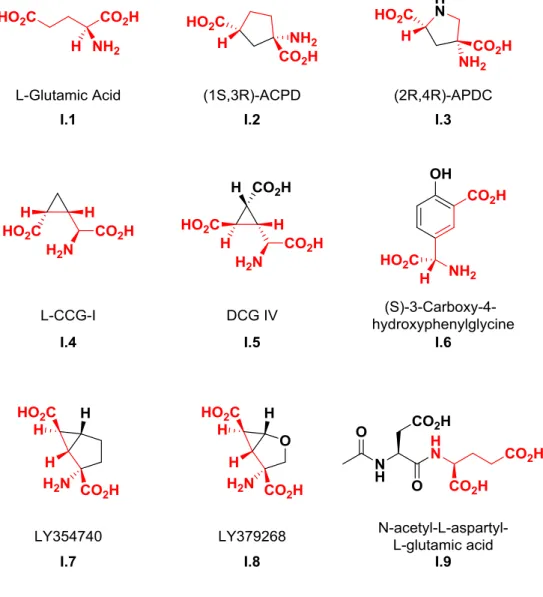
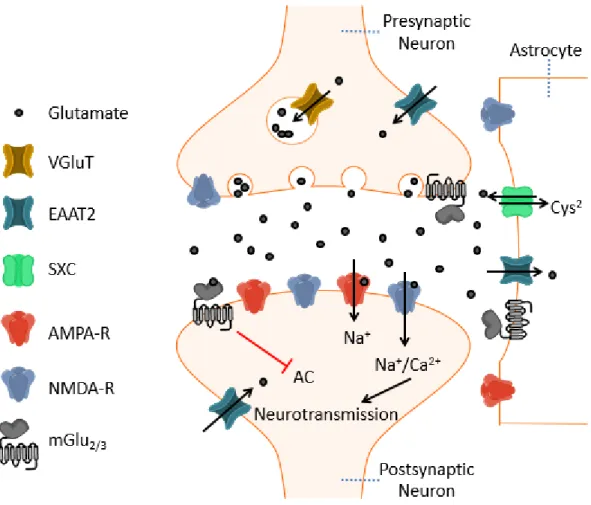
Targeting the group II mGlu receptors, mGlu2 and mGlu3, has been of particular interest in schizoaffective, depressive, anxiety and substance abuse disorders (Hovelsø et al., 2012; Nicoletti et al., 2011). The group II mGlu receptors, like all members of the mGlu receptor family, are class C GPCRs. These dimer pairs can be either homo- or heteromeric – in the case of the group II receptors, mGlu2/3.
These compounds are allosteric ligands that activate the receptor in the absence of an orthosteric ligand. One of the drugs that has been studied to treat TRD is ketamine. Further investigation of the individual roles of mGlu2 and mGlu3 in this context will help answer such questions.
Determination of fraction unbound in plasma. Fraction unbound in brain can be calculated in the same manner by using brain homogenate rather than plasma
For locomotion experiments, mice were injected IP with a dose of Elacridar (20 mg/kg) or vehicle 1.5 h before initiation of the assay, then dosed with test compounds or vehicle 30 min before initiation of the assay. Analysis of the total distance traveled, time spent in areas within 5 cm of the edge and time spent in the center of the area were measured. On the day of the assay, mice were injected IP with a dose of Elacridar (20 mg/kg) or vehicle 1.5 h before initiation of the assay, then dosed with test compounds or vehicle 30 min before initiation of the assay.
Their behavior was monitored by a video camera placed directly in front of the cylinder for the duration of the test. For the tail suspension test, mice were injected intramuscularly with a dose of Elacridar (20 mg/kg) or vehicle 1.5 h before the start of the test and then given a dose of test compounds or vehicle 30 min before the start of the test. For the elevated plus maze, mice were injected intramuscularly with a dose of Elacridar (20 mg/kg) or vehicle 1.5 h before the start of the test and then given a dose of test compounds or vehicle 30 min before the start of the test.
The animals were then placed in the center of the elevated plus-maze apparatus, which consisted of four arms (each approximately 10 x 30 cm) connected in a plus configuration and elevated approximately 50 cm from the ground. Mice were injected IP with a dose of Elacridar (20 mg/kg) or vehicle 1.5 h before the start of the test, then dosed with test compounds or vehicle 30 min before the start of the test. Mice receiving the same drug were placed in cages on either side of the table to control for effects of lighting and context.
DEVELOPMENT OF THE FIRST HIGHLY SELECTIVE CNS-PENETATING NEGATIVE ALLOSTERIC MODULATOR OF MGLU3 SUITABLE FOR IN VIVO USE. PAM compounds known to bind in the 7TM region of mGlu receptors, rather than in the VFT as seen with orthosteric ligands.
Scheme III.1 Amide coupling and Sonogashira coupling sequence for the synthesis of analogues exploring potential replacements for the distal phenyl moiety in VU0092273
Glu Min %
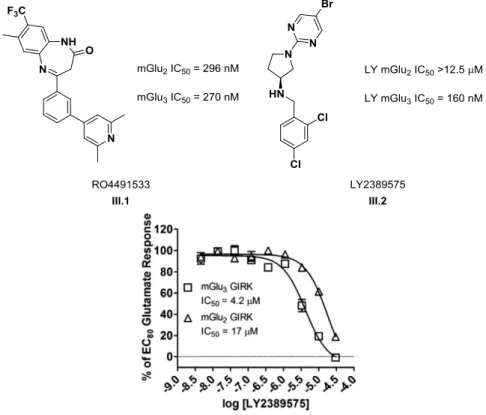
Based on these data, the next round of library synthesis kept the 4-methoxyphenyl moiety in III.31 (VU0457299) constant, and a broad spectrum of amines was employed to explore alternative amides.
Scheme III.2 Sonogashira coupling, saponification, and amide coupling synthesis of analogues to explore alternative groups as replacements for the amide head group in
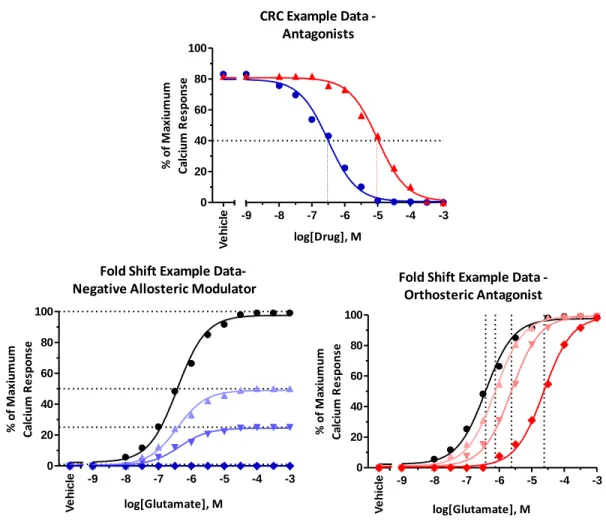
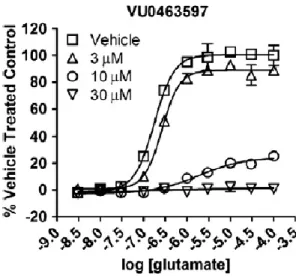
This result led us to the resolution of the racemic 3-hydroxymethyl analog III.65, which had an IC50 of 2.1 µM and provided complete EC80 block. Similarly, III.109 was inactive to potentiate the EC20 glutamate concentration or inhibit the EC80 glutamate concentration in our standard mGlu5 calcium assay. To test whether III.109 antagonizes mGlu3 through a noncompetitive (allosteric) mechanism of action, we next performed a progressive shift assay.
In these studies, III.109 dose-dependently induced a rightward shift and reduced maximal efficacy of the orthosteric glutamate agonist, consistent with a noncompetitive (allosteric) mechanism of action (Figure III.7). A Lead Profiling Screen by Ricerca revealed that III.109 had only limited off-target activity at 10 µM (Table III.3). The intrinsic clearance (CLint) for III.109 was determined in rat and human liver microsomes, and this assay showed that III.109 was rapidly cleared in vitro.
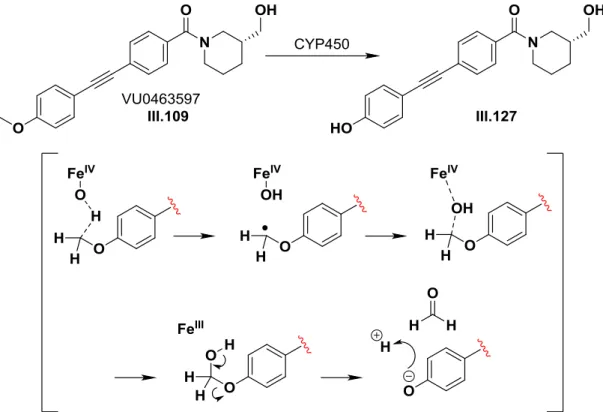
Intriguingly, III.109 was found to be a moderately cleared compound in the rat with a CL of 33 mL/min/kg after intravenous administration of 1 mg/kg of III.109. The more modest in vivo clearance rate may be attributed to the low Fu of the parent compound, limiting the presentation of unbound III.109 to microsomes. Metabolite identification studies in rat and human liver microsomes indicated that the major biotransformation pathway was P450-mediated O-demethylation of III.109 to generate phenol III.127, a metabolite subsequently shown to be mGlu3 inactive.
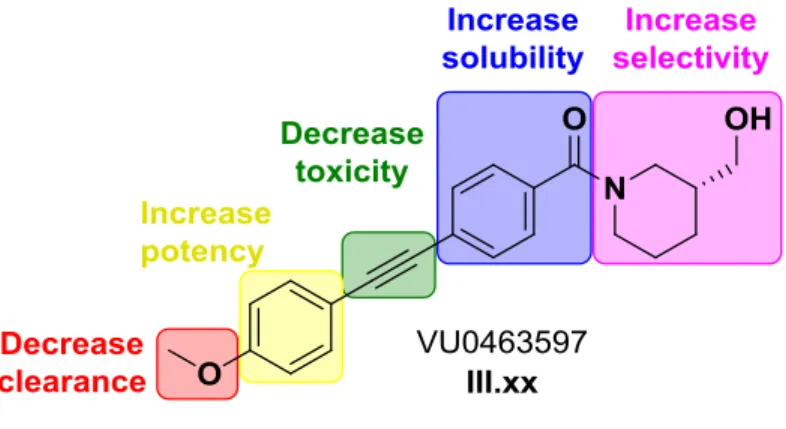
To pursue further chemical optimization, III.109 was divided into five sections for further SAR exploration and improvement of its properties to generate a tool that could be used in vivo (Figure III.9). The first libraries generated around III.109 focused on identifying a replacement for the p-methoxy group or electronically disrupting the aryl ring, making P450-mediated O-dealkylation less easy.
Scheme III.4 Sonogashira coupling, saponification, and amide coupling synthesis of analogues to explore replacements for the (R)-3-hydroxymethyl-piperidine of
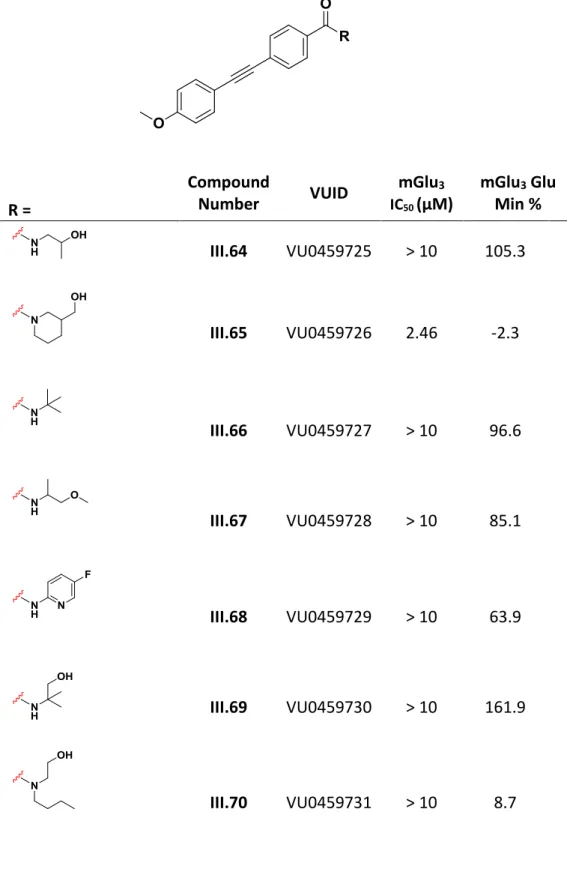
A racemic 3-hydroxy piperidine congener III.140 showed significant activity and after synthesis of pure enantiomers, enantioselective inhibition was observed. This also allowed a more direct comparison of the newer generation of amide analogs with acetylene-containing compounds previously synthesized during the generation of III.109. Among these analogs, there were two additional sub-micromolar mGlu3 NAMs worthy of further profiling: III.238 and III.239.
For further studies of the mechanism of action, III.239 (VU0469942) was selected as an exemplary compound. Importantly, III.239 was found to be inactive at all other mGlu receptors at concentrations up to 30 μM when tested in the calcium mobilization assay. Progressive fold shift analysis of III.239 revealed that it acts as a negative allosteric modulator of glutamate signaling (bottom left).
After evaluating the pharmacological and pharmacokinetic profiles of the lead analogs, we decided to proceed with III.270 (VU0477950) as our in vivo tool compound. During the resynthesis of III.270, all intermediates were purified and submitted to high-resolution mass spectroscopy (HRMS) and 1H and 13C nuclear magnetic resonance (NMR) analysis. The synthesis of III.270 proceeded in moderate to good chemical yields and led to the formation of the expected compounds v.
To test this hypothesis, III.270 binding was analyzed in the presence of [3H]-LY341495 to confirm that it did not bind to the orthosteric site. In summary, we have developed the first series of selective mGlu3 NAMs described to date, including III.270 (VU0477950). Application of III.270 to in vitro and ex vivo systems will allow dissection of the molecular mechanisms underlying mGlu3-mediated signaling and plasticity.
Administration of III.270 in rodent models of psychiatric diseases will allow observation of the impact of mGlu3 blockade in vivo.
Compound
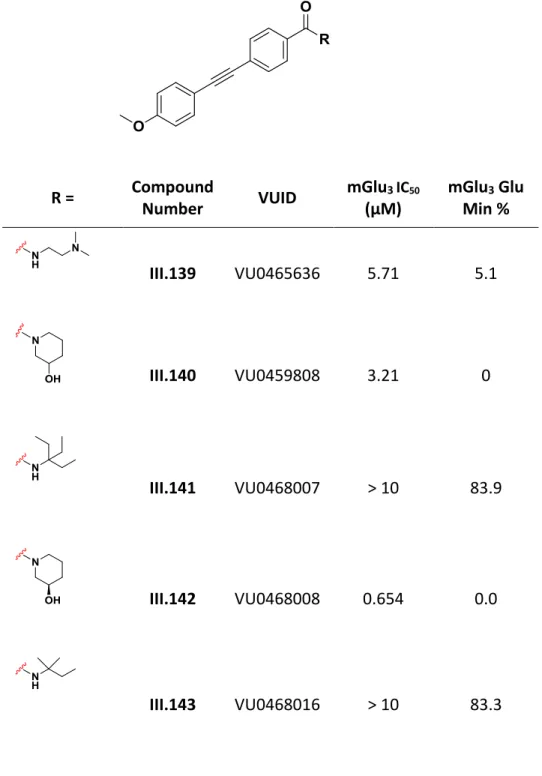
When the phenylene at R1 (IV.5 – IV.9) is replaced by a 3-fluorophenyl (IV.10 – IV.14), the analogous compounds generally show a reduction in potency at mGlu2 and mGlu3. In contrast, when a 3-methoxyphenyl is present at R1(IV.15 – IV.19), there is an increase in potency compared to their phenyl analogues. For the analogs screened here, having a 3-methylphenyl at R1(IV.20 – IV.22) results in an overall decrease in potency.
The general trend seen is that power decreases with increasing halogen size, with the IV.25 providing a notable exception. Notably, the presence of a 3-pyridyl group in R2 resulted in the most potent compound generated from this initial screen, IV.32, with an IC50 of 427 nM in mGlu2 and 67 nM in mGlu3. Given the trend seen with IV.5-IV.38, where the presence of a 3-pyridyl group at R2 resulted in compounds with elevated potency at mGlu2 and mGlu3, it was decided to explore the effects of additional heterocyclic substitutions at R2 by kept R1 as phenyl group (Table IV.3).
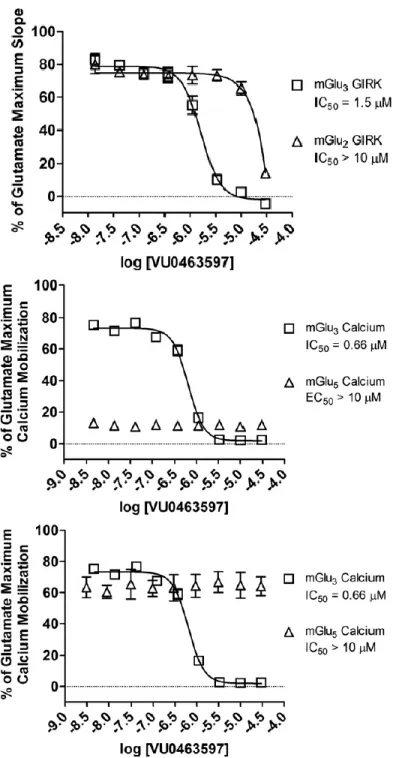
Compared to IV.8, analogs with benzo-fused heterocycles at R2 (IV.45 - IV.51) all had significantly reduced potency at mGlu2 and mGlu3. Because of its potency, IV.47 (VU0550418) was selected as an exemplary compound from this series for further pharmacological characterization. To determine its mechanism of action (competitive orthosteric antagonism or negative allosteric modulation), IV.47 was examined at several fixed concentrations against mGlu2 and mGlu3 versus an increasing concentration of glutamate (1 nM – 30 μM).
Overall, these results suggest that potent dual inhibitors of mGlu2 and mGlu3, such as IV.47, are rapidly accessible via modifications to a pyrazolo[1,5-a]quinazoline-5(4H)-ene scaffold. Fold-shifts of mGlu signaling for IV.47 at 10 μM across the group I and group III receptors.
Cyanation, Suzuki couplings, nitrile hydrolysis, and carboxamide formation to synthesize quinoline carboxamide compounds with the potential to generate
Glu Min %
Finally, a 10 μM concentration of IV.57 did not alter the response of the remaining mGlu receptors to glutamate (Figure IV.6). Previous studies have shown that pharmacological activation of group II mGlu receptors results in LTD of excitatory transmission in layer V of the rat medial prefrontal cortex (mPFC) (Huang & Hsu, 2008; Huang, Yang, Lin, & Hsu, 2007). When similar experiments were performed in the presence of the NMDA receptor antagonist AP5 (50 μM), LTD was still observed, with an average decrease in signal magnitude of.
Taken together, these findings from layer V of the mouse mPFC indicate that selective pharmacological activation of group II mGlu receptors produces an NMDA receptor-independent form of LTD that is expressed postsynaptically (Figure V.1). Next, we tried to evaluate the contribution of the mGlu2 and mGlu3 subtypes to this form of BPK. Quantification of the effects of mGlu3 NAMs on LTD measured 55–60 min after drug washout (average of shaded region).
To further assess the role of individual group II mGlu receptor subtypes, we compared LY379268-induced LTD in mGlu2 and mGlu3 KO mice. Quantification of the effects of IV.57 on LTD 55–60 min after drug washout (average of shaded region). Similarly, the lack of effect of IV.57 on LY379268-induced LTD and the maintenance of LTD in mGlu2 KO mice suggest that mGlu2 activation is not required to induce LTD, although it may play an important role in the transient depression of synaptic transmission in layer V of mouse mPFC.
Moreover, mice treated with the 100 mg/kg dose of the compound showed a significant increase in the number of trials to reach criterion compared to mice in the vehicle and 3-mg/kg groups. In the absence of radiolabeled versions of the compounds, the exact receptor occupancy at any given dose of each compound could not be directly determined.