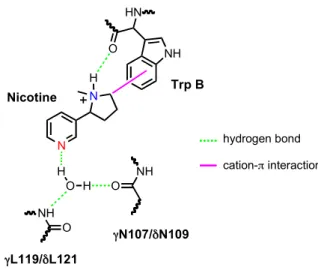
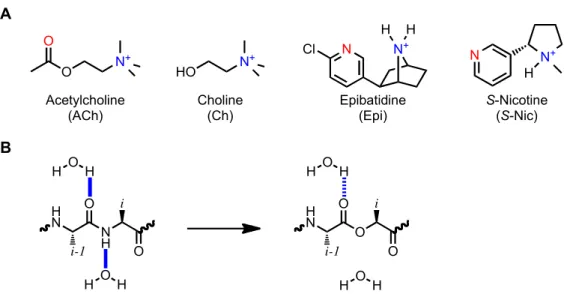
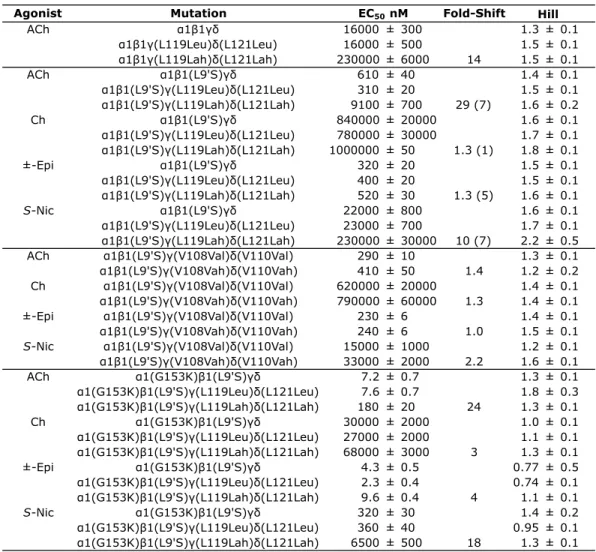
CHAPTER 3: Residues that Contribute to Binding of the Nicotinic Pharmacophore in the MuscleType Nicotinic Receptor
Bebas
22
0
0
Teks penuh
Gambar

Dokumen terkait
