Multiple of concentration 3.5 times Distillation temperature 42~48oC 2.3 Step III: Purification and packaging of the final product. Any lot of the product or intermediates during manufacture that do not meet the specification can be reprocessed by repeating the procedures described in the section on description of the manufacturing process and process control. Against this background, the development process has gone through three major phases: development and confirmation of the manufacturing process, trial production on a pilot scale and trial production on a commercial scale.
Trial Production at Pilot Scale
Trial Production at Commercial Scale
Refining Process Development
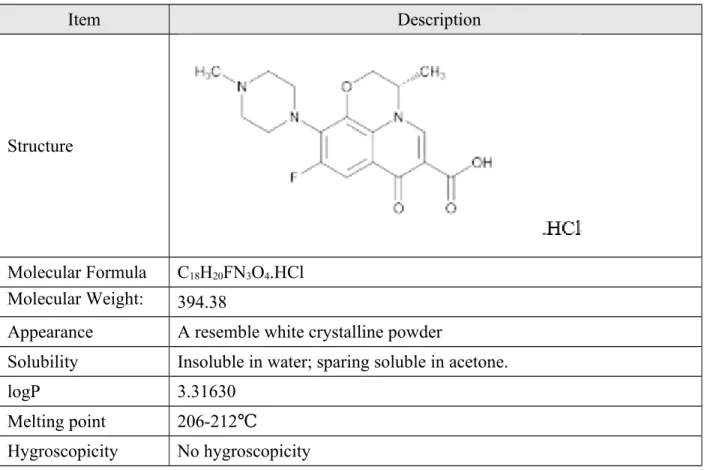
Detailed information on the manufacturing process and controls is presented in Section 3.2.S.2.2, Description of the Manufacturing Process and Process Controls. d) The chemical structure of levofloxacin API is elucidated using infrared (IR) spectroscopy, nuclear magnetic resonance (NMR) spectroscopy, mass spectrometry (MS) elemental analysis and thermal analysis. f) Specific absorption peaks of -F, saturated -CH, -CO- and -N are found in the IR spectra. These observed results are consistent with the structure assignment of levofloxacin. j) The UV, IR and NMR spectra of the levofloxacin sample and USP Levofloxacin RS are consistent. l) The information obtained from the above UV, IR and NMR spectra provides supporting evidence that the chemical structure of levofloxacin produced in JIUZHOU PHARM is consistent with the same structure of the reference standard USP levofloxacin. There are no polymorphic forms. o) Summary of studies conducted to determine the particle size distribution of the active ingredient:.

Organic Impurities
Inorganic Impurities
- S.4.1 Specifications
- S.4.2 Analytical Procedures
- S.4.3 Validation of Analytical Procedures
- S.4.5 Justification of Specifications
- S.7.2 Post-approval Stability Protocol and Stability Commitment
- S.7.3 Stability Data
- P Body of Data – Finished Drug Product (Levofloxacin Tablets 500mg)
- P.1 Description and Composition of the Drug Product (Levofloxacin Tablets 500mg)
- P.2 Pharmaceutical Development (Levofloxacin Tablets 500mg) P.2.1 Component of Drug Product
- P.2.1.1 Drug Substance
- P.2.1.2 Excipients
- P.2.2 Finished Pharmaceutical Product/Drug Product P.2.2.1 Formulation Development
- P.2.2.2 Overages
- P.2.2.3 Physicochemical and Biological Properties Refer Section P.2.1.1
- P.2.3 Manufacturing Process Development
The retention time of the main peak of the sample corresponds to that of the standard solution taken in the analysis. The retention time of the main peak of the sample corresponds to that of the standard solution taken internally. The retention time of the main peak of the sample corresponds to that of the standard.
The retention time of the main peak of the sample solution corresponds to that of the standard solution, as obtained in the analysis. Critical process parameters of the manufacturing method were identified and the process was validated.
Preformulation characterization of the drug substance identified particle size and solubility as critical factors for product performance. The compatibility of the drug substance was evaluated with all inactive ingredients and no evidence of incompatibility was observed. During process development, the manufacturing steps and critical process parameters that controlled the critical factors for product performance were identified and taken care of.
The formulation was optimized using a trial and error method considering the decay time as a response factor.
P.2.4 Container Closure System
For a device supplied with a multi-dose container, a summary of study results demonstrating reproducibility of the device (e.g., consistent delivery of the target volume for the lowest target dose):
P.2.5 Microbial Attributes
P.2.6 Compatibility
P.3 Manufacture (Levofloxacin Tablets 500mg) P.3.1 Manufacturer(s)
P.3.2 Batch Formula
After setup, check the initial plates for certain standard parameters and record the observations in the initial parameter check - record and sign it if necessary. Collect the tablets in a HDPE container with a double polyethylene coating, weigh them and mark them accordingly, enter the data on the weighing of the tablets, when weighing the tablets - record and, if necessary, sign. Enter the details of the inspection in the tablet inspection process - record and the details of the weighing of tablets inspected, in the weighing of tablets inspected - record and sign it if necessary.
Enter the name of the people who performed the tablet inspection process, and the length of time, in the tablet inspection process. Record this and sign it as required. Calculate the percentage yield of tablets by entering the yield data at all stages, in the yield reconciliation summary – record and sign this if necessary. Weigh the above coated tablets and label them appropriately, enter and record the weighing details of coated tablets when weighing coated tablets and sign them if necessary.
Enter the Inspection Details in Tablet Inspection Process – record and weigh details of inspected tablets, in weighing of inspected tablets – record and record them as required. Enter the name of the persons who did the tablet inspection process and length of time in tablet inspection process record and sign it as required. Calculate the % yield of tablets by entering yield details at all stages, in yield. Reconciliation Summary – record and sign it as required.
Set the machine speed to 20 to 30 cuts per minute and record the observations on the initial checks before starting the machine - record and sign as necessary.

P.3.4 Controls of Critical Steps and Intermediates
Start the machine and check the blisters Strips for blister quality such as empty blisters, missing, cracked, broken tablets, color change, sealing, stains and appearance & for leak test as per SOP and record the observations on initial checks before starting the machine – note from BMPR , and sign it as required.
P.3.5 Process Validation and/or Evaluation
Three validation batches of commercial-scale Levofloxacin tablets were produced using the current manufacturing process. Critical parameters identified during the development phase are checked at appropriate process intervals with acceptance criteria.
General Information
Final Product Analysis
Conclusion
- P.4 Control of Excipients (Levofloxacin Tablets 500mg) P.4.1 Specifications
The specification of Starch
The specification of Hypromellose
The specification of Titanium Dioxide
The specification of Povidone K-30
- P.4.2 Analytical Procedures
- P.4.3 Validation of Analytical Procedures
- P.4.4 Justification of Specifications
- P.4.5 Excipients of Human or Animal Origin
- P.4.6 Novel Excipients
- P.5 Control of Drug Product (Levofloxacin Tablets 500mg) P.5.1 Specifications
- P.5.2 Analytical Procedures
- P.5.3 Validation of Analytical Procedures
All excipient specifications are in accordance with the USP monograph. a) Summary of validation information for analytical procedures for additional tests (where applicable):. All analytical procedures for excipients are presented based on the relevant USP monograph and general chapter. Solution: Dilute a portion of the clear filtrate used to prepare the test preparation and a portion of the stock solution used to prepare the standard preparation prepared in the test with acidified methanol prepared according to the dissolution instructions to obtain solutions containing approximately 10 mg of levofloxacin per ml .
The retention time of the largest peak of Levofloxacin in the chromatogram of the assay preparation corresponds to that in the chromatogram of the standard preparation as obtained in the assay. Dissolve an accurately weighed amount of the powdered tablets, corresponding to approx. 0.1 g Levofloxacin, in water in a 50 ml volumetric flask and shake thoroughly. Dilute to volume with water, mix well and filter. Standard resolution – Transfer approx. 90 mg USP Levofloxacin RS, accurately weighed, into a 250 mL volumetric flask, add 10 mL acidified methanol and shake to dissolve.
Procedure: Transfer 10.0 ml of a filtered portion of the solution to be tested into a 250 ml volumetric flask, dilute with 0.1 N sodium hydroxide to volume and mix. Simultaneously determine the absorbances of this solution and the standard solution at the wavelengths of maximum and minimum absorbance at approximately 308 nm and 350 nm, using 0.1 N sodium hydroxide as a blank. Calculate the amount, in mg, of C12H15N3O2S dissolved using the formula:. where C is the concentration, in mg per ml, of USP Levofloxacin RS in the standard solution; and AU and AS are the differences in absorbance between 308 nm and 350 nm obtained from the solution under test and the standard solution, respectively.
Procedure—Determine simultaneously the absorbances of the standard solution and the test solution at the wavelength of maximum and minimum absorption at about 308 nm and 350 nm, using 0.1 N sodium hydroxide as blank.
Summary of the Verification Results of Assay System Precision
- P.5.4 Batch Analyses
- P.5.5 Justification of Specifications
- P.6 Reference Standards or Materials (Levofloxacin Tablets 500mg)
- The specification for Printed aluminium foil
- The test data of PVC Tray
- The test data of Carton
- P.8 Stability (Levofloxacin Tablets 500mg)
- P.8.2 Post-approval Stability Protocol and Stability Commitment
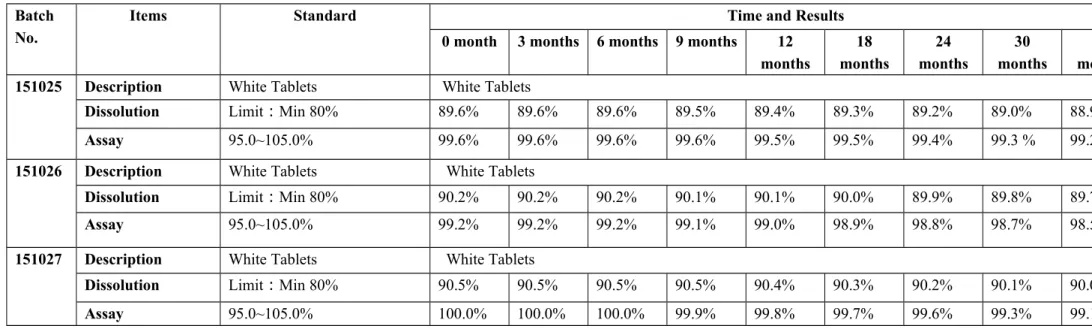
The production information relating to three commercial batches of Levofloxacin Tablets 500 mg is summarized in the table below. The specifications and analytical procedures for Levofloxacin tablets 500 mg correspond to those in the USP monograph for Levofloxacin tablets 500 mg. The identification method by UV is intended to identify the molecular structure of the compound and is described as a specific identification test in accordance with ICH.
The retention time of the main peak of the sample solution should correspond to that of the standard solution as obtained in the test. A specification is proposed to meet the requirement that is consistent with the acceptance criteria of the Levofloxacin USP Tablet Monograph. USP Reference Standards are used as primary standards in the testing of levofloxacin 500 mg tablets.
Please see Table P7-1 below for a summary of the container/closure system and packaging configuration for delivery batches of Levofloxacin 500 mg tablets, including package size, container/closure. -House twist Must be wrapped in suitable rolls with . proper core diameter and outer diameter. Stress studies were performed on a single batch of the product with batch no. Three batches of Levofloxacin 500 mg tablets were subjected to stability studies under conditions of accelerated and long-term stability.
The test will facilitate the establishment of a retest period and appropriate storage conditions and will allow the effect of short-term excursions outside the label storage conditions to be evaluated.
Stability Protocol 1 Selection of Batches
- P.8.3 Stability Data
Twelve-month stability results show that levofloxacin tablets remain chemically and physically stable at 25±2oC and 75%±5% RH. There is only one package configuration for levofloxacin 500 mg tablets and each batch submitted was fully packaged in three pack sizes of 10/box. We undertake that the first three batches of levofloxacin tablets in commercial scale will be subjected to a stability study under long-term RH conditions), and then one commercial batch will be subjected to a stability study every year under the same storage conditions.
All studies not completed prior to submission will continue during the proposed retest period and an amendment to this document containing the additional results will be submitted.
Accelerated Stability Test Results
- P.8.3 Stability Data
They will provide information on determining the retest period and storage conditions, as well as evaluating short-term trips outside of the label storage conditions. Analytical procedures and acceptance criteria follow the protocol described in Section 3.2.P.8.2 Post-Approval Stability Protocol and Stability Commitment. The results after six months show that the Levofloxacin tablet remained chemically and physically stable at 40±2oC and 75%±5% RH.
Long Term Stability Test Results
- A Appendices (Levofloxacin Tablets 500mg)
- A.1 Facilities and Equipment Not applicable
- A.2 Adventitious Agents Safety Evaluation
- A.3 Excipients
- R Regional Information (Levofloxacin Tablets 500mg) There is no other Regional Information about the document
- Literature References (Levofloxacin Tablets 500mg) There is no other Literature References about the document
The study will continue until the final time point test is completed and the results will be submitted as an amendment to this DMF.