I also have to thank Scott Virgil for all the helpful discussions, as well as keeping the Catalysis Center running. I must also thank the rest of the members of my cohort, Eric Alexy, Sean Feng and Zainab Al Saihati. Research in the Stoltz group is focused on the development of synthetic methods for the preparation of stereochemically rich molecules and the total synthesis of complex natural products.
A major theme of our group's method development is the transition metal-catalyzed α-functionalization of carbonyl derivatives, with a particular focus on the development of allylic alkylation protocols. Figure 3.3.1 Precedent for Cu/Bis-Picolinamide complexes and the use of L38 in enantioselective allylic alkylations. Challenging substrates in the palladium-catalyzed enantioselective arylation of g-lactams with aryl chlorides and bromides.
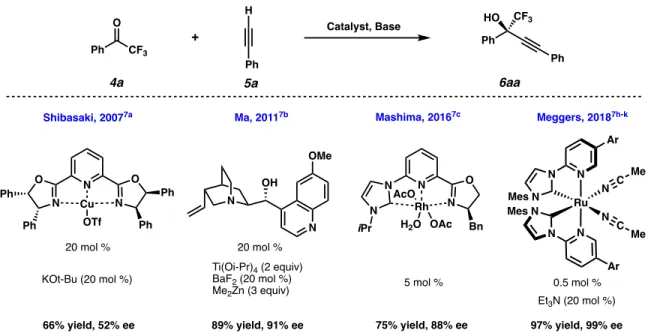
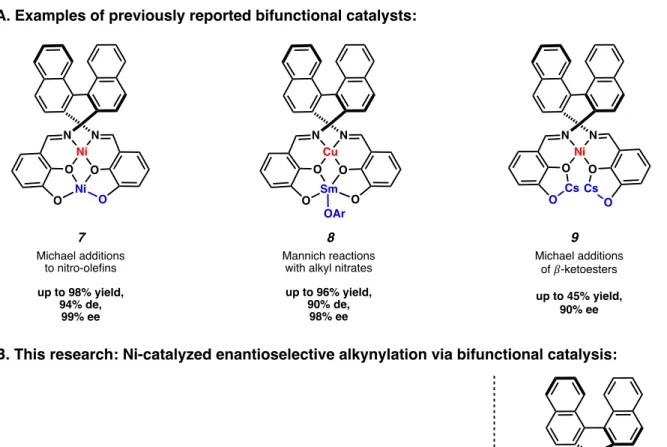
In two of these examples, the Brønsted base is the metal phenoxide incorporated into the framework (Figure 1.1.3. A, 7 and 9).

CATALYST DEVELOPMENT AND OPTIMIZATION OF REACTION CONDITIONS CONDITIONS
ARYL TRIFLUOROMETHYL KETONE SUBSTRATE SCOPE
We observed that in general, yields for electron-deficient trifluoromethyl ketones were slightly higher than with electron-rich substrates, with the latter requiring extended reaction times. However, we observed that the ee's for the electron-rich substrates were consistently higher than their electron-deficient counterparts. We found that the nature of the alkyne had a less pronounced effect on the overall reaction outcome, and alkynes possessing both electron-withdrawing substituents (6ab, 6ac, 6cb) and electron-donating substituents (6ad, 6ae, 6ee) in para . -The position of the aryl substituent was well tolerated.
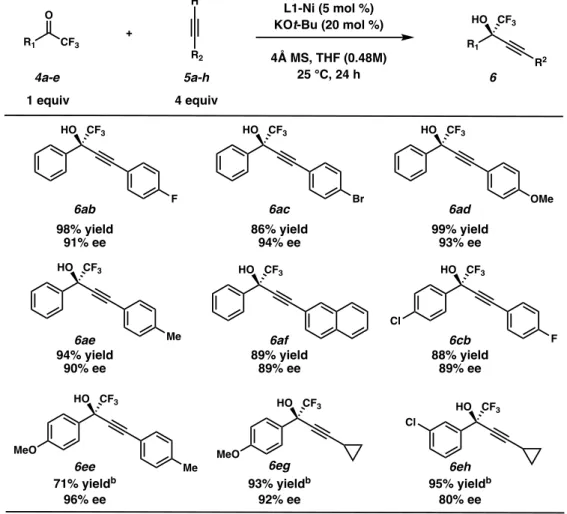
ALKYNYLATION OF UNSATURATED TRIFLUOROMETHYL KETONES
REACTION OPTIMIZATION FOR UNSATURATED TRIFLUOROMETHYL KETONES KETONES
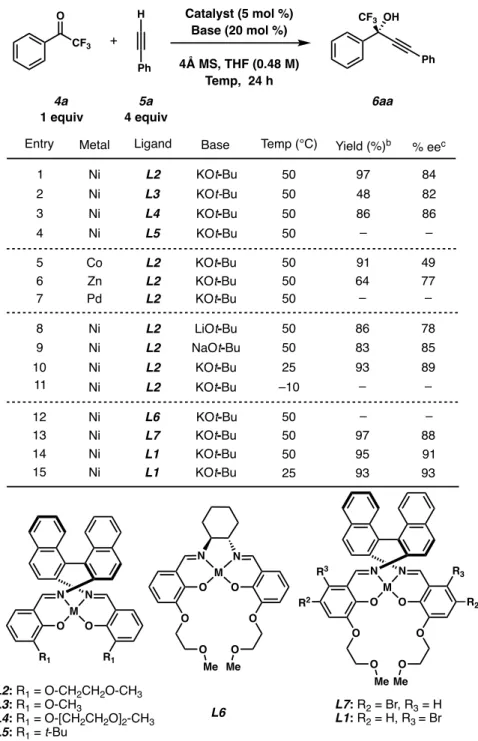
Finally, premixing the Ni-catalyst with the KOt-Bu base resulted in a slight improvement in yield (entry 6). With these results in hand, we examined two more reaction parameters; amount of KOt-Bu base and mole sieves used in the reaction. The original reaction conditions of Hong and co-workers did not include a moisture-free reaction setup, and we hypothesized that this could be the reason for the large excess of 4 Å molecular sieves.
Having switched to a glovebox setup, the last parameter we decided to investigate was the amount of molecular sieves in the reaction. Reducing the amount of molecular sieves even more (point 6) or completely excluding them (point 7) led to no improvements.
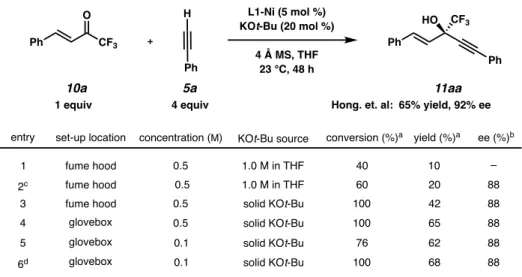
SCOPE OF ENANTIOSELECTIVE ALKYNYLATION OF UNSATURATED TRIFLUOROMETHYL KETONES TRIFLUOROMETHYL KETONES
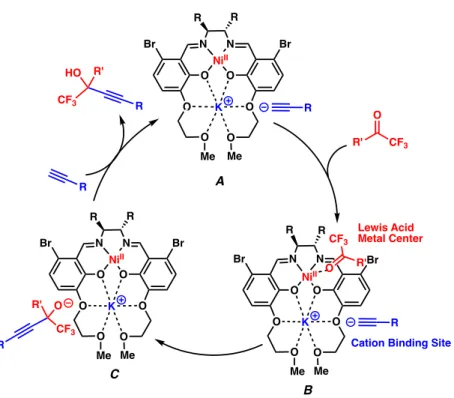
PRELIMINARY MECHANISTIC INVESTIGATIONS
The job plot analysis (Figure 1.7.1.B) agreed well with the titration study and clearly confirmed that L1-Ni binds to K+ with 1:1 binding stoichiometry. From the titration data based on the 1:1 binding model, the association constant (Ka) for binding of L1-Ni and K+ ions was calculated to be 6.6 x 105 M-1 using nonlinear regression analysis by DynaFit.15 . The mechanism is expected to begin with the deprotonation of the terminal alkyne to form potassium acetylide A.
When the nucleophile and electrophile are attached to the catalyst, they are close enough to bond. The resulting propargyl alcohol can then bind to the potassium cation (C) and act as a base for the deprotonation of the next terminal alkyne molecule.
CONCLUSIONS
EXPERIMENTAL PROCEDURES AND SPECTROSCOPIC DATA
- Synthesis and Spectroscopic Data for Salen-Crown Ether Ligands
- Complexation Procedure (Ni-L Synthesis) and HRMS for Salen-Ni Complexes
- Synthesis of Trifluoromethyl Ketones
- Procedure for Enantioselective Alkynylation of Aryl Trifluoromethyl Ketones
- PROCEDURE FOR ENANTIOSELECTIVE ALKYNYLATION OF VINYL TRIFLUOROMETHYL KETONES
After completion of the reaction, the crude reaction mixture was poured into ice water and stirred for 1 hour. The combined extracts were washed with water and dried over sodium sulfate and concentrated by rotary evaporation. The reaction was quenched with water and the pH was checked and adjusted to pH = 7.
The aqueous layer was extracted with methylene chloride three times, and the combined organic extracts were washed with 1 M HCl, dried over Na2SO4, and then concentrated. While still hot, the crude mixture was transferred to a 20 mL vial, rinsing with a small amount of additional EtOH. The mother liquor was then reduced via rotary evaporator and the crude reaction mixture was refrigerated and centrifuged a second time.
The combined precipitates were washed with hexanes and dried in vacuo to give a yellow solid (2.2 g, 2.58 mmol, 77% yield). The reaction was stirred at 0°C for 2 hours and then stirred at room temperature for 24 hours. The reaction was quenched with saturated ammonium chloride solution and the aqueous layer was extracted three times with ethyl acetate.
After the solution was stirred at room temperature for 30 min to give a dark yellow mixture, ketone (0.242 mmol, 1.0 equiv) was added dropwise to the solution. After the resulting mixture was stirred at room temperature for 24 h, saturated ammonium chloride (2 mL) was added to quench the reaction. The combined organic layers were washed with brine, dried over anhydrous magnesium sulfate, and concentrated by rotary evaporation.
UV-VIS DATA FOR METAL TITRATION
The DynaFit program was used for non-linear regression fitting of the titration data based on absorbance changes observed at 403 nm. Based on the result of the Job plot, the adjustment of the titration data was performed as a 1:1 binding mode.
NOTES AND REFERENCES
INTRODUCTION AND BACKGROUND
Palladium-catalyzed enantioselective arylation of g-lactams with aryl chlorides and bromides†. Figure 2.1.1).8 However, methods describing the asymmetric α-arylation of substituted pyrrolidinones to produce α-quaternary γ-lactams have remained elusive.
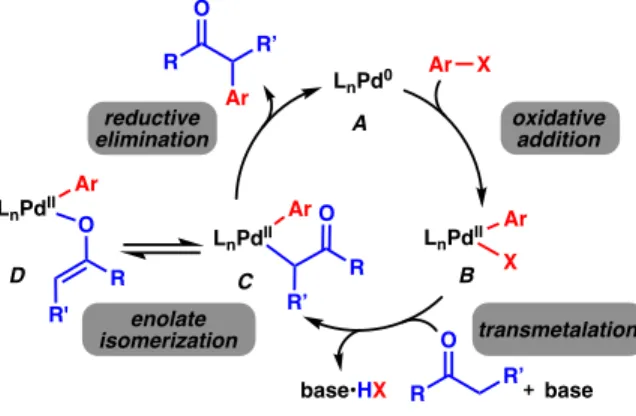
OVERVIEW ON a -ARYLATION
Finally, reductive elimination results in the desired product and regeneration of the initial Pd(0) species A. Although the efficiency of the reaction depends on many factors, the nature of the carbonyl compound used has the greatest influence on the reaction conditions. The acidity of the a-proton and the subsequent reactivity of the resulting enolate will dictate what types of bases should be used or whether a less reactive silyl or zinc enolate is needed.
Furthermore, the steric burden and reactivity of the generated enolate will then determine what types of aryl halides, ligands, solvents, and reaction temperatures will be required to form the product in synthetically useful amounts. One of the challenges is the need for enolization with a strong base, which, in addition to catalyst degradation, can lead to the formation of unwanted aryne intermediates from the aryl halide14,15. In general, where yields of the desired product were high, the amount of dechlorinated product 33 was low and vice versa.
Concerned about consumption of the starting material via formation of the lactam-dba adduct, a product which proved difficult to separate from 32aa, we chose to investigate a few more Pd sources in combination with L9. Using Pd2(pm-dba)3 as the Pd source, the effect of the solvent on the reaction outcome was then tested. Having identified two Pd sources that could be used interchangeably under these conditions,18 as well as the appropriate solvent, we then turned our attention to the nature and equivalents of the base, nucleophilic and electrophilic stoichiometry, and the reaction temperature (Table 2.3. 4).
One possible explanation for this could be that LiHMDS, which has a pKa of 26 in THF,31 may not be strong enough to fully deprotonate the g-lactam, thereby limiting the amount of reactive enolate present during the course of the reaction. Quantitative enolization is expected when using LDA, which may lead to degradation of the lactam enolate via other undesirable pathways. Finally, the nature of the cation also affected the HMDS; LiHMDS appears to be the best base for this reaction, as switching to Na or KHMDS resulted in significant drops in yield and ee (entries 6 and 7).
REACTION OPTIMIZATION WITH ARYL BROMIDES
With this new set of conditions, the reaction proceeds at a lower temperature; the desired product is obtained in 84%. Moreover, the product is still obtained at 54 °C in good yield and high enantiomeric excess (entry 7).
REACTION SCOPE
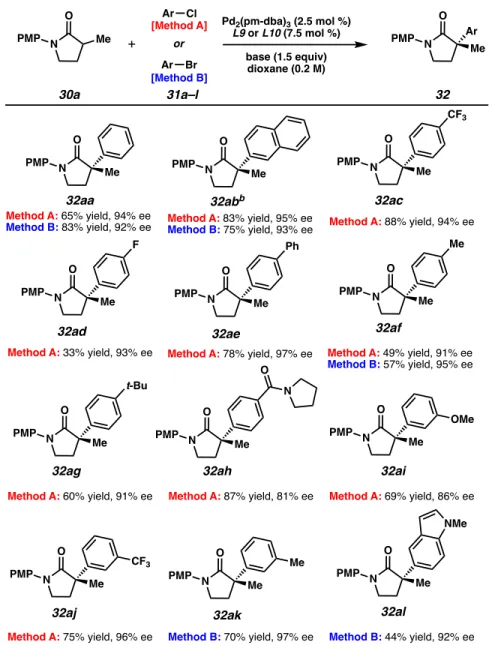
L10 was the best ligand when bromobenzene was used, and contrary to what was observed with chlorobenzene (Table 2.3.4), NaHMDS appeared optimal (entries 4 and 5). Switching to ortho-methoxyphenyl-substituted 30c or electron-deficient trifluoromethylphenyl 30e led to decreased yield and enantioselectivity (32ca and 32ea). Although N-phenyl 30d does not outperform 30a in Method A, it does show higher reactivity and enantioselectivity when exposed to Method B, yielding it.
Benzyl-protected lactam 30f provides the quaternary lactam 32fa in high enantiomeric excess in both methods. We found that aryl bromides and aryl chlorides with a variety of substitution patterns are amenable to arylation. Aryl halides possessing electron-deficient substituents (see products 32ab, 32ad, 32ah,22 32ae) and electron-rich substituents (32af, 32ag) at the para position led to products with excellent enantioselectivities using Methods A and B, respectively.
Unfortunately, only trace product is observed when ortho-substituted aryl halides are subjected to our reaction conditions.23 Happily, an N-methylindole was also tolerated as we obtained 5-indolyl lactam 32al in moderate yield and excellent enantioselectivity. Examples with ethyl (32ha, 32hb), benzyl (32ga, 32gb), propyl (32ia), phenethyl (32ja) and 2-naphthylmethyl (32ka) substitution give all the α-arylated products with good enantioselectivity. Even with a more hindered substrate, similar patterns of reactivity and selectivity to α-methyl substituted 30a were observed.
When an electron-deficient aryl chloride coupling partner is used in method A or m-bromotoluene is used in method B with 1 g, the desired product is formed with high enantioselectivity and good yield (32gc and 32gd).
PRODUCT TRANSFORMATIONS
Reduction of the lactam carbonyl with lithium aluminum hydride yields the corresponding medicinally valuable pyrrolidine (35). Partial reduction of the lactam with lithium triethyl borohydride and capture of the resulting iminium ion with potassium cyanide affords chiral aminonitrile 36 in moderate yield but with high diastereoselectivity.
CONCLUSION
EXPERIMENTAL PROCEDURES
- Synthesis of a -Substituted g -Lactams
- General Procedures for Pd-Catalyzed a -arylation of g -lactams
- Spectroscopic Data for Products from Catalytic Reactions
The reaction was then cooled to ambient temperature, filtered through a plug of silica and concentrated. The crude reaction mixture was quenched with saturated NH4Cl solution, extracted three times, then the combined organic layers were washed with saturated NaHCO3 solution. The reaction was stirred at –48 °C for 30 min, then allowed to warm to ambient temperature overnight.
The reaction mixture was quenched with a saturated NH4Cl solution and extracted with EtOAc three times. The solution was cooled to ambient temperature, quenched with saturated NH4Cl solution, and extracted with EtOAc five times. The crude reaction mixture was purified by silica gel flash chromatography to give the product.
The solution was cooled to ambient temperature, quenched with saturated NH4Cl. solution, and extracted five times with EtOAc. The crude reaction mixture was purified by silica gel flash chromatography to provide the product. After consumption of the starting material, the reaction was diluted with water and extracted three times with EtOAc.
The resulting solution was stirred at -78 °C for 2 h, then warmed to ambient temperature and allowed to stir for 8 h. The reaction was stirred at 0°C for 5 hours, quenched with anhydrous K 2 CO 3 and filtered through celite.