yang terkena lahir dengan kornea yang nampak seperti gelas kaca dan disertai dengan nistagmus [25].
Gambar 18. Stroma kornea nyata buram akibat edema sekunder untuk sel-sel endotel yang rusak
Gen bertanggung jawab untuk CHED1 terdapat di daerah pericentromeric kromosom 20 (20p11.2-q11.2) di daerah ysng overlapping dengan satu jenis gen PPCD dan ini berbeda dari lokus untuk CHED2 [116]. Kebanyakan kasus disebabkan oleh mutasi homozigot. Untuk CHED2 terjadi mutasi pada gen SLC4A11 [25].
Kekeruhan kornea CHED bilateral sangat parah, keratoplasty penetrating satu-satunya harapan untukmeningkatkan penglihatan. Perjalanan penyakit CHED1 bergerak perlahan-lahan selama 5 sampai 10 tahun, tetapi tetap seumur hidup CHED2 [25].
2.3.3.4.X-linked endothelial corneal dystrophy (XECD)
X-Linked distrofi kornea endotel bermanifestasi sebagai kabut difus mengaburkan kaca bawaan kornea kornea atau [149]. Pada pria, ini sering dikaitkan dengan penglihatan kabur. Lanjutan kasus memiliki keratopathy band yang berhubungan dengan perubahan endotel subepitel menyerupai kawah bulan. Pria lebih parah terkena dibanding wanita dan kekeruhan kornea dapat berat dan terkait dengan nistagmus. Para wanita tidak menunjukkan gejala tetapi memiliki Bulan endotel abnormal dan kawah. Dengan modus terkait warisan, laki-laki terkena menularkan penyakit ke anak perempuan mereka tapi tidak anak-anak mereka [26]
XECD telah dipetakan ke lengan panjang kromosom X (Xq25) [150]. Interval Kritis mengandung 72 gen yang kode untuk faktor transkripsi 7 putatif. Sebuah keratoplasty menembus kadang-kadang ditampilkan dalam XECD dan korupsi dapat tetap jelas selama 30 tahun. Karena begitu sedikit kasus telah dirawat, modus yang optimal terapi tidak pasti. Kursus XECD perlahan progresif dengan kabur intermiten dan keratopathy Band kornea subepitel berkembang pada masa dewasa dimulai pada kornea perifer [17].
2.3.4. Diagnosis Banding
Fuchs corneal dystrophy
Guttae kornea tidak spesifik untuk FECD dan dapat timbul sebagai bagian dari kornea penuaan atau sebagai respons terhadap keratitis interstisial. Mereka juga fitur dari MCD. Karena katarak yang umumnya terkait dengan FECD, diagnosis diferensial meliputi aphakic keratopathy bulosa dan keratopathy pseudophakic bulosa [2].
Posterior polymorphous corneal dystrophy
PPCD perlu dibedakan dari kekeruhan kornea posterior uveitis diperoleh setelah berulang dan keratitis (keratopathy polymorphous posterior). PPCD perlu dibedakan dari gangguan lain di mana endotelium kornea digantikan sebagian oleh epitel skuamosa berlapis, seperti sindrom endotel iridocorneal (ICE) dan ingrowth epitel berikut menembus luka dalam limbus corneoscleral [2].
Congenital hereditary corneal dystrophy
CHED dapat bingung dengan penyebab lain kekeruhan kornea bawaan, seperti CSCD dan anomali Peters. Seperti disebutkan di atas CHED juga perlu dibedakan dari PPCD tersebut [2].
2.3.5. Kriteria Diagnosis
Diagnosis klinis dystrophies kornea yang berbeda bervariasi dengan entitas yang berbeda, tetapi harus dicurigai bila kehilangan transparansi kornea atau kekeruhan kornea terjadi secara spontan, terutama pada kedua kornea terutama dengan adanya riwayat keluarga yang positif atau keturunan dari kerabat orang tua. Karena kemudahan memeriksa kornea, dokter biasanya dapat menentukan tingkat keterlibatan sifat anatomi dan morfologi kelainan dystrophi yang menentukan gejala yang terkait dengan semua jenis penyakit [2].
2.3.6. Konseling Genetik
individu yang terkena atau orang tua mereka biasanya dapat diberikan dengan informasi rinci tentang mereka distrofi kornea tertentu . Hal ini dapat sangat berharga dalam memberikan konseling genetik yang relevan dengan perawatan yang berbeda dan prognosis yang berbeda [2]
BAB IV
DAFTAR PUSTAKA
1. Ilyas, Sidharta. Ilmu Penyakit Mata. Edisi 3. Jakarta : Balai Penerbit FKUI : 2002. 2. Klintworth, dystrophies GK kornea. J Orphanet Langka Dis. 2009, 04:07
3. Szaflik JP, Oldak M, et al. Genetics of Meesmann corneal dystrophy. 2008, 27:S1-S42.
4. Meesmann A. Dominant Verterbte Dystrophia Epithelialis Corneae. Am J Hum Genet 1998, 63:1073-1077.
5. Klintworth GK, et al. : Identification of a new keratin K12 mutations associated with Stocker-Holt corneal dystrophy that differs from mutations found in Meesmann corneal dystrophy. Arch Ophthalmol 1955, 53:536.
6. Haddad R, et al. Unusual superficial variant of granular dystrophy of the cornea. Klin Monatsbl Augenheilkd 1949, 114:386-397.
7. Wittebol-Post D, Pels E. The dystrophy described by Reis and Bucklers. Separate entity or variant of the granular dystrophy?. Ophthalmology 1983, 90:1507-1511 8. Klintworth GK, et al. Familial subepithelial corneal amyloidosis (gelatinous drop-like
corneal dystrophy): exclusion of linkage to lactoferrin gene. Arch Ophthalmol 1980, 98:144-148.
9. Ren Z, et al. Mutations of the M1S1 gene on chromosome 1P in autosomal recessive gelatinous drop-like corneal dystrophy. Mol Vis 1998, 4:31.
10. Lisch W, et al. A new, band-shaped and whorled microcystic dystrophy of the corneal epithelium. Proc Internat Soc Eye Res 2000, 71:S108P.
11. Lisch W, et al. Lisch corneal dystrophy is genetically distinct from Meesmann corneal dystrophy and maps to Xp22.3. Am J Ophthalmol 1992, 114:35-44.
12. Hammar B, et al. A new corneal disease with recurrent erosive episodes and autosomal-dominant inheritance. Am J Ophthalmol 1994, 117:543-544.
13. Wales HJ. A family history of corneal erosions. Acta Ophthalmol (Copenh) 1967, 45:829-836.
14. Klintworth GK, et al : Macular corneal dystrophy. Lack of keratan sulfate in serum and cornea. Arch Ophthalmol 1993, 111:1106-1114.
15. Klintworth GK, et al. CHST6 mutations in North American subjects with macular corneal dystrophy: a comprehensive molecular genetic review. Mol Vis 2000, 6:261-264.
16. Klintworth GK, et al. Accumulation of β ig-h3 gene product in corneas with granular dystrophy. Acta Ophthalmol (Copenh) 1990, 68:384-389.
17. Munier FL, et al. Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Am J Hum Genet 1998, 62:320-324.
18. Akimune C, et al. Corneal guttata associated with the corneal dystrophy resulting from a betaig-h3 R124H mutation. Mol Vis 2007, 13:1390-1396
19. Holland EJ, et al. Avellino corneal dystrophy. Clinical manifestations and natural history. Cornea 1999, 18:424-429
20. Weiss JS. Visual morbidity in thirty-four families with Schnyder crystalline corneal dystrophy. Jpn J Ophthalmol 1998, 42:450-455.
21. Purcell JJ Jr, et al. Fleck corneal dystrophy. Am J Ophthalmol 1977, 83:554-60 22. Vithana EN, et al. SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Arch
Ophthalmol 1978, 96:2036-2039.
23. Biswas S, et al. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Orphanet J Rare Dis 2008, 3:28.
24. Harissi-Dagher M, et al. Keratoglobus in association with posterior polymorphous dystrophy. Albrecht von Graefes Arch Klin Exp Ophthalmol 1916, 91:363-379. 25. Toma NMG,et al. Linkage of congenital hereditary endothelial dystrophy to
chromosome 20. Cornea 2000, 19:570-573
26. Schmid E,et al. A new, X-linked endothelial corneal dystrophy. Klin Monatsbl Augenheilkd 1976, 169:717-727