The sequence of amino acids in a protein is determined by the sequence of nucleotides in the gene (DNA) that codes for it. Understand the relationship between properties of the amino acids and the structure and function of proteins. What differences are there between an A-T base pair and a C-G base pair in the major groove.
The most common protein-DNA binding motif is the so-called 'helix in the major groove' motif. Look at the helix in the major groove in the file CQT.pdb (provided by supervisor). The scales 1 and 4 are derived from the distribution of amino acids on the surface and in the core of proteins.
Why are there four scales in the diagram above (hint: look up how they were produced). Sort the 20 amino acids by the number of atoms in the side chain (we do not count hydrogens). Something you wouldn't expect once you understand the role of hydrophobicity in amino acid function, packing and placement.
Compare the role of monomers in HIV protease action with the role of each monomer in the TIM dimer.

For each of the following examples, determine which is the better alignment: right or left. CPISRTWASIFRCW CPISRTWASIFRCW CPISRT---LFRCW CPISRTL---FRCW CPISRTRASEFRCW CPISRTRASEFRCW CPISRTK---FRCW CPISRT---KFRCW CPISRTIASNFRCW CPISRTIASNFRCW CPISRT---FRCW CPISRT---HFRCW CPISRTEASDFRCW CPISRTEASDFRCW CPISRT---NFRCW CPISRTN---FRCW CPISRTASIFRCW CPISRT TSASIFRCW CPISRT---TFRCW CPISRTT---FRCW CPISRTGASIFRCW CPISRTGASIFRCW CPISRTA---FRCW CPISRT---AFRCW CPISRTEASNFRCW CPISRTEASNFRCW CPISRTQ---FRCW CPISRT---QFRCW CPISRTFASTFRCW CPISRTFASTFRCW CPISRT---YFRC W CPISRTY---FRCW Secondary Structure Prediction . This can either be the secondary structure as derived from the PDB file (which contains the 3D coordinates) or it can be the predicted secondary structure.
In the following sections, we will use the predicted secondary structure features for better alignment. You know that secondary structure elements fall into four categories: the helical strand turns the rest. If you look at the Chou and Fasman parameters (and other useful data), you will see that there is a relationship between residue type and secondary structure.
As always in bioinformatics, the rules suggested by these parameters are not exactly hard and fast, and there are numerous exceptions. The Chou-Fasman method for predicting secondary structure depends on assigning a set of prediction values to a residue and then applying a simple algorithm to those numbers. The numbers in the first three columns, P(α) P(β) P(turn), are approximately equal to the preferred parameters for the 20 amino acids for α-helix, β-strand, and β-turn, respectively, although there are 1.0 has been added for each preference parameter.
A preference parameter of 0.0 (which corresponds to 1.0 in this column) indicates that something happens as often as expected. Unfortunately, Chou and Fasman's math was not up to today's standards, so these parameters are not calculated correctly, but the correct numbers are not that different from these. You may not rename files, adjust dock size/icons, move items or files to the Recycle Bin or modify them.
Do not swap keyboards, mice, or other equipment between computers without notifying your supervisor.
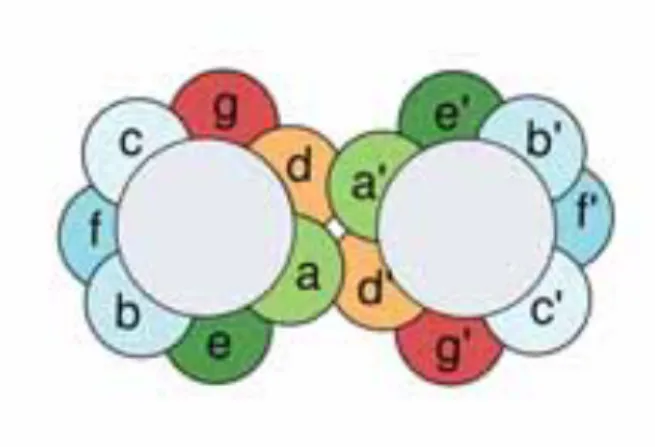
Follow the instructions in the analysis and discussion to better understand the role of structure in sequence alignment. We have already discussed that an alpha helix usually has a hydrophobic side and a hydrophilic side. The gray circles represent the central cores of the two packed helices, and the colored spheres represent the side chains.
Draw the spiral part on the spiral wheels (use the spiral wheel below); both on the circular wheel (a) and on the flat plot (b). In both cases, it is clear that the left part of the sequence does not have a regular structure, whereas the right part is helical. By moving the sequences back and forth, we can find several reasonably bad/good alignments.
We now use this table and indicate preferred positions of residues relative to the first position of the helix. So in the upper series the helix starts with PDQ and in the lower series with LET. Clustal will do well, but with only three identities it would not be satisfied with the result.
Do not bring food or drink into the lab unless it is in your backpack.
What kind of molecule(s) do you see in the active site of the gvbc enzyme. In step 4, gaps in the model are closed, and optionally loops are constructed ab initio. Furthermore, the growing number of structures collected in the PDB makes it easier to find a homologous one every year.
Take "My Beautiful Sequence" from the box above and use the BLAST software to find a protein structure in the PDB that can be used as a homology modeling template. Finally, see what the percent identity is between 'My beautiful sequence (model)' and the best template with known structure in the PDB. Where in the plot does the comparison between My Beautiful Sequence and the 1BQB template fall.
Molecular class-specific information systems (MCSISs) can be a great asset in the homology modeling process. When we build a homology model, we are in the fortunate situation of having an almost perfect profile: the known structure of the template. Alignment techniques such as SSALN make use of this structural knowledge found in the template (Qiu and Elber 2006).
Any insertion or deletion in the alignment represents a structural change to the backbone and therefore cannot be modeled in the previous step. The resulting models can ultimately be combined into a hybrid model, which consists of the strongest points of each individual model. In the second practice, you will validate the homology model and decide where it went wrong.
Amino acids in the binding pocket are colored light blue, the ligand is colored orange. Write the residue names and amino acid numbers in the protein with which the ligand forms hydrogen bonds. In which amino acid direction do you expect the newly inserted group to point.
Which amino acid in the Fleksy result has moved the most compared to the original 1kim structure. For the Fleksy result, you must write the residue names and numbers of the amino acids in the protein to which BTD forms hydrogen bonds.
