To the best of my knowledge, I have always enjoyed learning and working with my hands. For the last 5.5 years or so as a graduate student, I have had the unique privilege of being able to fill my days with these two things, and while these years have without a doubt been the most challenging, frustrating and difficult of my life I have also been one of the most exciting and rewarding and as much as I am ready to move on I am truly going to miss this place. At some point I received a helpful suggestion, insightful comment, or learned something new just by quietly listening from everyone I worked with in the lab and would like to thank all my fellow students as a whole for their part in my education .
I must also thank Mike Takase and Larry Henling in the X-ray crystallography facility for teaching me how to mount crystals and all their help in solving structures. I'll start with the people I've lived with for the past ~4.5 years in the dilapidated abode we affectionately refer to as. Playing music has been a big part of my life since I was very young and some of the most fun I've had in the last few years has been sweating and screaming in the music room with you all and my only regret is, that we could not have spent more time playing together.
There are probably more people who deserve thanks, but there are certainly many regulars at "The Lake House" who are great friends who I haven't named here because it's been a long time coming and I've been rambling for a while.
LIST OF CHARTS
LIST OF TABLES
XJAB NMR coupling constant between atoms A and B along X number of bonds J NMR coupling constant or magnetic coupling constant.
INTRODUCTION
- Motivation
- Catalysis with Earth-Abundant Metals
- Ligand Design and Metal Reactivity
- TPB and DPB ligand scaffolds
- TPB binding modes
- DPB binding modes
- TPB and DPB metal compounds where the borane acts as a hydride acceptor
- Appended Lewis Basic Functionalities as H + Shuttles: An additional strategy for imparting the desired reactivity at first row transition metal centers is to take inspiration
- Active sites of hydrogenase enzymes. Sites which may act as proton shuttles are shown in red
- Chapter Summaries
- Reversible Si-H bond activation and catalytic hydrosilylation of ketones and aldehydes by (DPB)Co(N 2 )
- H BOND ACTIVATIONS BY IRON AND COBALT METALLOBORANES AND THEIR APPLICATION IN
An additional degree of flexibility of the introduced TPB and DPB ligands is the ability of the borane to act as a hydride acceptor. Each of the three types of known hydrogenase enzymes contains a group that can act as an H+ shuttle (Figure 1.5). Similar groups have been successfully incorporated into synthetic systems and have improved their catalytic activity.
The reactivity of the N2 adducts of these new ligands [(DP*BPh)Ni]2(N2) and (DP*BMes)Ni(N2) with H2 was investigated and compared with the reactivity obtained with the previously studied (DPB) Ni was observed. (N2) and (PhDPBMe)Ni. It is possible that the pKa of the trialkylamine is too high and that this is the cause of the poor activity in H+ reduction and H2 oxidation. The protonation of the anionic Fe-N2 adduct supported by this ligand [(ArP3B)Fe(N2)][Na(12-C-4)2] led to observation of the first parent Fe-diazenido (ArP3B)Fe( NNH) (Scheme 1.3) via EPR, ENDOR, HYSCORE and 57Fe Mössbauer spectroscopies. Scheme 1.3) which showed similar spectroscopic signatures to the previously reported [(TPB)Fe(NNH2)][BArF24].22 The activity of (ArP3B)Fe species as catalysts for the conversion of N2 to NH3 under different sets of conditions is discussed using KC8, HBArF24•2Et2O and CoCp*2, [H2NPh2][OTf] as the reductant/acid cocktails.
The Organometallic Chemistry of the Transition Metals, 5th Edition; John Wiley & Sons Inc.: New York, 2009.
HYDROSILYLATION CATALYSIS
Select metal-borane compounds that facilitate bond activations via cleavage of the M-B interaction
The solid state structures of (DPB)Co(OPh) and (DPB)Co(SPh) are shown in Figure 2.5 and Figure 2.6. Cooling to low temperatures (−90 °C) results in the disappearance of most of the paramagnetically shifted features with no new diamagnetic signals (Figure 2.S3). The difference in spin states is also reflected in the solid state structure (Figure 2.9) of (DPBH)Fe(8-amidoquinoline); it adopts a five-coordinate distorted trigonal bipyramid geometry (τ5 = 0.82) as opposed to the six-coordinate pseudo-octahedral geometry.
Subjecting the reaction mixture to three freeze-pump-thaw cycles ensures complete conversion of (DPB)Co(N2) to the new species (Figure 2.11, spectrum D). Reintroduction of N2 into the J-Young NMR tube, followed by stirring overnight, regenerates the spectrum shown in Figure 2.11 spectrum C. After degassing the reaction mixture with three freeze-pump-thaw cycles, a slight color change from red-orange to orange was observed. and the 1 H NMR spectrum of the reaction mixture confirmed the complete consumption of (DPB)Co(N2) (Figure 2.11 spectrum D).
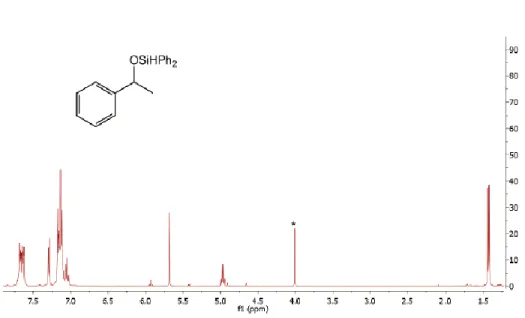
The EPR spectrum of the reaction mixture (Figure 2.12) showed clean formation of a new S = 1/2 species arising from the new paramagnetic species observed in 1H NMR: (DPBH)Co(SiHPh2) (Figure 2.11).
Seleted examples of previously reported Fe and Co carbonyl hydrosilylation catalysts
- Conclusions
- Supporting Information .1: Experimental Section .1: Experimental Section
- ray Crystallography. ray diffraction studies were carried out at the Caltech Division of Chemistry and Chemical Engineering ray Crystallography Facility on a Bruker three-
- Crystallographic Data Tables
The previously reported (PhDPBMes)Ni-benzaldehyde hydrosilylation system showed rate improvement with electron donating groups, in contrast to Chirik's (PDI)Fe(CH2SiMe3)2 system and other carbonyl reduction catalysts, which showed rate improvement with electron-withdrawing substituents.11,62 65 Oddly enough, we found that installing an electron-withdrawing or electron-donating group dramatically increased the rate of hydrosilylation by (DPB)Co(N2) with complete consumption of the aldehyde in less than 2 minutes when exposed to our standard conditions (Table 2.2). We also found that at lower catalyst loadings the selectivity for a 1:1 stoichiometry of silane to benzaldehyde was reduced and additional products from the reduction of multiple benzaldehyde equivalents were detected in the 1 H NMR spectra and GC-MS traces of the reaction mixtures. (Table 2.3, products A−E). Nevertheless, we speculate that the first step in the catalytic cycle is the replacement of the N2 ligand with silane, together with activation of the Si-H bond, resulting in (DPBH)Co(SiHPh2).
Insertion of the ketone into the Co−Si bond would then give a borohydrido-siloxylalkyl intermediate (Scheme 2.5, A) as shown for (PhDPBMes)Ni. It is interesting to note that in almost all cases (DPB)Co(N2) facilitates the fastest conversion of the carbonyl to the hydrosilylated product. 1H NMR, X-band EPR, and solution IR studies indicated that reversible activation of Si−H bonds in diphenylsilane is mediated by (DPB)Co(N2), prompting us to investigate [(DPB)Fe]2(N2) and ( DPB)Co(N2) for applications in hydrosilylation catalysis.
The isotropic displacement parameters of all hydrogen atoms were fixed at 1.2 (1.5 for methyl groups) times the Ueq of the atoms to which they are attached. Over ~1.5 h the solution turned from dark red brown to dark yellow-brown. Evaporation of saturated Et2O solution in HMDSO gave single crystals of (DPBH)Fe(benzo[h]quinolin-10-yl), which were lyophilized from C6H6 to give purple solids.
Evaporation of a saturated solution of Et2O in hexamethyldisiloxane (HMDSO) provided crystals of (DPBH)Fe(8-amidoquinline), which were lyophilized to give a red powder. A dark red-orange solution of (DPB)Co(N2), 67.6 mg (0.12 mmol), dissolved in 10 mL of THF was added to the stirring solution of DPB and CoBr2 via pipette, resulting in darkening of the mixture of the reaction. The EPR and NMR tubes were subjected to three freeze-pump-thaw cycles in a Schlenk line to generate a mixture of S = 1/2 (DPBH)Co(SiHPh2) species observed by 1H NMR and X-band EPR and some unidentified diamagnetic impurities.
Hydrosilylation products were identified by comparison of 1H NMR spectra and GC-MS traces with those reported in the literature To aid characterization, (1-(4-trifluoromethylphenyl)methoxy)diphenylsilane was converted. 1H NMR and GC-MS analysis of the reaction mixtures showed complete conversion of starting materials to products in the expected time.
![Table 2.1: Comparison of [(DPB)Fe] 2 (N 2 ) and (DPB)Co(N 2 ) to ( Ph DPB Mes )Ni for Benzaldehyde Hydrosilylation by Ph 2 SiH 2](https://thumb-ap.123doks.com/thumbv2/123dok/10402204.0/57.918.163.810.249.441/table-comparison-dpb-dpb-dpb-mes-benzaldehyde-hydrosilylation.webp)
SYNTHESIS AND REACTIVITY OF NICKEL METALLOBORANES WITH AMINE GROUPS IN THE SECONDARY COORDINATION
SPHERE
Introduction
Active sites of hydrogenase enzymes. Sites which may act as proton shuttles are shown in red
- Results and Discussion
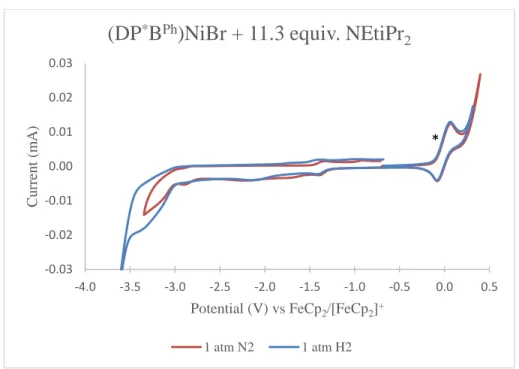
- Reactivity with H 2 and catalytic hydrogenation of styrene. Despite the unfortunate lack of electrocatalytic activity towards H + reduction and H 2 oxidation
- Conclusions
- Experimental Section
- ray Crystallography. ray diffraction studies were carried out at the Caltech Division of Chemistry and Chemical Engineering ray Crystallography Facility on a
- Introduction
- Results and Discussion
- Conclusion
- Supporting Information .1 Experimental: .1 Experimental
- Ray Crystallography: XRD studies were carried out at the Beckman Institute Crystallography Facility on a Bruker AXS KAPPA APEXII diffractometer (Mo Kα
- Synthetic Procedures
- Optimized Structure Coordinates for [( Ar P 3 B)Fe(N 2 )] - (TPSS) P 2.123882 0.308698 0.836816
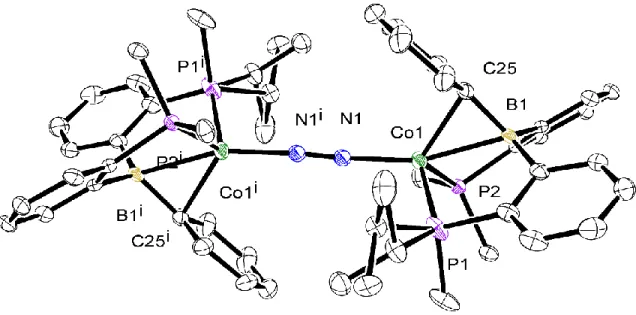
Reduction of the (DP*B)NiBr species with Na/Hg in THF gave [(DP*BPh)Ni]2(N2) similar to the dimeric iron species [(DPB)Fe]2(N2) (see Chapter 1) we previously reported (DP*BMes)Ni(N2) (Scheme 3.4).24 The IR spectrum of [(DP*BPh)Ni]2(N2) similar to that of [(DPB)Fe]2(N2 ) does not show an N-N stretching frequency, and the dimeric structure was confirmed by single-crystal X-ray crystallography (Figure 3.1). The product remained diamagnetic and a new broad resonance in the hydride region of the 1H NMR spectrum was observed at -5.8 ppm. Crystallization of the green compound by vapor diffusion of Et2O in HMDSO produced crystals suitable for single-crystal X-ray diffraction studies, which revealed the resulting compound to be the dimeric species shown in Scheme 3.6 and Figure 3.7.
A cooled solution of PhB(OMe)2 (532.8 mg, 2.77 mmol) in 5 mL Et2O was added dropwise to the stirred solution and allowed to slowly warm to room temperature overnight (~14 h) to give a cloudy pale yellow solution. the stirred solution and stirred cold for 2 h, followed by stirring at room temperature for ~12 h, yielding a cloudy pale yellow suspension. Herein, we report the EPR, ENDOR, HYSCORE, and 57Fe Mössbauer spectroscopic characterization of the first directly observed Fe(NNH) species and its. Geometric constraints derived from the spectroscopic data are supported by Density Functional Theory optimized geometries of the proposed Fe(NNH) and [Fe(NNH2)]+.
However, a comparison of the ENDOR spectrum of (ArP3B)Fe(NND) with that of (ArP3B)Fe(NNH) clearly reveals the single 1H hyperfine coupling (Figure 4.6). A thin film IR spectrum of the crude reaction mixture shows an intense stretch at v NN = 1717 cm -1 . Much information about the electronic structure of the NNHx (x = 0.1, 2) can be obtained from investigations of the nuclear hyperfine couplings, nuclear quadrupole couplings (e2Qq/h) and asymmetry parameters (η) for Na and Nβ around the world . the series of [(ArP3B)Fe(N2)]-, (ArP3B)Fe(NNH) and [(ArP3B)Fe(NNH2)]+.
One would expect the magnitude of the hyperfine coupling to Na to increase with protonation of Nβ with [(ArP3B)Fe(N2)][Na(12-C-4)2] with the smallest ANα, [(ArP3B) Fe(NNH2) )][BArF24] with the largest ANα and ANα for (ArP3B)Fe(NNH) distributed somewhere between [(ArP3B)Fe(N2)]- and [(ArP3B)Fe(NNH2)]+. A phosphine-protonated species as shown in Diagram 4.1 C would likely have an extremely small Aiso for hyperfine coupling to the acid-derived 1H, which is inconsistent with the magnitude of the observed 1H hyperfine coupling with Aiso = 16.5 MHz. 4.2.2 57Fe Mössbauer Spectroscopic Studies: 57Fe Mössbauer spectroscopy was also used to characterize the iron speciation of the reactions.
This additional steric shielding present in other Fe-N2 adducts allows the observation and characterization of the first Fe(NNH) species which are converted to [Fe(NNH2)]+ in the presence of 2-5 equiv of acid. The isomer shifts quoted are relative to the center of the spectrum of a sheet of α-Fe at room temperature. After freezing the solution, the cup was quickly removed from the glove box and immersed in liquid N2 until mounted in the cryostat.
Further cooling of the mother liquor to -35 ºC overnight yielded an additional small crop of white solids. The two layers were then mechanically mixed over a 30 minute period with a stainless steel needle inside the EPR tube, maintaining intermittent contact with the chilled sides of the cold well to maintain solutions at or near -135 ºC. The two layers were then mechanically mixed over a period of 15 minutes with a stainless steel needle inside the EPR tube, maintaining intermittent contact with the cooled sides of the cold well to maintain the solutions at or near -135 ºC.
![Figure 3.1: ORTEP representation of [(DP * B Ph )Ni] 2 (N 2 ), H atoms have been omitted for clarity](https://thumb-ap.123doks.com/thumbv2/123dok/10402204.0/100.918.167.799.164.515/figure-ortep-representation-dp-ph-atoms-omitted-clarity.webp)
![Figure 2.8: ORTEP representation of of (DPBH)Co(benzo[h]quinolin-10-yl).](https://thumb-ap.123doks.com/thumbv2/123dok/10402204.0/47.918.321.689.144.538/figure-ortep-representation-dpbh-benzo-quinolin-10-yl.webp)
![Table 2.S2: XRD experimental parameters for (DPB)Co(N 2 ), (DPB)Co(OPh), (DPB)Co(SPh), (DPBH)Co(benzo[h]quinolin-10-yl), (DPB)CoBr](https://thumb-ap.123doks.com/thumbv2/123dok/10402204.0/77.918.166.812.224.1014/table-xrd-experimental-parameters-dpbh-benzo-quinolin-cobr.webp)



![Figure 3.3: Cyclic voltammograms of (DP * B Mes )NiBr in the presence of 2,6- 2,6-dichloroanilinium tetrafluoroborate in THF (0.1 M [TBA][PF 6 ])](https://thumb-ap.123doks.com/thumbv2/123dok/10402204.0/102.918.160.808.137.585/figure-cyclic-voltammograms-mes-nibr-presence-dichloroanilinium-tetrafluoroborate.webp)
![Figure 3.6: 1 H NMR spectra of the reaction between [(DP * B Ph )Ni] 2 (N 2 ) and H 2 in d 8 -THF](https://thumb-ap.123doks.com/thumbv2/123dok/10402204.0/105.918.302.697.314.691/figure-nmr-spectra-reaction-dp-ph-ni-thf.webp)
