I would like to thank all my collaborators over the years, including Doug Behenna and, most recently, Jen Stockdill, for their diligent work on zoanthenol. I'd like to thank Eric Ashley for pretending not to have a photographic memory and for shining a light on me during those early years as my friend.

Progress Toward the Total Synthesis of Zoanthenol
Introduction
For example, norzoanthamine 2, norzoanthaminone 3, epinorzoanthamine 4, and oxyanthamine 5 are all inhibitors of murine leukemia P388. Norzoanthamine 2 is also a potent antiosteoporotic due to its inhibitory activity against interleukin-6 and has been shown to have an effect of low in weight. in ovariectomized rats.3 Moreover, zoanthenamide 6, zoanthamide 7, and 28-deoxyzoanthenamide 8 all inhibited phorbol myristate acetate (PMA)-induced inflammation in a mouse ear model. Despite the great structural diversity of zoanthus alkaloids, the fact that several years have passed since their structural elucidation and a number of relevant synthetic studies have been published, only a single total synthesis of a zoanthus alkaloid has been reported to date.4 total synthesis of norzoanthamine was completed in 2004 by Miyashita and co-workers.As shown in Scheme 1, they solved the structure of norzoanthamine 2 by a number of bond cleavages, with the key step in their synthesis relying on the formation of the ABC ring system.
Miyashitas’ intramolecular Diels-Alder reaction
The intramolecular Diels–Alder substrate 9 was synthesized in 14 steps from the known chiral enone 12 by successive C–C bond-forming reactions, including a diastereoselective cuprate addition with cuprate 13 and an aldol reaction with aldehyde 14 ( Scheme 2 ), to provide furanol 15 . .
Synthesis of the Diels-Alder substrate
Endgame for Miashita’s norzoanthamine synthesis
Williams’ synthetic studies on norzoanthamine AB-rings
Williams’ tandem aminal cyclization
- Results and Discussion
Finally, Kobayashi has completed a diastereoselective synthesis of the CDEFG ring system 31 in good yield by demonstrating the biogenic acid-catalyzed tandem aminal cyclization to the protected amino-alcohol 30 (eq 2).9. The iminium ion is then quenched upon nucleophilic attack of the adjacent carbonyl, closing the last six-membered ring to provide zoanthenol (1).
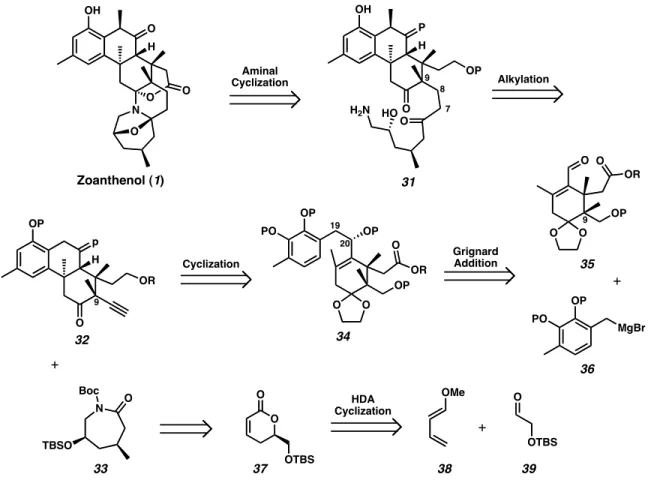
Synthetic route from glycidol
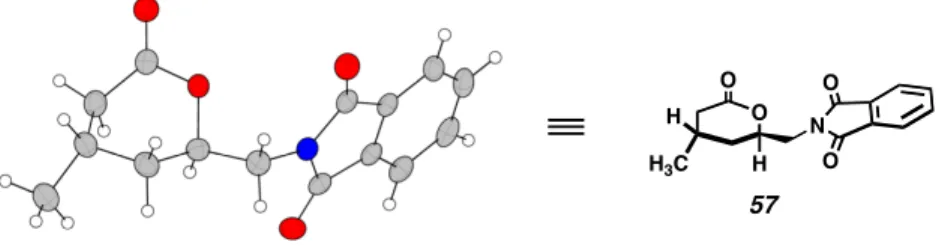
Installation of the primary amine was achieved by hydrogenolysis of the benzyl ether followed by Mitsunobu reaction with phthalimide to provide crystalline intermediate 57. Protection of the amide as t -butylcarbamidate provided 33 , the key retron for the overall synthesis of 1 .
Advancing enantiopure synthetic intermediates
Mild desilylation in the presence of a resin-bound acid source provided the unprotected alcohol (–)- 65 , which was converted to caprolactam (–)- 33 as described previously ( Schemes 7–8 ). All intermediates in the enantioselective pathway were analytically identical to those in the racemic pathway by 1H NMR and 13C NMR analysis.
Endgame for zoanthenol
- Conclusion
After all reagents were dissolved, the reaction mixture was cooled to 0 °C and DEAD (1.707 mL, 10.79 mmol) was added dropwise to the stirred solution. The biphasic mixture was allowed to warm to room temperature with vigorous stirring, then transferred to a separatory funnel.
EtOAc:hexane) as a waxy solid: m.p. The Chemistry of Enamines, Part 1 John Wiley & Sons, Inc.:. The weighted R-factor (wR) and goodness of fit (S) are based on F2, conventional R-factors (R) are based on F, with F set to zero for negative F2.
Uemura’s protocol for the oxidation of alcohols
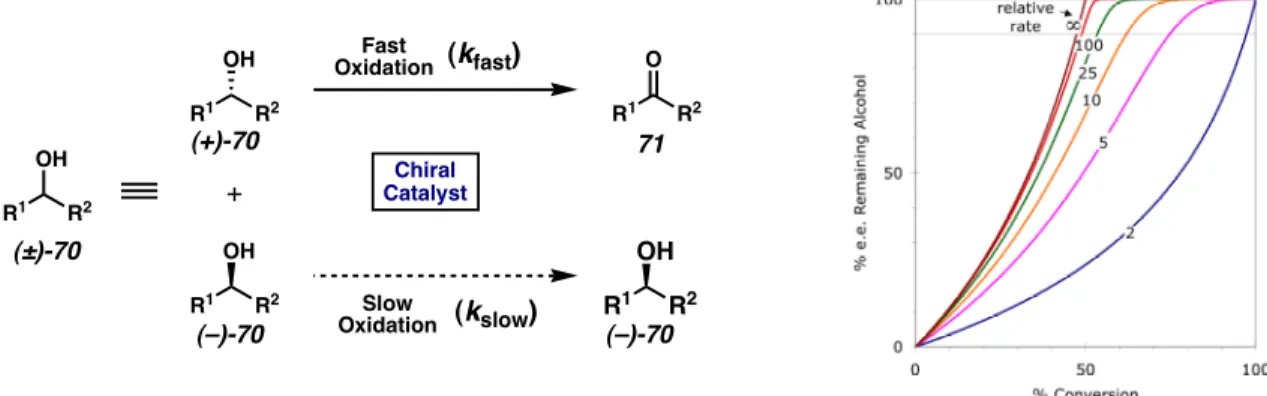
In particular, the reaction conditions require heated mixtures of the flammable solvent toluene under a pure atmosphere. Interestingly, the reactivity of the system could be restored after introducing 3 equivalents of sparteine towards the Pd(sp)Cl2 complex (entry 3). However, the catalytic activity was initially restored upon incorporation of the achiral base triethylamine (entry 3).
As shown in Table 4, increased amounts of sparteine still have a positive effect on the selectivity and overall reaction rate (entries 1-3). That a combination of the exogenous bases sparteine and Cs2CO3 gives the optimal reaction rate and selectivity is interesting.
Tandem oxidation/intermolecular Si-transfer
Titration of the exogenous non-stabilized alcohol n-BuOH in the reaction (Table 5) appears to suppress the formation of TMS ether 84 (entries 2–4). After observing a dramatic dependence of the rate of the oxidative kinetic resolution with the inclusion of the primary alcohol n-BuOH, a wide range of alcohol additives were evaluated for their ability to improve catalysis. In all cases, inclusion of 1.0 equiv. of the exogenous alcohol additive increased reactivity compared to the deprived system (point 1).
In the case of n -BuOH, the inclusion of 2.0 equiv results in a decrease in the overall selectivity (entry 3), while the increase to 8.0 equiv appears to negatively affect both the selectivity and the reactivity of the system (entry 4). While the purpose of the carbonate base fit well with our mechanistic model for oxidative kinetic resolution, the effect of t-BuOH remained more subtle.
Formation of a (sp)Pd(II)carbonate complex
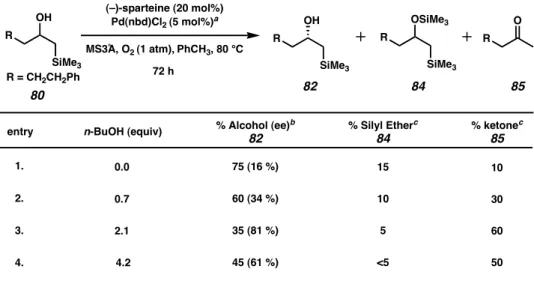
A number of possible H-bond/catalyst and H-bond/halide interactions are outlined in Figure 6 and include A) hydrogen bonding to, or solvation of, a chlorine substituent on the [Pd(sparteine)Cl2] complex, leaving the complex is activated toward alcoholysis, B) then aids in the solvation of another chloride, opening a coordination site for β-hydride elimination to occur, C) facilitates the release of the product, D) reductive elimination of HCl causes, generates a palladium (0) species and its reoxidation to palladium (II) by E) which helps in the opening of a peroxopalladium species for a palladium hydroperoxide, and F) which participates in the turnover exchange of the palladium hydroperoxide to the palladium alkoxide complex. When using preformed Pd(sp)Cl2 with excess sparteine under an atmosphere of molecular oxygen and in the presence of 3A molecular sieves, the reaction rate and catalytic efficiency were found to be very sensitive to the nature of the solvent. Particularly striking is the improvement in selectivity of the non-benzyl alcohol shown in point G.
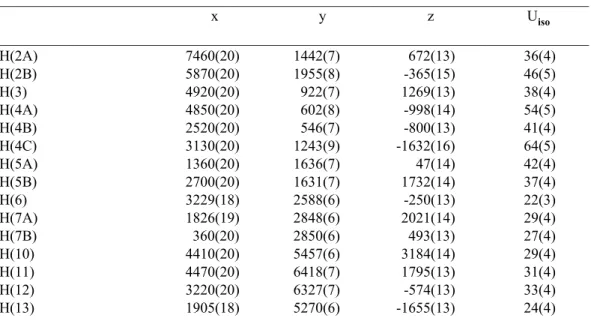
Previously, this substrate was difficult to resolve at high ee, exhibiting a low selectivity in the oxidative kinetic resolution process (s=6.6). Although the enantioselective oxidative kinetic resolution developed in our laboratories offers one of the most convenient methods for obtaining a secondary benzyl alcohol in very high enantiomeric excess (>95% ee), a potential disadvantage with the resolution is the limited theoretical yield.
Retrosynthesis of a polymethoxydiene natural product
An abundance of potential targets accessible from an oxidative desymmetrization reaction is found in polyol/polyether natural products. For example, the isotactic polymethoxydienes30 such as 91 (Scheme 5) are a class of marine natural products well suited for synthesis by an oxidative desymmetrization approach. Often complex, these pseudo-symmetric stereochemical arrays are excellent targets for bidirectional synthesis followed by enantioselective desymmetrization.31,32 A common approach to isotactic polymethoxydiene 91 may involve a late-stage oxidative desymmetrization of meso-diol 92.
A palladium-catalyzed oxidative addition via the diene moiety with 2 equiv of benzyl alcohol via the diene functionality directly provided the anti-tris-ether 96 as the single observed diastereomer. Alternatively, the syn-tris-ether series 98 is obtained according to known preparations34 by formal Diels–Alder addition of singlet oxygen across diene 97, followed by reduction of the peroxy-ether and benzylation of the intermediate diol.
Variable ether arrays from a common source
To demonstrate our desymmetrization technique, we developed a synthetic approach that allows access to a variety of meso-diol stereochemical arrays from a common synthetic intermediate. As shown in Scheme 6, the readily available alcohol 9433 can be protected in various ways as the TBS ether 97 or the benzyl ether 95. Further development of the anti-tris ether 96 continues in Scheme 7 with dihydroxylation and oxidative cleavage of the intermediate. diol with Pb(OAc)4, to generate the meso-dialdehyde 99.
Importantly, no epimerization of the sensitive α-benzyloxy stereocenter is observed using this two-step oxidation protocol. Mesodialdehyde 99 is converted to a variety of mesodiols by a diastereoselective, chelate-controlled addition of an aryl nucleophile, yielding 100 or 101 as a single observed diastereomer.
Advancing the anti-ether array
Advancing the syn-ether array
Establishing relative stereochemistry
Enantioselective oxidative meso-diol desymmetrization
Oxidative desymmetrization of the complex meso-diols and 104 was achieved with a catalytic amount of Pd(sparteine)Cl2 in the presence of excess (–)-sparteine under an atmosphere of oxygen (Scheme 10). These aldehydes are excellent starting materials for the generation of complex series of mesopolyols using known methods.37 Subsequent oxidative desymmetrization of mesodiol products generated from aldehydes 113 and 114 will yield extended chiral ketoalcohol motifs.
Homologation to the skipped framework
To date, we have synthesized a number of relevant benzyl alcohols.29 For example, the protected amino alcohol 115 is an intermediate used in the synthesis of the antidepressants fluoxetine·HCl (Prozac®) 116 and tomoxetine 117 (eq 1).38,39. In one of the most selective palladium-catalyzed oxidative kinetic solutions observed to date, the enantiopure allylic alcohol 120 was synthesized (eq 3). Specifically, our analysis focused on diamine chain length, hybridization, and flexibility, taking into account the properties of the parent ligand, (–)-sparteine.
Although several of the diamines examined yielded only very low conversions, some trends in the impact of ligand structure on reaction productivity can be observed. It should be noted that the facile synthesis of effective sparteine mimics has been a challenge for the broader community.61 However, in the course of these studies, we have outlined some structural requirements for a competent diamine ligand for the palladium-catalyzed OKR. .
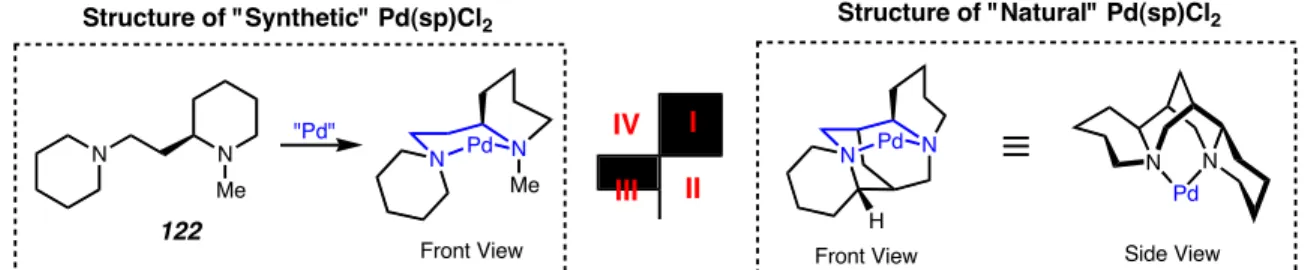
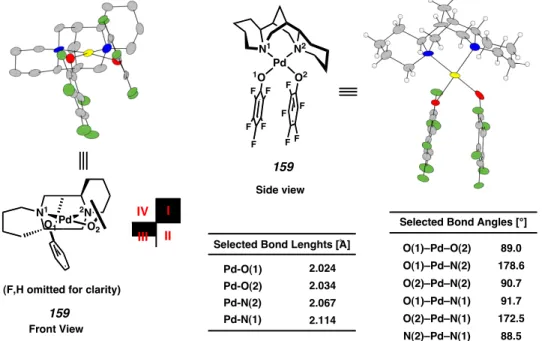
Regioselective formation of a palladium-alkoxide
Dissection of the subsequent steric interactions involved for β-hydride elimination of the diastereomeric Pd alkoxides 155 and 156 provides the basis for a predictive model. In this way, Pd alkoxide 155 can achieve a low-energy reactive conformation for β-hydride elimination by placing the RS group in a minimized eclipse interaction with the Cl group, which has now migrated into the open quadrant II as shown in the transition structure 157 .In the absence of a significant destabilizing steric interaction, β-hydride elimination proceeds to generate the ketone.
In contrast, Pd alkoxide 156 experiences a severe steric interaction between the RL group and the axial Cl group, destabilizing a reactive conformation for β-hydride elimination, as evidenced by the unfavorable transition structure 158. We attempted to investigate the sterics of the X-type ligand by preparing (sp)Pd(II)Br and (sp)Pd(II) variants using different methods.
A palladium phenoxide
The isolated products are obtained after filtration of the reaction mixture through celite, concentration of the solvent under reduced pressure and flash chromatography on silica gel. Anhydrous Cs2CO3 powder (0.4 mmol, 0.4 eq.) and a chloroform (2.0 mL) solution of the racemic alcohol (1.0 mmol, 1.0 eq.) were introduced and the reaction mixture was maintained at room temperature. The reaction flask was sealed with a septum and the contents were stirred at room temperature for 5 min before anhydrous powdered Cs2CO3 (0.4 mmol, 0.4 equiv) and a chloroform solution (2.0 mL) alcohol (1.0 mmol , 1.0 equivalent).
Powdered anhydrous Cs 2 CO 3 (0.4 mmol, 0.4 equiv.) and a chloroform solution (2.0 mL) of the alcohol (1.0 mmol, 1.0 equiv.) were introduced and the reaction mixture was kept at room temperature. The reaction was kept at room temperature for 1 hour and then quenched by the addition of saturated aqueous NaHCO3. The reaction was warmed to room temperature for 2 hours, then diluted with Et 2 O (30 mL) and washed with water (2 x 20 mL).
The reaction was kept at room temperature for 3 h, then quenched by the addition of aq saturated NH 4 Cl (5 mL), then diluted with water (5 mL).