The second part of this thesis investigates the use of niobium- and pyridine-exchanged heteropolyanions as catalyst precursors for the selective oxidation of light alkanes with dioxygen. Niobium- and pyridine-exchanged heteropolyanions (HPAs) have been shown to produce highly active and selective catalysts for the oxidation of propane and n -butane to acrylic acid and maleic acid, respectively, by Davis and co-workers. Previous work demonstrated the incorporation of an acidic site into OFMS and the use of the material for shape-selective acid catalysis.
The second part of this thesis investigates the use of niobium- and pyridine-exchanged heteropolyanions as catalyst precursors for the selective oxidation of light alkanes with dioxygen. The use of the exchanged heteropolyanions for the selective oxidation of light alkanes has been previously reported by Davis and co-workers. The source of the acetic acid is investigated by examining the oxidation of ethylene and D2O at low conversions.
Heterogeneous Base Catalysis
Leaching of active sites over a long period of time is also a concern and may limit the industrial utility of the catalyst.12.
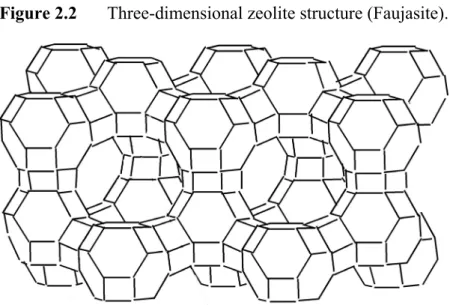
Zeolites
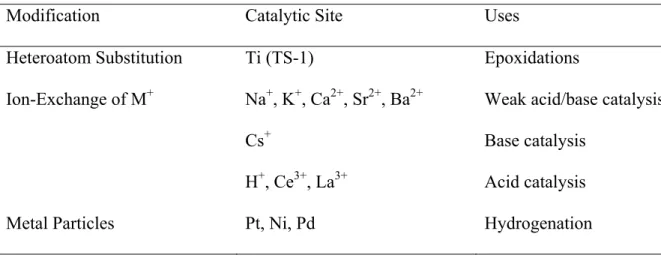
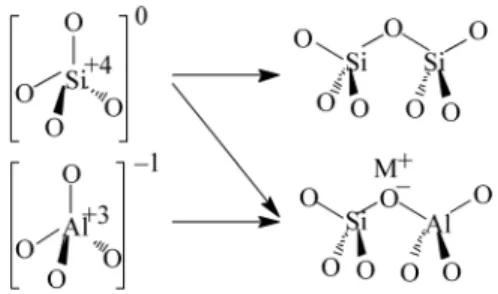
Subsets within each structure code identify the specific composition of heteroatoms (e.g., Al) that share the same structure. The catalytic activity of zeolites for reactions can be tailored by appropriately selecting the structure, cations and framework composition (see Table 2.1). Post-synthetic treatment of the zeolite can also be performed (ion exchange with inorganic cations, calcination and addition of guest molecules) to modify the zeolite for catalytic use.
Objectives
The SDA, TEAF, was removed from the pores by a variety of solutions while maintaining the integrity of the zeolite structure and the organic moiety. Acid-containing OFMSs were shown to be useful as acidic catalysts in the reaction of cyclohexanone and ethylene glycol and also demonstrated shape selectivity in the reaction of 1-pyrenecarboxaldehyde with ethylene glycol. The goal of this project was to incorporate a strong base site within the zeolite pore space for base catalysis.
Sol-Gel Science: Die Fisika and Chemistry of Sol-Gel Processing; Academic Press, Inc.: Boston, 1990.
Introduction
Here we specifically address the use of organic-inorganic hybrid materials for use as catalysts. A recent special issue of Chemistry of Materials has brought together selected topics concerning organic-inorganic composites and deals with both materials science and catalytic applications of these materials.3.
Organic-Inorganic Hybrid Materials
The formation of these sites was attributed to the breaking of the framework Si-O bonds. 13C CP/MAS NMR confirmed the integrity of the proline functional groups after grafting and modification. IR spectroscopy and elemental analyzes were used to confirm the complete removal of the SDA.
13C CP/MAS NMR data confirmed the integrity of the vinyl groups, ie. the functionality of the vinyl was not changed during the synthesis and extraction steps. The presence of the methacrylate part was observed by IR spectroscopy in already produced samples. 13C CP/MAS NMR was used to verify the presence of ethane and ethylene functional groups.
Loss of order was observed in the materials that had higher concentrations of the organosilane. 13C CP/MAS NMR data was used to prove the integrity of the imprint in the silica samples.
Future Directions of Organic-Inorganic Hybrid Materials
The one-aminopropyl functionalized silica showed the expected amide (1710 cm-1) and carboxylic acid (1647 cm-1) peaks in IR spectroscopy after adsorption of azelaoyl chloride, while the two-aminopropyl functionalized silica had only a single, broad amide band in the IR , in accordance with the two-point bond (Scheme 3.34).
Acknowledgements
Example of grafting organosilanes onto silanol-containing surface
Knoevenagel condensation of benzaldehyde and ethyl cyanoacetate
Example of a nitroaldol condensation
Example of a Michael addition reaction
Monoglyceride synthesis from glycidol and fatty acids
Immobilization of imidazole on ordered, mesoporous materials
Aldol condensation and intramolecular Michael addition of benzaldehyde and 2’-hydroxyacetophenone to flavanone
Synthesis of coumarins and chromenes via base catalyzed condensations
Preparation of silica-supported phase transfer catalysts. Adapted from ref
Acid catalyzed addition of 2-methylfuran and acetone
Synthesis of (R)-1-phenyl-propan-1-ol from benzaldehyde and diethylzinc
Co-condensation of organosilanes and tetraalkoxysilanes for the assembly of ordered, mesoporous, organic-inorganic hybrid solids
Phosphorous-containing organosilane and mesoporous material syntheses
Shape-selective behavior of OFMS catalysts
Interaction modes of phenylphosphonic acid with guanidine-
One (A) and two (B) point interactions of aminopropyl-imprinted silicas
Acid and base catalyzed reactions in one-pot reactions. Adapted from ref
Introduction
Rodriguez and colleagues demonstrated the incorporation of a covalently bonded ammonium into a mesoporous material to enable the exchange of hydroxide within the pore space.1 The work of Tsuji et al. Modifications to the previously reported synthesis of OFMSs to adopt this strategy would making it possible to place a strong Brønsted base in the pore space of a zeolite.
Experimental Section .1 Silica Zeolite Beta Preparation .1 Silica Zeolite Beta Preparation
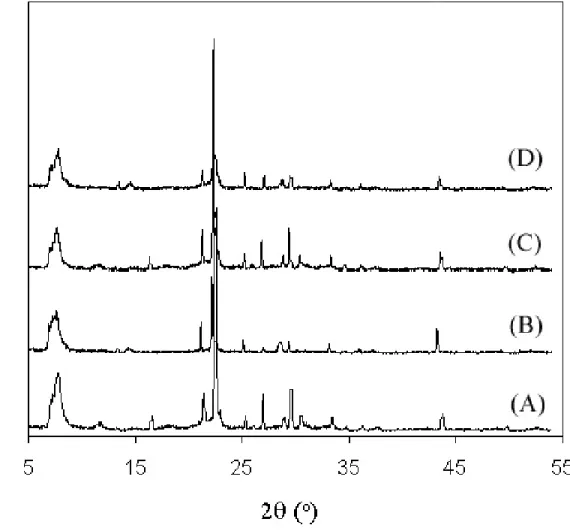
The ammonium functionalized organosilane (1.195 g) was then added to the ethanol solution. (It is important to note that without the use of ethanol, an inhomogeneous gel is formed, which may result in a phase separation of the ammonium functionality in the resulting solids.) The silane/ethanol solution was added to the aqueous TEAF solution dropwise via pipette with vigorous stirring . The silane mixture was added to the aqueous TEAF solution dropwise via pipette with vigorous stirring. The crystals of bromopropyl functionalized *BEA (Br-*BEA) were recovered by filtration and washing as above.
OFMSs were also made with a covalently bonded phosphonium group to compare the stability of the ammonium and phosphonium groups. The silane/ethanol solution was added dropwise via syringe to the aqueous TEAF solution with vigorous stirring. The removal of the SDA from zeolites is usually accomplished by calcination at elevated temperatures under air.
However, since such a treatment would also remove the organic functionalities in the OFMS materials, extraction was used to remove the TEAF from the pores of the zeolite.2 The as-produced OFMS crystals isolated from the autoclave were overnight in an acidified stirred aqueous pyridine solution (1 part aqueous 1M HCl to 1 part water) at 80oC to extract the SDA. Phosphonium-functionalized OFMSs can also be prepared by nucleophilic displacement of a halogenated moiety after preparation and extraction of the zeolite. This technique allows one to study the behavior of the organic parts during various treatment methods, such as extraction, by 13C CP/MAS.
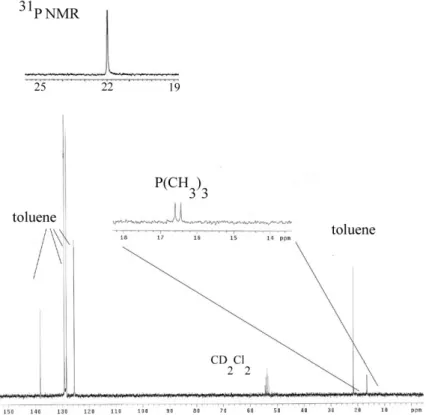
The materials were analyzed by 13 C and 29 Si CP/MAS NMR and 31 P MAS NMR to determine the success of the reaction. After extraction and quaternization, phosphonium-containing *BEA samples were ion exchanged to investigate the presence of the phosphonium group and to exchange the hydroxide in the materials for basic catalysis testing. Quantitation of halide removal (and nitrate uptake) by ion exchange was performed by titration of the filtrate with silver nitrate.
After nitrate exchange, phosphonium-containing materials were also exchanged with sodium tetraphenylborate (NaBPh4) to determine whether functional groups were accessible; i.e., if the phosphonium moieties were within the zeolite pore space.
Results and Discussion
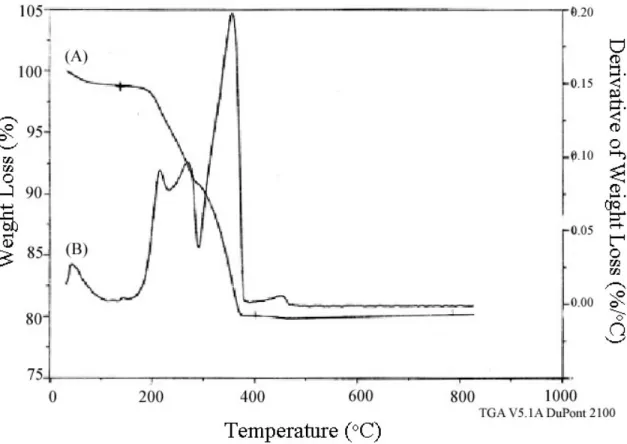
100oC, 13C CP/MAS NMR showed the onset of decomposition of the ammonium moiety (see Figure 4.14C). Phosphonium-containing materials with different anions were prepared by the nucleophilic displacement of the halogenated propyl-organosilane and. After nucleophilic displacement (P+Br-CPG), 13C CP/MAS NMR indicated the presence of the expected shifts from the phosphonium moiety,7 although some of the original material remained unreacted.
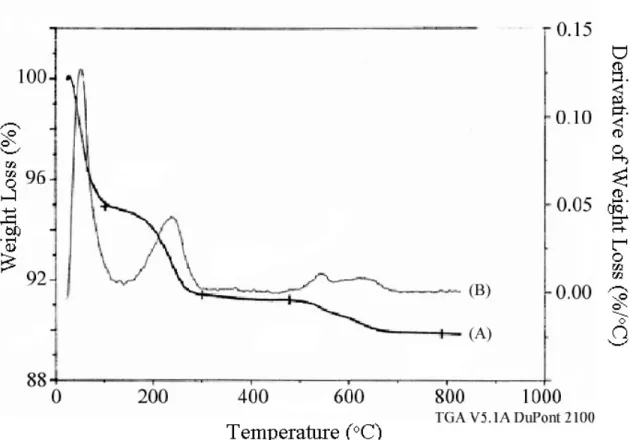
Due to the improved stability of the phosphonium moiety on CPG-240, efforts have been made to prepare phosphonium-containing OFMSs using Br-. Extracted Br-*BEA and Br-CPG were also reacted with triphenylphosphine to investigate whether the bromopropyl functionality was located within the pores of the zeolite or on the surface (see Scheme 4.6). P+Ph3Br--*BEA or P+Ph3Cl--*BEA, indicating that the organic moieties are located within the pore space of the zeolite (see Figure 4.21).
After extraction with acetic acid, only 0.16 mmol/g remained in Pquat-*BEA (removing 61% of the phosphonium species). Binding of the phosphonium organosilane to CPG (Pquat-CPG) resulted in an increase in phosphonium load of 0.83 mmol/g (see Figure 4.23). To investigate the location of the phosphonium group, P+Br--*BEA materials were ion exchanged with NO3- and BPh4- (see Scheme 4.7).
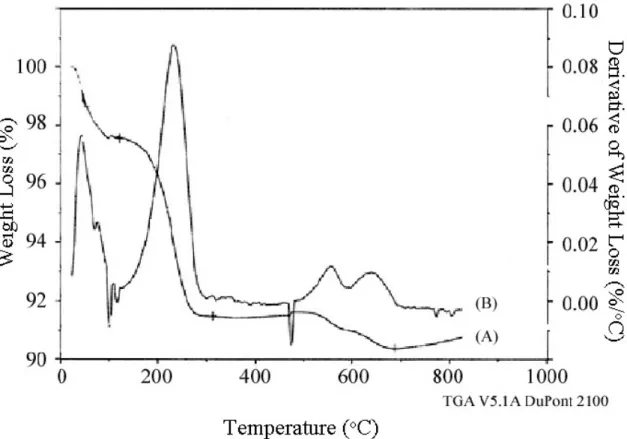
Quantitative titration of the filtrate for Br- indicated that the ion exchange to nitrate in P+Br--CPG was only 56% exchanged (see Table 4.2). No bromides could be detected with the AgNO3 assay in the P+Br--*BEA exchange, despite the appearance of the nitrate in the IR. This nitrate recovered may simply be due to the removal of physisorbed nitrate, as no Br- was observed in the nitrate exchange filtrate.
After the hydroxide exchange, part of the P+OH-*BEA and P+OH-CPG was added to a 0.0126 N NaBr solution.
Introduction
The reactions (see scheme 5.1) took place at 60oC and were observed from a few hours to a few days, depending on the catalyst. The ability of the P+OH--*BEA material to perform shape-selective base catalysis was tested by the Knoevenagel condensation reaction of 1-pyrenecarboxaldehyde, 4, and malononitrile (see Scheme 5.2). The reactions proceeded similarly, except that dichloroethane (purchased from Alfa Aesar, reflux dried with calcium hydride, distilled under vacuum, and degassed by a freeze-pump-thaw cycle with liquid N2) was used as the solvent because 4 has limited solubility in acetonitrile .
The GC/FID analysis for these reactions used a temperature program from 40oC to 280oC at a rate of 15o/min to elute the pyrene compounds. Note that the pyrene reaction product, 5, was not reliably eluted from the column, so reaction calculations were based on the conversion of 7 over time. 2-Cyclohexen-1-ene, 6, was purchased from Aldrich and nitromethane, 7, was purchased from Aldrich. The reactants 6 and 7. about 15 mmol each) were measured and sealed in a glass vial with acetonitrile.
The solution is added to the reaction vessel containing the dehydrated catalyst under argon via a cannula. The reactions (see Scheme 5.3) were carried out at 60 °C and monitored for several hours to several days, with sample preparation for GC/FID analysis as described above. The sample was analyzed by GC/FID with a temperature ramp from 40oC to 200oC at a rate of 10o/min.
As controls, tert-butylphosphonium hydroxide, (TBPOH, purchased from Gelest, Inc. as a 40% solution in water, homogeneous reaction), hydroxide-containing ion exchange resin (purchased from BioRad), extracted Si-*BEA, and CPG were also tested. The Michael Addition reaction of methyl vinyl ketone, 9, (purchased from Aldrich and stored in a freezer before use) and ethyl cyanoacetate, 10, (purchased from Aldrich) was also used as a base reaction for the materials (see Scheme 5.4). The vials were capped and allowed to stir at room temperature for several hours to days.
The sample was analyzed by GC/FID with a temperature ramp from 40oC to 200oC at a rate of 10o/min.
Results and Discussion
The catalysts (70 mg) were not dehydrated before reaction for this reaction series, and the reactions were carried out in air.