Therefore, these enzymes named as PLDs are not classified as members of the PLD superfamily. These enzymes exhibit PLD-like activity and are no less physiologically relevant than members of the PLD superfamily. Historically, there has been a dispute over the classification of some or all of these HKD enzymes as members of the PLD superfamily.
Ponting and Kerr proposed that enzymes with these four conserved motifs are members of the PLD superfamily, as described above [18].
![Figure 1. A schematic of the various enzyme-catalyzed reactions that results in the formation of phosphatidic acid (PA) and some of the cellular functions mediated by PA (figure from [19])](https://thumb-ap.123doks.com/thumbv2/123dok/10730649.0/10.918.198.762.329.717/schematic-catalyzed-reactions-formation-phosphatidic-cellular-functions-mediated.webp)
PMF PLD side view
PMF PLD top view
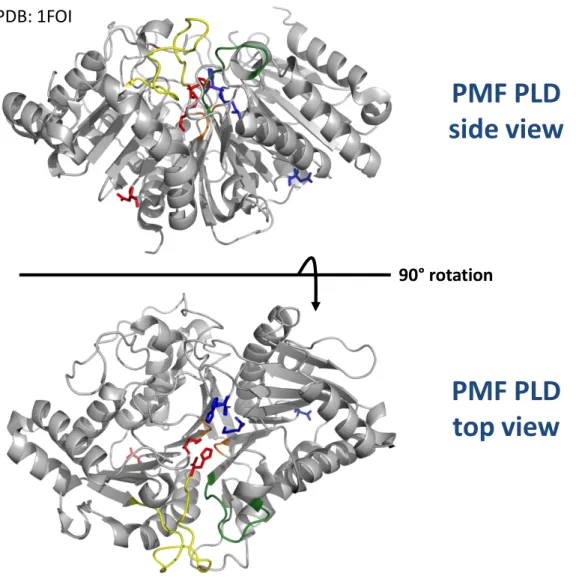
Histidine residues in the double HKD motifs are adjacent to each other at the interface of the Salmonella typhimurium Nuc homodimer. The biophysical properties of the lipids in phospholipid liposomes significantly influence the PLD activity of scooting enzymes (Figure 6). Plant PLD enzymes contribute to the rich history of the PLD superfamily, as the first description of a PLD enzyme was made from carrot. The PLD hydrolytic and transphosphatidylation activities were originally described in plants in 1947 [14], respectively.
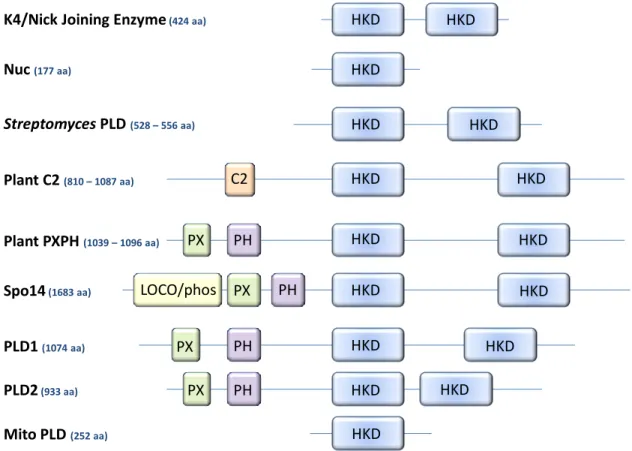
In the mid to late 1990s, following the cloning of the castor bean PLD [109], a wave of plant PLD enzymes were identified, sequenced, and characterized by genetic and biochemical approaches [108]. PLDδ1 and PLDδ2 also retain four of the five basic residues in the conserved PI(4,5)P2-binding motifs flanking the 2nd HKD [113]. Spo14 is predicted to be the only HKD-PLD in this organism and is a member of the PLD superfamily.
PLD activity has been described in Dictyostelium for different growth stages of three different reproductive cycles. However, conserved residues in the predicted α-helix at the C-terminal end of the PH domain are required for catalysis in the liposome activity assay [177] (unpublished data, Henage, Selvy and Brown). For mammalian PLD activity, the specific identity of the residues in this sequence must be conserved.
This enzyme may not be a member of the PLD superfamily because it cannot perform transphosphatidylation [194]. Caspase cleavage of PLD isoforms appears to divergently regulate these enzymes during apoptotic signaling. A critical characterization of the multiple and sometimes overlapping mechanisms by which EGFR signaling activates PLD activity has been performed.
A large body of knowledge about the role of PLD in signaling pathways has been determined using small molecules as tools.

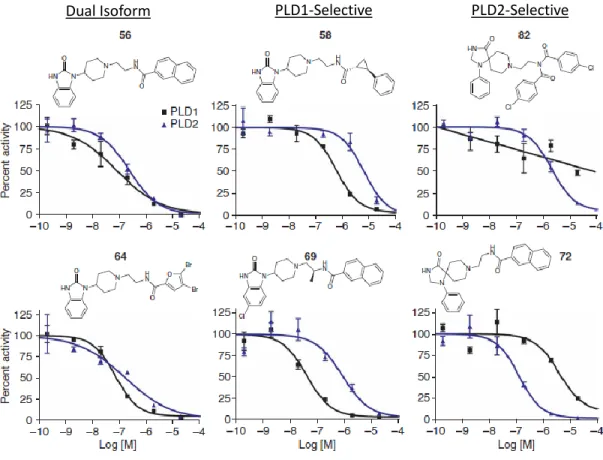
I DENTIFICATION AND D EVELOPMENT OF N OVEL I SOFORM -S ELECTIVE PLD I NHIBITORS
An increase in the overall potency of the benzimidazolone-containing compounds was observed by adding a halogen, such as a chlorine group, at the 5-position of the benzimidazolone. Regardless, rigorous characterization of the VU series compounds to demonstrate both biochemical and cell-based efficacy suggests their potential power as a tool for studying PLD enzymology and signaling. Characterization of the Structure-Activity Relationship of Novel PLD Inhibitors Following the initial description of the VU series inhibitors, a larger library of compounds was generated in efforts to improve the potency and selectivity of these VU series inhibitors.
SAR from this larger library tested the identity and position of the halogen substitution on the benzimidazolone, the available chemical space around the ethyldiamine linker, and experimented with modifications or substitutions to the amide cap in the eastern part of the molecule. Overall potency was almost always increased by halogen substitution of the scaffold, but the most significant increase in potency of PLD1-selective compounds was achieved by bromine rather than chlorine substitution of the 5-position on the benzimidazolone. In contrast to PLD1-selective inhibitors, which were identified from direct modifications of the halopemide lead compound, potent PLD2-selective compounds have been more elusive.
There is no change in the potency of the VU series PLD inhibitors, showing that these compounds act directly to inhibit the enzyme. The specificity of these compounds, taken together with their ability to directly inhibit a highly purified amino-terminally truncated mammalian PLD, demonstrates that the compounds allosterically inhibit the enzyme at a site that does not require the presence of the amino terminus. It is important to note that the potency of the compounds is shifted to the right for PLD1.d311, which may suggest a different compound binding site in the amino terminus, or that the amino terminus increases potency through conformational change or provides support or stability to the small molecule binding site.
Since the first report of the halopemide and VU-series PLD inhibitors, several groups have continued to demonstrate their utility in studying the signaling roles of PLD. In the initial report of the VU series compounds, our laboratory showed that invasive migration was significantly blocked for three highly metastatic cancer cell lines (MDA-MB-231, 4T1 mouse metastatic breast cancer, and PMT mammary tumors).
M OLECULAR M ECHANISM OF I SOFORM -S ELECTIVE P HOSPHOLIPASE D I NHIBITORS
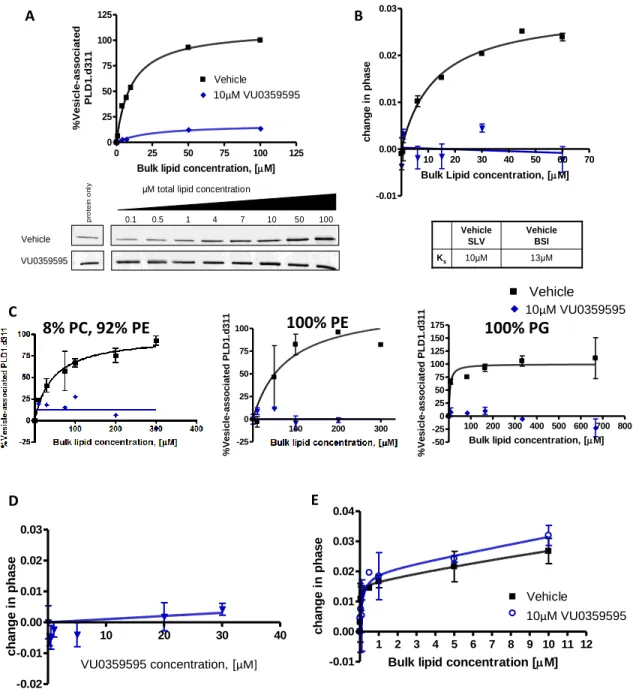
Subsequent studies, in our own laboratory and in others, using these small molecule inhibitors have. This suggests that either the roles of PLD, as distinguished by primary alcohols, have been widely overemphasized, or the mechanism of action of these small molecule PLD inhibitors extends beyond the inhibition of PA formation. To address this discrepancy, we used backscatter interferometry (BSI), a highly sensitive and novel method for measuring protein-small molecule and protein-lipid binding affinities, to characterize the mechanism of action of these small molecule PLD inhibitors.
As such, the nature of the lipid interface has a major influence on the binding affinity for the lipid surface (1/Ks), the accessibility and affinity for substrate (Km) and the catalytic activity of the enzyme. BSI has been validated as a method for measuring picomolar receptor-ligand binding affinities using nanomolar protein concentrations and was used here to measure protein small molecule binding affinities for PLD1. A shift in edge spots is quantified as a phase change of a wave function using Fourier transform.
Recombinant expression of full-length PLD1 is weak and due to the multitude of protein binding partners, full-length protein remains a heterogeneous population after several chromatographic purification steps. This construct consists of a truncation of the PX and PH domains, leaving 16aa of an alpha helix at the C terminus of the PH domain that is required for lipid binding (Henage, Selvy, and Brown, unpublished data). Corrected PLD1c maintains the same biochemical and cellular features of the natural PLD1a and PLD1b, including protein activator response and robust receptor stimulation.
Using BSI and silver-stained pure preparations of the amino-terminally truncated PLD1c splice variant, referred to here simply as PLD1.d311, we demonstrate that this construct directly binds the compounds in a one-site binding model in the absence of lipid (Figure 21). To study PLD kinetics and the mechanism of action of these small molecules.
![Figure 19. Backscattering interferometry (BSI) technique optimized for measuring protein-small molecule and protein-lipid binding affinities (figure modified from[295])](https://thumb-ap.123doks.com/thumbv2/123dok/10730649.0/134.918.200.801.288.692/backscattering-interferometry-technique-optimized-measuring-molecule-affinities-modified.webp)
Interfacial EnzymesInterfacial Enzymes
Compound Concentration, Log [M]
The absence of any interaction with an unrelated phospholipase demonstrates that the VU series of compounds do not non-specifically disrupt protein-lipid binding. This demonstrates the VU series of compounds inhibit catalytic activity at a site allosteric to the substrate binding site. Based on the somewhat hydrophobic nature of the VU series of compounds, it is not surprising that a low affinity binding site remains for the PXPH construct even in the absence of the coupling loop.
Truncation constructs suggest that VU series PLD inhibitors bind PLD directly in a loop region C-terminal to the PH domain. These binding data are consistent with the proposal that His338 and Trp354 contribute to the small molecule binding site in the coupling loop. In vitro studies will further demonstrate that these conserved residues in the coupling loop are integral components of the high affinity binding site for the VU series of compounds.
Point mutations confirm VU-series PLD inhibitors directly bind PLD in a loop region C-terminal to the PH domain. Taken together, these data confirm that the binding loop contributes to the high-affinity small molecule binding site. Truncation studies suggest that the high-affinity small molecule binding site lies in the binding loop (a loop region C-terminal to the PH domain and N-terminal to the start of the catalytic domain).
The VU series compounds bind PLD directly at a loop region C-terminal to the PH domain to allosterically block both bulk lipid interface interaction and catalytic activity. Preliminary in vitro data from several PLD2 constructs suggest that the VU series PLD inhibitors bind the enzyme in the linker loop.
C ONCLUSIONS AND F UTURE D IRECTIONS
It is important to be aware of the mechanistic effects of applying a small molecule to a whole cell, and caution is necessary when interpreting signaling results. Still to be characterized are the protein structural changes that occur in response to small molecule binding. NMR studies of small molecule binding in the binding loop, both alone and in the context of the PH domain, were initiated in collaboration with Chuck Sanders.
However, given the position of the binding site of the small molecule and the noncompetitive bimodal mechanism of inhibition, it is proposed that the conformational change induced by the small molecule results in a closed enzyme. Biochemical data suggest that His338 and Trp354 are integral for binding of small molecules to the binding loop, but these residues are quite far apart according to linear sequence and preliminary homology modeling of the binding loop (approx. 19, figure 29). Based on the significant conformational changes that occur in Akt upon binding of inhibitor VIII (figure 28), it is proposed that the tertiary binding of small molecule PLD1.
Highlighted in blue, the small molecule binding loop is loosely modeled on an amino-terminal region of the protein. Early SLV binding studies with raoxifene further support differences in the effects of the small molecules on the catalytic versus PXPH domains (Figure 30c). Other interesting small molecule mechanistic findings include differences in the composition of lipids that co-purify with PLD or differences in gel filtration elution profiles depending on the small molecules present.
The completion of these studies leaves us with a new class of potent and isoform-selective small molecules that specifically target mammalian PLD, for which we have determined the mechanism of action and the region of the protein that provides the binding site for high-affinity small molecules includes. In characterizing these new compounds, we optimized and validated the use of BSI for measuring both protein-small molecule and protein-lipid binding affinities.

R EFERENCES
Raghu, P., et al., Findings from studies of phospholipase D in model organisms (and a brief update on phosphatidic acid effectors).

![Table 2. Alignment of Catalytic motifs for PLD superfamily (table from [19]) ENZYME SOURCE CATALYTIC MOTIF ACTIVITY](https://thumb-ap.123doks.com/thumbv2/123dok/10730649.0/21.918.153.829.337.758/table-alignment-catalytic-superfamily-enzyme-source-catalytic-activity.webp)
![Figure 6. Substrate presentation in the liposome is highly dependent on the interfacial lipid composition due to biophysical properties of the lipid and headgroup exposure for lipid binding cofactors and substrate (figure from [19])](https://thumb-ap.123doks.com/thumbv2/123dok/10730649.0/34.918.169.797.273.721/substrate-presentation-dependent-interfacial-composition-biophysical-properties-headgroup.webp)

![Table 5. Mammalian PLD regulatory proteins (table from [19])](https://thumb-ap.123doks.com/thumbv2/123dok/10730649.0/89.918.214.757.298.866/table-5-mammalian-pld-regulatory-proteins-table-19.webp)
![Figure 8. G protein coupled receptor activation of PLD through Gα q , and protein kinase C (panel A), Gα 12 and RhoA (panel B), and Gα 13 and Arf (figure from [19])](https://thumb-ap.123doks.com/thumbv2/123dok/10730649.0/100.918.230.730.117.1017/figure-protein-coupled-receptor-activation-protein-kinase-figure.webp)
