The author participated in performing the calculations, considering the results and preparing the draft part. All implicit hydrogen atoms have the same index of the atom to which they are attached. The gray line indicates (𝐸(𝑡) −𝐸avg)/(KEavg), while the black line indicates the cumulative average of the same quantity.
The harmonic correction consists of the thermal contribution to the free energy difference with respect to the ideal gas approximation (equation 5), and the anharmonic correction reflects the contribution of conformational sampling of the isolated catalyst to the free energy difference (equation 6). In particular, the computationally expensive mean field method is only applicable to a subset of the molecular system. The binding energies of ethylene and propylene to the catalyst systems are within 1 kcal/mol of the high-level B3LYP result, while the wall clock time of the SCF cycle is reduced by up to 20-fold.
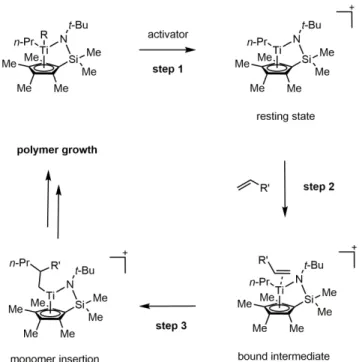
The dissociation of ethylene from the catalyst is caused by the proximity of the counterion to the metal complex. These contributions are quantified via constrained MD trajectories along the minimum energy path of the insertion barrier.
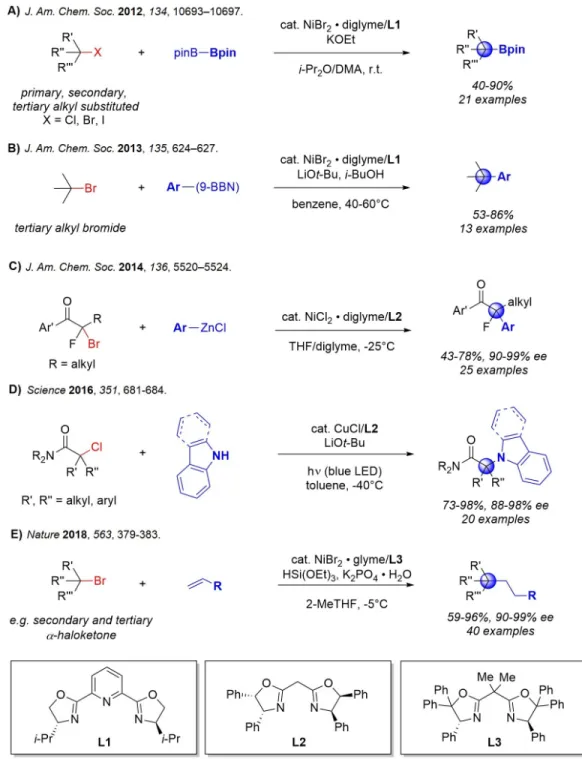
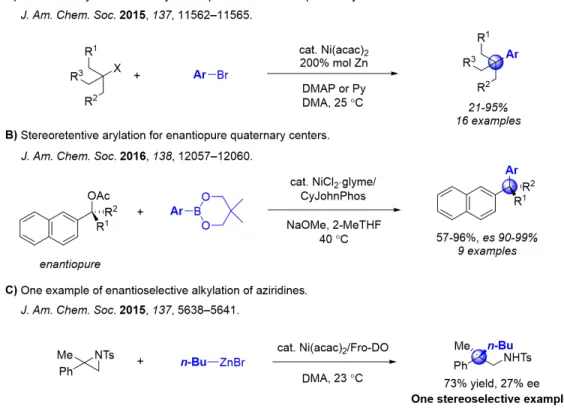
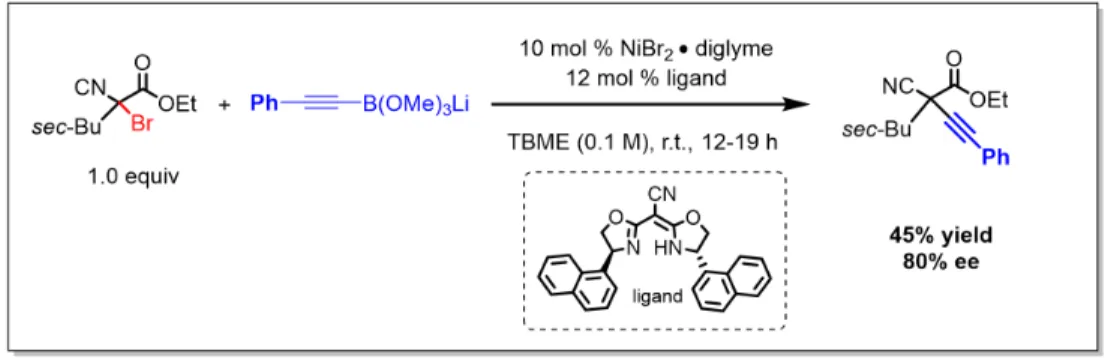
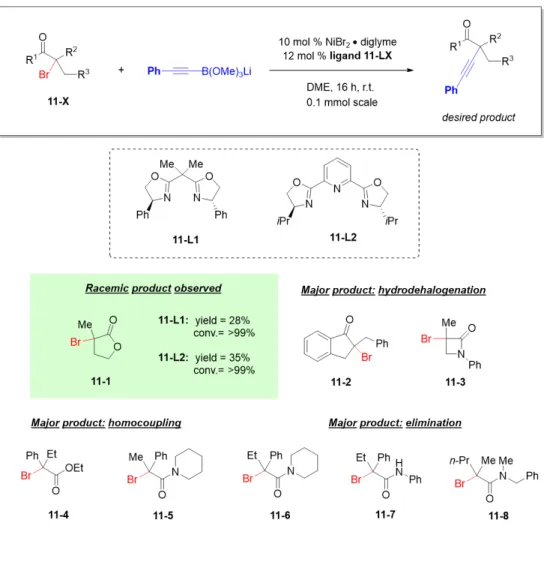
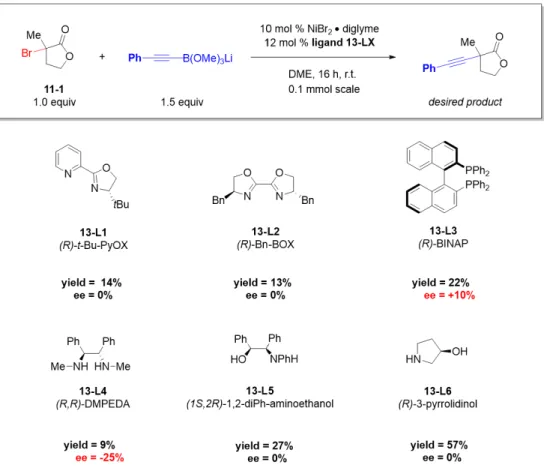
CROSS-COUPLING OF TERTIARY ALKYL ELECTROPHILES
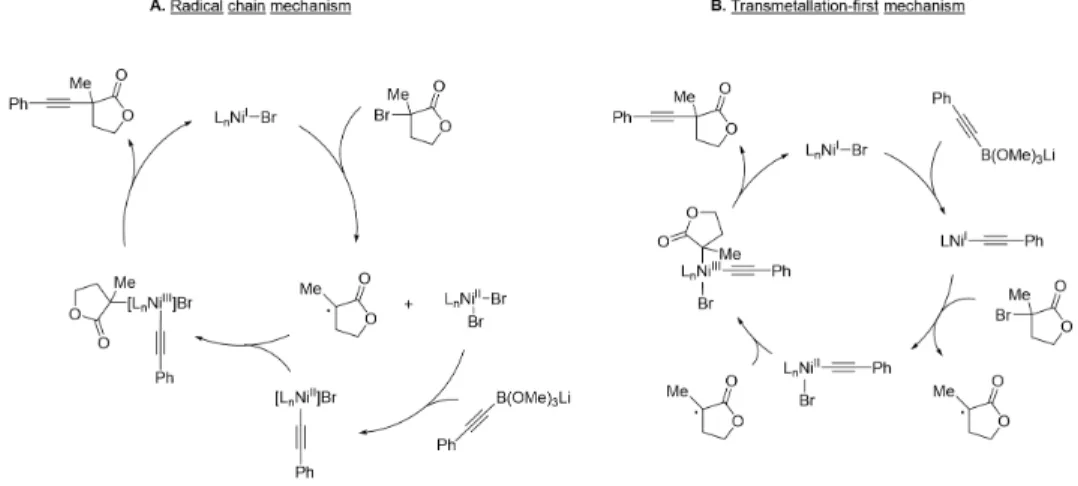
In the previously described mechanism for the nickel-catalyzed arylation of propargyl halides, the radical chain mechanism was supported [46]. Addition of the tertiary radical to nickel(II) generates a nickel(III) species that undergoes reductive elimination to give product (Figure 2.8b). The remainder of the filtrate was concentrated and the product was isolated by preparative TLC (1:4 hexanes/EtOAc).
The enantiopurity of the product was determined by chiral HPLC (OD column, 2% i -PrOH in hexanes, 30 min). Therefore, managing the cost of QM computation is an important objective for developing QM/MM simulations at the computationally efficient solution stage. For EMFT energy estimates of the resting state and associated catalyst intermediates, the system partition is illustrated in Figure 3.3 and Figure 3.4.
To compactly label the high-level regions for each subsystem size, we assign a label from 1 to 4 to each atom in the catalyst resting state structure (Figure 3.3), with the hydrogen atoms sharing the index of the heavy atom to which they are bound. We now further investigate the quality of the EMFT-optimized structures for the resting state of the catalyst in all. The rows of the table are grouped into distances between atoms that are within the high-level region of the EMFT description (upper unshaded block), distances between atoms that span the high- and low-level regions (shaded block), and distances between atoms that are within the region low levels of EMFT description (bottom unshaded block).
For bonds involving high- and low-level subsystems, it is seen in the table that these distances are generally more consistent with the description of low-level DFT theory, as expected; In Figure 3.9a, we plot the root mean square error (RMSE) for the difference between the EMFT-optimized bond lengths and the high-level or low-level DFT-optimized bond lengths for atoms located within the high-level region of minimum EMFT separation. In Figure 3.9b, we present the corresponding results for bond lengths involving atoms that lie within the low-level region of the EMFT splitting.
These results clearly illustrate that EMFT offers improved accuracy in the high-level region, while the rest of the system is described with the quality of low-level DFT theory. For each of the catalysts, timings are reported for the energy calculation, so that 11 atoms are described in the high-level region (e.g., metal, propyl side chain). These results, in combination with the previously shown benchmarks of EMFT accuracy, illustrate that the method maintains the high-level accuracy of the DFT method while vastly reducing computational costs.
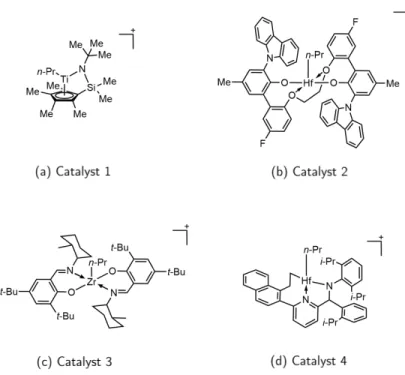
As a demonstration of robustness, Figure 3.11 illustrates the energy conservation of the EMFT/MM microcanonical trajectories for each of the four counterions considered. Specifically, Figure 3.12 presents the RDF with respect to the distance between the Ti atom of Catalyst 1 and the center of mass of each of the four counterions considered from separate EMFT/MM simulations.
SOLUTION-PHASE CONFORMATIONAL/VIBRATIONAL ANHARMONICITY IN CO-MONOMER INCORPORATION
POLYOLEFIN CATALYSIS
Our studies show that the competition for metal binding between the auxiliary ligands present in the precatalyst, pyridine or phosphine, and olefin monomers significantly favors the former. Therefore, we investigated the effect of removing auxiliary ligands (e.g., pyridine) on the nickel-catalyzed copolymerization of ethylene and polar monomers. Stable, coordinatively saturated metallocyclic precatalysts prevent the use of the auxiliary ligand and have shown promising results in palladium-catalyzed ethylene polymerization [17].
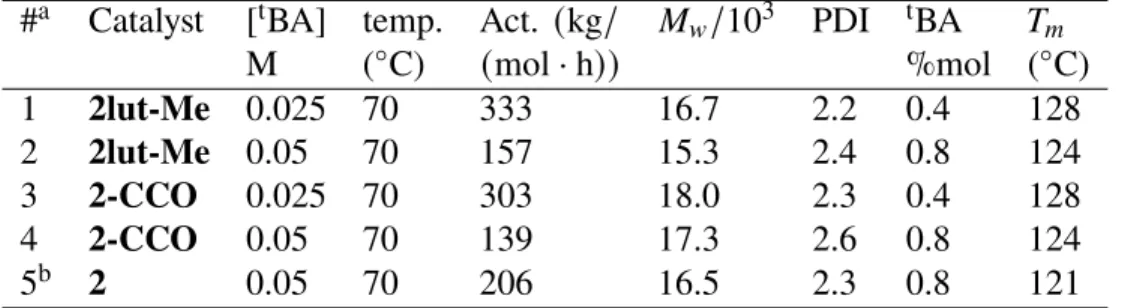
Examples of the nickel analogue are missing; however, the effect of weaker complementary ligands has been investigated in Ni-phenoximine catalysts and weaker donor coordination leads to higher activity in ethylene polymerization and suppresses 𝛽-H elimination [42]. Since 2-CCO is the first structurally characterized, thermally robust four-membered chelate complex generated post-BA insertion, we investigated its application as a precatalyst in ethylene/tBA copolymerization. Both the nickel complex with a weak auxiliary ligand (lutidine,2lut-Me) and auxiliary donor-free nickel complexes (2-CCO) are highly active in ethylene/tBA copolymerization (Table 1, Entry 1 4), producing polymers with moderate molecular weight and tBA incorporation.
Compared to 2 (Table 1, Entry 5), both are slightly less active overall, which is in contrast to the previously reported effects of the L ligand on ethylene polymerization. Notably, the corresponding ethylene uptake curves revealed that 2lut-Meand2-CCO consumes ethylene much faster than 2 in the first 5 min of ethylene/tBA copolymerization. However, a significant decrease in the rate of ethylene consumption was observed in a longer time, which may be related to the decrease in their stability (Figure 5.14).
Overall, our observation shows that the absence of the strong auxiliary ligand L indeed accelerates the rate of monomer insertion, but may also lead to a lower thermal stability. Ligand binding studies have provided quantitative thermodynamic data regarding the influence of precatalyst structure on the binding of donors such as olefins, pyridines, and phosphines. A relatively compressed degree of binding was observed with the introduced acrylate species (1-CCO and 2-CCO) compared to the Ni-CH2SiMe3 and Ni-CH3 complexes (1, 2 and 2lut-Me), which is related to an increased acidity Lewis of Ni in enolate complexes. , as determined by computational studies.
Addressing the influence of the supporting phosphine phenoxide, the large donors were shown to have a higher binding affinity to the 1-CCOthan2-CCO complex, likely due to the rigid steric profile close to the 2-CCO phenoxide. This mechanism is consistent with the isomerization mechanism in ethylene/acrylate copolymerization found by calculation. Finally, both 2-CCO and 2lut-Me serve as a competent one-component catalyst in ethylene/acrylate copolymerization.