Accepted Manuscript
Title: Polyoxomolybdates Catalysed Cascade Conversions of Cellulose to Glycolic Acid with Molecular Oxygen via Selective Aldohexoses Pathways (an Epimerization and a [2+4] Retro-aldol Reaction)
Authors: Asep Bayu, Surachai Karnjanakom, Akihiro
Yoshida, Katsuki Kusakabe, Abuliti Abudula, Guoqing Guan
PII: S0920-5861(18)30620-5
DOI: https://doi.org/10.1016/j.cattod.2018.05.034
Reference: CATTOD 11464
To appear in: Catalysis Today Received date: 10-3-2018 Revised date: 7-5-2018 Accepted date: 15-5-2018
Please cite this article as: Bayu A, Karnjanakom S, Yoshida A, Kusakabe K, Abudula A, Guan G, Polyoxomolybdates Catalysed Cascade Conversions of Cellulose to Glycolic Acid with Molecular Oxygen via Selective Aldohexoses Pathways (an Epimerization and a [2+4] Retro-aldol Reaction), Catalysis Today (2018), https://doi.org/10.1016/j.cattod.2018.05.034
This is a PDF file of an unedited manuscript that has been accepted for publication.
As a service to our customers we are providing this early version of the manuscript.
The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
1
Polyoxomolybdates Catalysed Cascade Conversions of Cellulose to Glycolic Acid with Molecular Oxygen via Selective Aldohexoses Pathways (an Epimerization and a [2+4] Retro-aldol Reaction)
Asep Bayu a,Surachai Karnjanakoma,Akihiro Yoshida,a,b Katsuki Kusakabec, Abuliti Abudulaa, and Guoqing Guana,b*
aGraduate School of Science and Technology, Hirosaki University, 1-Bunkyocho, Hirosaki 036- 8560, Japan.
bDepartment of Renewable Energy, Institute of Regional Innovation (IRI), Hirosaki University, 2-1-3, Matsubara, Aomori 030-0813, Japan.
cDepartment of Nanoscience, Sojo University, 4-22-1 Ikeda, Nishi-ku, Kumamoto 860-0082, Japan
*Corresponding author. Tel.: +81-17-762-7756; Fax: +81-17-735-5411
Email-address: [email protected] (G. Guan)
ACCEPTED MANUSCRIPT
2 Graphical abstract
Highlights
Polyoxomolybdates selectively promotes cascade conversion of cellulose into glycolic acid.
Molybdate in Keggin structure of polyoxomolybdates is the main active site.
The reaction route is via aldohexoses pathway, i.e. an epimerization and a [2+4] retro-aldol.
The catalytic activity of polyoxomolybdates is increased with addition of O2.
ACCEPTED MANUSCRIPT
3
O2 can re-oxidize the reduced heteropoly-blue back to polyoxomolybdates.
Abstract
Selective cascade conversion of cellulose into valuable C2 or C4 products over acid catalysts is still not fully explored because glucose-fructose isomerization followed by fructose conversions is thermodynamically easy to occur during the reaction, leading the formation of C6, C5, C3 and C1 products such as 5-hydroxymethylfurfural (HMF), levulinates, lactates, formates, and so on.
In this study, phosphomolybdates (PMo) with the Keggin structure was found to promote cascade oxidation of cellulose conversion via selective aldohexoses pathways, i.e. an
epimerization and a [2+4] retro-aldol of glucose/mannose, rather than aldo-ketohexoses routes, i.e. a glucose-fructose isomerization and a [3+3] retro-aldol of fructose, which produced glycolic acid (GlycA) (C2) as the main product (~50% selectivity). Either in aerobic or anaerobic state, PMo selectively catalyzed glucose epimerization into mannose at <100 oC (~95% selectivity) and effectively promoted the [2+4] retro-aldol reaction of these aldohexoses at >100 oC. This behavior is completely different from that of molybdate in MoO3 which is only effective for the epimerization reaction (the Bilik reaction). In this cascade oxidation reaction, PMo was reduced into heteropoly-blue (PMored) as observed by the color change of the solution, UV-VIS and FT- IR measurements. In this case, the molecular oxygen was found to reoxidize PMored into PMo, leading the catalytic activity to be remained stable. The results shown in this study provide an insight for the catalyst development on selective synthesis of C2, C4 and/or other novel valuable chemicals from carbohydrates via the aldohexose pathways.
Keywords: Cellulose; Glycolic acid; Heteropolyacids; Epimerization; Retro-aldol reaction.
ACCEPTED MANUSCRIPT
4 1. Introduction
Biomass is regarded as a promising alternative carbon resource for the sustainable production of fuels and chemicals [1, 2]. One of the techniques to convert biomass into those carbon-based valuable products is a chemo-catalytic process [3]. However, a bulk biomass is recalcitrant toward most of biological as well as chemical reactions because of its rigid structure and the complex chemical compositions [4]. Furthermore, the organic components in biomass have different physico-chemical properties which cause them to have different reactivity and thermal decomposition behaviors. For instances, hemicellulose and cellulose were significantly decomposed (i.e. 60-90 wt%) with the mass loss rate of ~1.0 and ~2.8wt% oC-1 into gas (i.e. CO2,
CO, H2, H2O, CH4, C2H4, C2H6) at 220-315 oC and 315-400 oC, respectively, and left solid char with yields of ~20 and ~6.5wt% at 900 oC, respectively, under inert atmosphere [5]. Meanwhile, lignin was slowly decomposed (i.e. <0.1wt% oC-1) [5] and about 50wt% of solid char was obtained at 900 oC [6]. Moreover, each of biomass components has unique chemical structure and produces specific valuable chemicals under different reaction conditions, e.g. cellulosic materials to sugar derivatives [7, 8], lignin to aromatics [9]; triglycerides to glycerol and fatty acid derivatives. Therefore, the development of effective and selective chemo-catalytic conversion of biomass-derived components into a desired set of chemicals is one of the main topics in current biorefinery research area [7].
Cellulose is the most abundant component with a content as high as 50wt% in dry basis of biomass [10]. It is a polymer of D-glucose linked by -1,4-glycosidic bonds with extensive intra- /intermolecular hydrogen bonds and has a certain level of crystallinity [4]. To date, effective and
ACCEPTED MANUSCRIPT
5
selective transformation of cellulose into valuable chemicals is interesting but also full of challenging. Generally, acidic catalysis systems are applied for this purpose because of their higher effectiveness on converting cellulose to the desired chemicals selectively when compared with alkaline systems [3]. In the acid-catalyzed cellulose conversion, the obtained valuable chemicals are mainly derived via the glucose-fructose (aldo-ketohexoses) isomerization pathway (Scheme 1). For instances, cellulose conversion over Brønsted/Lewis acid catalysts can produce 5-hydroxymethylfurfural (HMF) (C6) and/or levulinic acid (LevA) (C5) via successive reactions of cellulose depolymerization into glucose, glucose-fructose isomerization, fructose dehydration into HMF and hydrolytic cleavage of HMF into LevA and formic acid (FA) (C1) [11]. Lewis acid metal ions such as Pb2+ and La3+ were reported to be active for synthesizing lactic acid (LacA) (C3) [12-14]. These metal ions are able to generate a bifunctional catalytic system, i.e.
the in situ generated Brønsted acid and Lewis acid metal ion, which can effectively catalyze cellulose-glucose depolymerization, glucose-fructose isomerization and a [3+3] retro-aldol reaction of fructose. In the acid-catalyzed systems with an oxidation environment, FA is commonly obtained via glucose as well as fructose fragmentations [15-17].
Heteropolyacids are known as the efficient acid catalyst for direct transformation of
cellulose into high-value added chemicals [18]. Owing to their strong Brønsted acidity, they are commonly applied to depolymerize cellulose into monomeric sugars such as glucose [19-21] or alkyl glucosides [22]. Moreover, the combination of their Brønsted acid properties with their Lewis acidity and/or oxidation potency can enhance their catalytic activities on catalyzing multi- reactions in the cellulose conversion [23]. For instances, H3PW12O40 was active for direct
transformation of cellulose into 5-hydroxymethylfurfural (HMF) with a yield of 49% in biphasic systems (i.e. MIBK/H2O 10:1, 140 oC, 8 h) [24]. Due to its highly strong Brønsted as well as
ACCEPTED MANUSCRIPT
6
Lewis acidities, H5AlW12O40 was found to be effective for cascade conversion of cellulose into levulinic acid (LevA) with a yield of 72.1% by using a similar biphasic system (120 oC, 10 h) [25]. Meanwhile, the strong oxidation potency of H4PVMo11O40 was reported to be effective to oxidize cellulose directly into FA with a yield of 35-67% in the presence of 0.5-2 MPa molecular oxygen [17].
Recently, Zhang et al. reported that H3PMo12O40 was an active and stable catalyst for the oxidation of cellulose into glycolic acid (GlycA) (C2) at relatively mild operation conditions (180 oC, 0.6 MPa O2) [26]. GlycA is one of valuable -hydroxy acids (AHAs) in polymer and cosmetics industries with a high market demand, i.e. ~11.5% annual growth rate in the period 2017-2021 [27]. They proposed that the catalytic conversion of cellulose into GlycA over H3PMo12O40 involves three main routes, i.e. (1) depolymerization of cellulose into glucose and glucose-fructose isomerization catalyzed by Brønsted acid of H3PMo12O40; (2) fragmentation of glucose and fructose into smaller sugars (<C6) via retro-aldol reaction; (3) oxidation of retro- aldol products into the final organic acids [26]. However, controlling of sugar fragmentation via a retro-aldol reaction is full of challenge because it can involve two types of C-C bond scissions, i.e. a [2+4] and a [3+3] retro-aldol of glucose and fructose, respectively [28]. As such, it can give different proportions of C2, C3 and C4 products. In fact, glucose-fructose isomerization followed by a [3+3] retro-aldol reaction is thermodynamically favored in such a kind of acidic reaction system and C3 compounds are commonly generated as the main product [12]. Furthermore, the role of oxygen on this cascade oxidation of cellulose is still unclear because oxygen can act as an oxidant in some organic reactions [29, 30].
ACCEPTED MANUSCRIPT
7
In this study, selectively catalytic conversions of cellulose to C2 or C3-compounds with Keggin-based molybdates, especially PMo12O403- (PMo) as the catalyst, were explored in details.
Unlike the previous work [26], it is found that these catalysts selectively promoted aldohexoses pathways (i.e. an epimerization and a [2+4] retro-aldol reaction) rather than aldo-/ketohexoses ones (i.e. an isomerization and a [3+3] retro-aldol reaction) on the cascade conversion of
cellulose into GlycA. Particularly, the molybdate in Keggin structure of PMo served as not only an oxidation function on the oxidation of the retro-aldol products, but also a Lewis acid catalyst for the selective epimerization as well as the [2+4] retro-aldol reaction at <100 oC and >100 oC, respectively. This behavior is completely different from that of molybdate in MoO3,in which it is effective only for the sugar epimerization in the Bilik reaction but ineffective for GlycA
formation. Furthermore, the presence of molecular oxygen is found to be beneficial on maintaining the activity of the catalyst during the conversion by continuous re-oxidation of PMored, i.e. a reduced form of PMo resulted from the oxidation step and is known as
heteropolyblue, into PMo. These findings on the effective catalysis of PMo toward the [2+4]
retro-aldol reaction at a moderate temperature (<160 oC) and selective promotion of aldohexose pathways provide a new insight for the catalyst development on biomass valorization into valuable C2 and C4 chemicals.
2. Experimental
2.1. Chemicals.
Cellulose powder, glucose, fructose, mannose, glycolic acid (GlycA), L-lactic acid (LacA), 5- hydroxymethylfurfural (HMF), pyruvic acid (PyrA), erhytrose, levulinic acid (LevA), formic
ACCEPTED MANUSCRIPT
8
acid (FA), DL-glyceraldehyde, phosphoric acid (H3PO4), 12-molybdo(VI)phosphoric acid hydrate (H3PMo12O40∙nH2O), 12-molybdosilicic acid hydrate (H4SiMo12O40∙nH2O), nickel(II) chloride hexahydrate (NiCl2∙6H2O), aluminium chloride hexahydrate (AlCl3∙6H2O), tin(IV) chloride pentahydrate (SnCl4∙5H2O), copper(II) chloride hexahydrate (CuCl2∙6H2O) and copper(II) nitrate trihydrate (Cu(NO3)2∙3H2O) were purchased from WAKO Chemicals, Japan.
Glycolaldehyde dimer, sorbose, silver nitrate (AgNO3) were supplied from SIGMA while dihydroxyacetone dimer was obtained from NACALAI TESQUE. All chemicals were used without any further pretreatments. The hydrate content of HPAs was calculated based on a thermogravimetric analysis measurement (TGA, DTG-60H, Shimadzu, Japan). The anhydrous form of HPAs was obtained after calcined at 250 oC for 4 h with a heating rate of 1 oC min-1. 2.2. Synthesis of Ag-based PMo Catalysts.
Ag-substituted PMos were prepared via the ion-exchange method according to the previous literature [31]. Briefly, a 20 mL aqueous solution of H3PMo12O40 and AgNO3 with an
appropriate molar ratio, depending on the Ag content (x) to remove proton (equation 1), was stirred at room temperature for 24 h in dark. Thereafter, water was evaporated in vacuum at 40
oC and the remained solid was dried in oven at 60 oC for 12 h. Finally, calcination was conducted at 250 oC for 4 h with a heating rate of 1 oC min-1 to remove the bounded water in the crystal structure of material. Characterizations were performed by using a FT-IR spectrophotometer (JASCO FT/IR-4200), a scanning electron microscope (SEM, Hitachi SU8010) equipped with a Horiba Scientific energy dispersive spectrophotometer (EDS) analyzer and a X-ray
diffractometer (Smartlab, Rigaku, Japan) (Figs. S1-S3)
H3PMo12O40 + xAgNO3 H(3-x)AgxPMo12O40 + xHNO3 (1)
ACCEPTED MANUSCRIPT
9 2.3. Catalytic Reaction
Hydrothermal reaction was performed in an autoclave reactor (BR-25, BERGHOF, Germany).
In brief, required amounts of the substrate and catalyst were added into 7 mL of water and heated at the designated temperatures either in autogenous or aerobic condition. The reaction was stopped by cooling down the reactor in an ice bath. The remained cellulose was separated by filtration (ADVANTEC, 0.2 m) and the filtrate was diluted with water before injected to HPLC.
The samples were analyzed by using a Shimadzu Prominence HPLC equipped with a RID-10A as well as a UV detector (210 nm) as described in our previous study [32]. The yields of the products were calculated based on carbon weight of the component and cellulose. In this context, carbon content of cellulose was determined by using an elemental analyzer (Vario EL cube elemental analyzer).
3. Results and Discussion
3.1. Conversion of Cellulose to Glycolic acid with PMo-based catalysts
Firstly, the product distributions of cellulose conversion over the Keggin-based molybdate catalysts were investigated in either aerobic or autogenous states. No significant products were detected after cellulose was heated in the absence of catalyst (Table S1, entry 1). In contrast, GlycA and monomeric hexoses such as glucose and mannose were generated when Keggin-
ACCEPTED MANUSCRIPT
10
based molybdate was used as the catalyst (Fig. 1). This result is different from the work of Zang et al., where they only found glucose and fructose in the product [26]. Since cellulose is only composed of glucose units as the monomer, these results indicate that the catalytic conversion of cellulose to GlycA by Keggin-based molybdates involved the glucose-mannose epimerization in its reaction pathway. Considering the significant proportion changes of glucose as well as mannose in the products of autogenous and aerobic reactions (Fig. 1), GlycA should be mainly formed from the cascade conversion of aldohexoses (glucose and mannose) rather than that of ketohexose (fructose). Detail explanations will be further stated in the next section.
In addition, a color change of the solution was observed when Keggin-based molybdate was used as the catalyst. In the autogenous reaction with H3PMo12O40, the color was changed from yellow to dark blue. In contrast, it became a dark green when the reaction was performed under an oxygen atmosphere (Fig. S4A). The formation of dark blue color could be associated with the reduction of PMo into PMored (equation 2), i.e. a Keggin phosphomolybdate anionwhich
contains Mo5+ together with Mo6+ [33]. It is supported by the UV measurement of the product solution where a new absorption peak was found at the relatively higher wavelength region (c.a.
310-335 nm) (Fig. S5). This absorption has been reported to attribute to the charge-transfer change of molybdenum ions [16]. The FT-IR measurement confirmed no destruction of the Keggin structure of PMo after it was reduced into PMored (Fig. S6). On the other hand, the appearance of green color indicated that the reduction level of PMo in an aerobic reaction was lower than that in an autogenous state [23, 34]. In this case, the molecular oxygen should play an important role on reoxidizing PMored into PMo and thus, decreasing the amount of PMored in the solution. This is supported by the fact that no color change of H3PMo12O40 solution was observed after heating it with a trace amount of glycolaldehyde (GlycAld) under an aerobic condition (Fig.
ACCEPTED MANUSCRIPT
11
S4B). The effect of oxygen molecules on reoxidizing PMored into PMo in this system was also indicated from the intensity of the chromatogram corresponding to the PMored ions (Fig. S7).
PMoVI12O403- + ne- PMoVnMoVI(12-n)O40(3+n)- (2) (PMo, yellow) (PMored)
The enhancements of cellulose conversion and GlycA selectivity in the aerobic state should be related with the higher proportion of PMo than PMored in such a condition (Fig. 1). Here, oxygen molecules re-oxidized PMored into PMo during the conversion so that the activity of this catalyst was maintained continuously. This was confirmed by replacing oxygen atmosphere with nitrogen one where a similar result as that in the autogeneous state (i.e. 28-30% selectivity) was obtained. Nevertheless, the excess introduction of molecular oxygen can promote side reactions, especially over-oxidation, which reduced the selectivity of the reaction (Fig. 2). This was
identified from the appearance of multiple peaks in the HPLC chromatograms and the generation of some organic acids such as glyxolic acid, glyceric acid and pyruvic acid. Furthermore, the instability of the retro-aldol products (e.g. erthyrose, glycolaldehyde (GlvAld), glyceraldehydes and pyruvaldehyde) can also reduce the selectivity toward GlycA formation, which will be discussed in the next section.
3.2. Aldohexose Retro-Aldol Routes for Glycolic acid Formation reduction
oxidation
ACCEPTED MANUSCRIPT
12
As shown in Fig. 1, the yield of GlycA was related with the formation of glucose and mannose as the intermediates besides fructose. It is supported by the result that a significant amount of mannose appeared at the low reaction temperature but completely disappeared with the increase of the reaction temperature (Fig. 3). These observations indicate that GlycA formation should involve the oxidation of the fragmented products of mannose besides glucose and fructose. Particularly, GlycA was found to be more preferably produced from the cascade oxidation of fragmented sugars via the [2+4] retro-aldol reaction of aldohexoses (i.e. glucose and mannose) pathways.
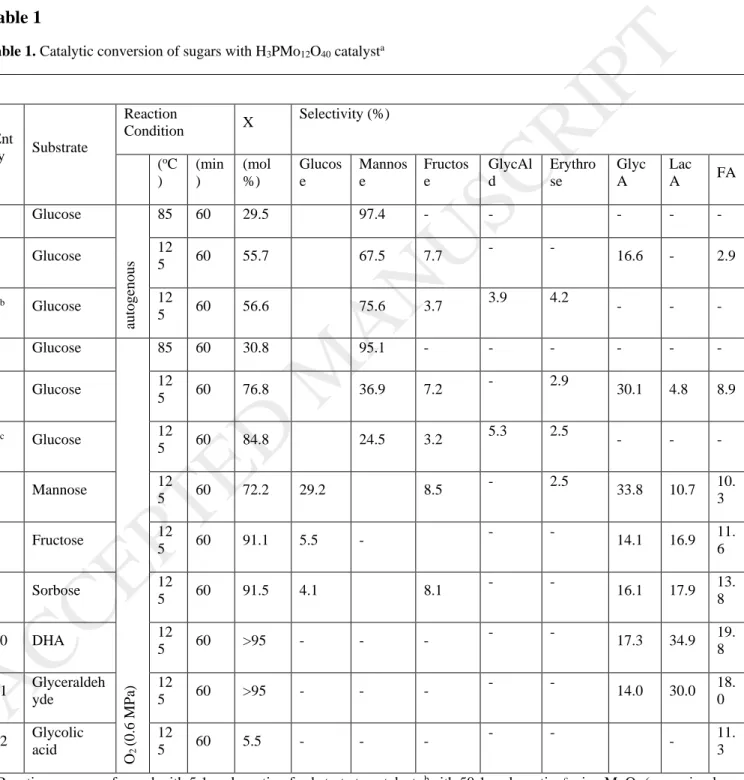
To verify the above hypothesis, the catalytic conversions of various sugars were investigated by using H3PMo12O40 as the catalyst. It is clearly observed that H3PMo12O40 promoted the
selective epimerization of glucose to mannose at a low temperature (ca. 85 oC) (Table 1, entries 1 and 4). Although the selective sugar epimerization over H3PMo12O40 under an autogenous state was reported recently [33], in this study, this catalyst also remained its selectivity under an aerobic state (Table 1, entry 4). Meanwhile, a significant amount of GlycA was obtained from glucose conversion at 125 oC (Table 1, entries 2 and 5). This is an interesting result since an elevated condition (>160 oC, >1MPa) is usually required for selective C-C bond scission of glucose via the [2+4] retro-aldol reaction [35-37]. Herein, the higher fraction of mannose than that of fructose in the obtained product solution, the similar high yield of GlycA from glucose as well as mannose, and the lower yield of GlycA from fructose (Table 1) indicated that the
pathway of glucose or cellulose conversion into GlycA over H3PMo12O40 involved glucose- mannose epimerization. However, the isomerization reaction cannot be hindered since in general the glucose-fructose isomerization is easily realized in a hot acidic condition [38], as indicated by the formation of small fraction of fructose.
ACCEPTED MANUSCRIPT
13
In order to investigate the reaction behavior of fructose in this reaction system, the catalytic conversion of ketohexoses was also carried out. Unlike glucose and mannose, the conversions of ketohexoses over H3PMo12O40 resulted in a higher yield of LacA than GlycA (Table 1, entries 8 and 9). Moreover, HPLC analysis obviously showed the significant formation of C3 degradation products (Fig. S8). These results imply that fructose was mainly fragmented via [3+3] retro-aldol reaction in this catalytic system. It is aligned with the reported results by Orazov and Davis, in which H3PMo12O40 promoted fructose conversion into lactate esters (C3) in alcoholic reaction systems [39].
Thusly, although H3PMo12O40 is active on catalyzing retro-aldol reactions of hexoses, the selective activity of PMo on promoting epimerization reaction suppressed the formation of fructose and its derivatives in glucose or cellulose conversion. As such, their cascade oxidations should mainly occur via the [2+4] retro-aldol of aldohexoses rather than the [3+3] retro-aldol of aldo-ketohexoses. It was confirmed by performing glucose conversion with a low concentration of H3PMo12O40, in which an equimolar of [2+4] retro-aldol products, i.e. GlycAld (C2) and erythrose (C4), were produced while C3 products were not detected (Table 1, entry 3). Moreover, the addition of strong Lewis acid metal ions such as Al3+, Sn4+ and Zn2+ as the co-catalyst, which are active for isomerization as well as [3+3] retro-aldol reactions [40-42],reduced the formation of GlycA (C2) in the cellulose conversion over H3PMo12O40 (Fig. S9).
Furthermore, the conversions of trioses were also examined in order to investigate the possibility of further fragmentation of these [3+3] retro-aldol compounds into smaller molecules, especially GlycA. Although the reaction was performed at the mild condition, all trioses were completely converted in the presence of H3PMo12O40. However, the products were dominated with C3 compounds such as pyruvic acid, LacA, glyceric acid and other oxidized products
ACCEPTED MANUSCRIPT
14
(Table 1, entries 10-11, Fig S8). Hayes et al. also reported that heating of [13C1]glyceraldehyde with molybdic acid resulted in a major product of [13C1]dihydroxyacetone [43]. As such, the formation of GlycA from trioses or their sugar source (fructose) should be restricted by their high reactivity to isomerization or to oxidization in the PMo catalyzed system.
The oxidation potency of PMo also played an important role on oxidizing the intermediates of retro-aldol products into GlycA. Besides from the higher yield of GlycA produced in the aerobic state (Fig. 1), it is supported from the fact that the selectivity was decreased after the co- addition of metal ions with strong oxidation potency such as Fe3+ and Cu2+ (Fig. S9). The results were different when the relatively weak oxidation potency metal ions such as Ni2+ and Co2+ were co-added. In addition, the presence of a strong oxidizing agent of oxyanions of nitrate and sulfate also resulted in a negative effect on the selectivity to GlycA (Table S1, entries 5-7).
3.3. Optimization of Reaction Conditions
The product distribution of cellulose conversion over H3PMo12O40 catalyst was investigated in order to get more information on this catalytic system (Fig. 4A). In agreement with the above discussion, GlycA was observed in parallel with mannose at the beginning of the reaction. Subsequently, the amount of GlycA was reached to the maximum after 1 h reaction and then dropped with the increase of reaction time. In this case, a significant amount of FA and multiple peaks in HPLC analysis were observed. Apparently, the intermediates of this reaction (e.g. sugars, retro-aldol products) were transformed into undesired products with the prolonged reaction time. Moreover, the over-oxidation of GlycA possibly occurred as observed from its conversion into FA, glyoxylic and oxalic acids on the HPLC analysis after it was reacted at a
ACCEPTED MANUSCRIPT
15
mild temperature (Table 1, entry 12). Meanwhile, a higher conversion rate was achieved with the increased loading amount of the catalyst (Fig. 4B). This is resulted by the more sufficient
amounts of Brønsted acid sites and PMo species which are benefit for promoting GlycA formation on these cascade reactions, i.e. depolymerization/hydrolysis, epimerization, [2+4]
retro-aldol and oxidation. Here, the maximum yield of GlycA obtained from cellulose was ~50%.
In order to investigate the reaction behaviors of the intermediates in the cellulose conversion, the reactions of glucose and mannose with different amounts of H3PMo12O40 were also performed at 125 oC. Since their equilibrium states were achieved [33], both of these aldohexoses showed a similar conversion rate (Fig. S10A and B). Interestingly, the formation of GlycA was found to be significantly depended on the catalyst loading amount. A shorter
induction period of GlycA formation was observed when 5:1 molar ratio of hexoses:catalyst was loaded compared with that of 50:1 molar ratio (Fig. S10C). A high loading catalyst should provide more protons as well as PMo ions in the system. Particularly, high loading of PMo would be benefit on promoting cascade oxidation into GlycA formation as previously discussed.
It should be noted that the kinetic rate of the glucose retro-aldol reaction over homogenous tungstate was also increased non-linearly with the increase in catalyst amount [36].
Moreover, from the following results, the side reactions due to the high reactivity of the retro-aldol products can be considered as the main reason for the limited selectivity to GlycA.
For instances, heating GlycAld with H3PMo12O40 at 125 oC (1 h, 0.6 MPa) resulted the
formations of GlycA (29.5%), FA (26.4%) and some multiple peaks in the HPLC chart with the conversion of ~90%. Meanwhile, considering the similar reaction rates as well as the activation energy of erythrose and GlycAld [36], a successive [2+2] retro-aldol and oxidation reaction of erythrose with a high selectivity seems to be difficult to occur, especially at a high temperature.
ACCEPTED MANUSCRIPT
16
It can be seen from the reported result that the maximum yield of ethylene glycol (C2) was only
~50% in the cascade hydrogenation of eryhtrose [44]. Hence, the high reactivity of the
intermediate sugars or retro-aldol products leads them to undergo undesired transformations at the high temperature. However, a high temperature is generally required to convert robust cellulose effectively. As such, the selectivity of GlycA in the cascade conversion of hexoses or cellulose over PMo should be limited to some extent.
3.4. Cellulose-Glycolic acid Pathways with Phosphomolybdate
Based on the above results, it can be concluded that the catalytic conversion of cellulose into GlycA with H3PMo12O40 should involve four major reactions: depolymerization,
epimerization, [2+4] retro-aldol reaction and oxidation. The depolymerization of cellulose into glucose was clearly observed to be catalyzed by the Brønsted acid of PMos as indicated by using Ag-based PMos (Table S1, entries 8-10). Here, the complete substitution of proton with Ag+ on Ag3PMo12O40 decreased the Brønsted acidity of the catalyst [45]. Although no proton is
contained in Ag3PMo12O40 structure, this PMo salt was hydrolyzed at hot conditions [33, 46] as observed from the acidic property of its solution (pH ~3). Meanwhile, molybdate in Keggin PMo played an important role on catalyzing glucose conversion into GlycA via successive reactions of epimerization, [2+4] retro-aldol and oxidation (Table 1). It is supported from the following results: (i) only Mo in Keggin of HPAs showed the catalytic activity to convert cellulose directly into GlycA (Table S1, entries 2-4) and (ii) no decomposition of its primary Keggin structure was observed after the reaction (Fig S11). In addition, MoO3, i.e. an active and selective catalyst for sugar epimerization in the Bilik reaction [39, 47], was found to be ineffective on catalyzing of
ACCEPTED MANUSCRIPT
17
[2+4] retro-aldol from glucose when compared with H3PMo12O40 (Table 1, entries 3 and 6, respectively).
Sugar epimerization over Keggin structured PMo was reported to occur via the 1,2-Carbon shift (1,2-CS) mechanism similar as that of MoO3 in the Bilik reaction [33], in which a tricentric complex structure was formed between hexose and MoO [43]. However, in this study, it is found that only PMo was effective for the generation of [2+4] retro-aldol products and GlycA (Table 1, entries 3, 5-6, respectively). Herein, [2+4] retro-aldol products were not detected at a reaction temperature <100 oC, but it was observed at >100 oC in parallel with the epimerization reaction (Table 1). These results suggested that the selective C−C bond scission of hexoses for [2+4]
retro-aldol reaction more effectively occurred on MoO structure of PMo when compared with that on MoO3 (the detail explanation can be found in Supplementary Information, Schemes S1- S2). The ineffectiveness of MoO3 on catalyzing retro-aldol reaction was also seen from the low selectivity of ethyl lactate synthesis from fructose in ethanol (13% selectivity at 100 oC) [39].
Considering the typical catalytic behavior of molybdate species as described above, PMo is proposed to exhibit bifunctional catalysis for sugar conversion: (i) as a Lewis acid for catalyzing an epimerization as well as a retro-aldol reaction; (ii) as an oxidant for oxidizing the fragmented sugars resulting from the retro-aldol reactions. Here, the higher selectivity of PMo to promote aldohexose (i.e. glucose-mannose) epimerization rather than aldo-ketohexose (i.e. glucose- fructose) isomerization leads the [2+4] retro-aldol reaction of glucose/manose to be more easily taken place. Meanwhile, molecular oxygen has bifunctional effects: (i) a stoichiometrically consumed oxidation agent to produce GlycA and (ii) maintaining an oxidative condition by
ACCEPTED MANUSCRIPT
18
keeping the concentration of active PMo species through re-oxidation of PMored into PMo. As such, the mechanism on the cascade catalytic conversion of cellulose can be illustrated in Fig. 5.
4. Conclusions
Catalytic conversions of cellulose over acid catalysts especially with the Lewis acidic properties involve multiple reaction networks where the cascade reaction generally proceeds via the aldo-ketohexoses pathway; i.e. depolymerization of cellulose into glucose, isomerization of glucose into fructose and then fructose dehydration or fragmentation via the [3+3] retro-aldol reaction. These routes always lead the reaction to generate the chemicals of C6, C5, C3 and C1 like HMF, levulinates, lactates, and formates. However, in this study, Keggin structured PMo derived catalysts were found to be effective for selective conversion of cellulose to C2/C3 products. Particularly, it was active to promote the reaction via the aldohexoses pathway (i.e.
epimerization and [2+4] retro-aldol reaction) rather than the aldo-ketohexoses pathway (i.e.
isomerization and [3+3] retro-aldol). This property allows to yield GlycA (C2) selectively as the final oxidized product (~50% from cellulose). It is confirmed from the results that (i) PMo selectively epimerized glucose into mannose at a temperature <100 oC (~95% selectivity) and promoted [2+4] retro-aldol reaction in parallel with the glucose-mannose epimerization at a temperature >100 oC, in either aerobic or anaerobic conditions; (ii) molybdate of MoO3, i.e. an active and selective catalyst for the epimerization in the Bilik reaction [47, 48], was ineffective for the [2+4] retro-aldol reaction; (iii) glucose and mannose were converted to GlycA with a similar selectivity (30-34% at 125 oC); (iv) fructose was converted to GlycA with a low
selectivity (14% at 125 oC) but with a high selectivity to C3. These cascade oxidation reactions
ACCEPTED MANUSCRIPT
19
reduced PMo into PMored without destructing its primary Keggin structure as confirmed by the color changes of the solution, UV-VIS and FT-IR measurements. Herein, the presence of molecular oxygen was beneficial to re-oxidize PMored into PMo, which maintained the catalytic activity continuously and resulted in more GlycA production than the reaction in the anerobic state. Nevertheless, the high reactivity of the intermediates especially retro-aldol products to undergo undesired reactions limited the catalytic cascade conversion of cellulose into GlycA over Mo-HPAs. The effectivity of molybdate in Keggin catalyst of Mo-HPAs on catalyzing sugar epimerization and [2+4] retro-aldol at a moderate temperature provided an insight on the catalyst development for the selective aldohexoses pathways to generate C2, C4 and/or other novel oxygenated compounds besides the current strategy for the selective carbohydrates transformation via aldo-ketohexoses routes.
5. Acknowledgments
This study is supported by Aomori City Government and President Fund of Hirosaki University, Japan. A. Bayu and S. Karnjanakom gratefully acknowledge the scholarship from Ministry of Education, Culture, Sport, Science, and Technology (MEXT) of Japan.
ACCEPTED MANUSCRIPT
20 6. References
[1] J. Bozell, Clean, 36 (2008) 641-647.
[2] T. Werpy, G.R. Petersen, A. Aden, J. Bozell, D. Elliot, L. Lasure, S. Jones, M. Gerber, K.
Ibsen, L. Lumberg, S. Kelley, in, U.S. Department of Energy, 2004, pp. 1-67.
[3] C.-H. Zhou, X. Xia, C.-X. Lin, D.-S. Tong, J. Beltramini, Chem. Soc. Rev., 40 (2011) 5588-5617.
[4] M.E. Himmel, S.-Y. Ding, D.K. Johnson, W.S. Adney, M.R. Nimlos, J.W. Brady, T.D.
Foust, Science, 315 (2007) 804-807.
[5] H. Yang, R. Yan, H. Chen, D.H. Lee, C. Zheng, Fuel, 86 (2007) 1781-1788.
[6] I. Kurnia, S. Karnjanakom, A. Bayu, A. Yoshida, J. Rizkiana, T. Prakoso, A. Abudula, G.
Guan, Fuel Process. Technol., 167 (2017) 730-737.
[7] M. Dusselier, M. Mascal, B.F. Sels, Top Chemical Opportunities from Carbohydrate Biomass: A Chemist's View of the Biorefinery, in: K.M. Nicholas (Ed.) Selective Catalysis for Renewable Feedstocks and Chemicals, Springer International Publishing, 2014, pp. 1-40.
[8] F.W. Lichtenthaler, in: B. Kamm, P.R. Gruber, M. Kamm (Eds.) Biorefineries - Industrial pocesses and products, status quo and future directions, Wiley-VCH Verlag GmbH & Co, KGaA, 2006.
[9] J.J. Bozell, in: K.M. Nicholas (Ed.) Selective catalysis for renewable feedstocks and chemicals, Springer-Verlag Berling Heidelberg, 2014, pp. 229-256.
ACCEPTED MANUSCRIPT
21
[10] L.D. Klass, in: Biomass for Renewable Energy, Fuels and Chemicals, Academic Press, 1998, pp. 51-90.
[11] D.J. Hayes, S. Fitzpatrick, M.H.B. Hayes, J.R.H. Ross, in: K. B., P.R. Gruber, M. Kamm (Eds.) Biorefineries-Industrial Processes and Products: Status Quo and Feature
Directions, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2006.
[12] Y. Roman-Leshkov, M.E. Davis, ACS Catal., 1 (2011) 1566-1580.
[13] Y. Wang, W. Deng, B. Wang, Q. Zhang, X. Wan, Z. Tang, Y. Wang, C. Zhu, Z. Cao, G.
Wang, H. Wan, Nat. Commun., 4 (2013) 1-7.
[14] F.-F. Wang, C.-L. Liu, W.-S. Dong, Green Chem., 15 (2013) 2091-2095.
[15] Z. Tang, W. Deng, Y. Wang, E. Zhu, X. Wan, Q. Zhang, Y. Wang, ChemSusChem, 7 (2014) 1557-1567.
[16] T. Lu, M. Niu, Y. Hou, W. Wu, S. Ren, F. Yang, Green Chem., 18 (2016) 4725-4732.
[17] J. Zhang, M. Sun, Y. Han, Catal. Today, 233 (2014) 77-82.
[18] W. Deng, Q. Zhang, Y. Wang, Dalton Trans., 41 (2012) 9817-9831.
[19] K.-i. Shimizu, H. Furukawa, N. Kobayashi, Y. Itaya, A. Satsuma, Green Chem., 11 (2009) 1627-1632.
[20] J. Tian, J. Wang, S. Zhao, C. Jiang, X. Zhang, X. Wang, Cellulose, 17 (2010) 587-594.
[21] Y. Ogasawara, S. Itagaki, K. Yamaguchi, N. Mizuno, ChemSusChem, 4 (2011) 519-525.
ACCEPTED MANUSCRIPT
22
[22] W. Deng, M. Liu, Q. Zhang, Y. Wang, Catal. Today, 164 (2011) 461-466.
[23] I.V. Kozhevnikov, Chem. Rev., 98 (1998) 171-198.
[24] X. Zhang, D. Zhang, Z. Sun, L. Xue, X. Wang, Z. Jiang, Appl. Catal. B: Environmental, 196 (2016) 50-56.
[25] Z. Sun, L. Xue, S. Wang, X. Wang, J. Shi, Green Chem., 18 (2016) 742-752.
[26] J. Zhang, X. Liu, M. Sun, X. Ma, Y. Han, ACS Catal., 2 (2012) 1698-1702.
[27] R.a. Markets, in, 2017.
[28] S. Yamaguchi, T. Baba, Molecules, 21 (2016) 937-956.
[29] T. Punniyamurthy, S. Velusamy, J. Iqbal, Chem. Rev., 105 (2005) 2329-2363.
[30] Z. Shi, C. Zhang, C. Tang, N. Jiao, Chem. Soc. Rev., 41 (2012) 3381-3430.
[31] S. Borghese, A. Blanc, P. Pale, B. Louis, Dalton Trans., 40 (2011) 1220-1223.
[32] A. Bayu, G. Guan, S. Karnjanakom, X. Hao, K. Kusakabe, A. Abudula, ChemistrySelect, 2 (2016) 180-188.
[33] F. Ju, D. VanderVelde, E. Nikolla, ACS Catal., 4 (2014) 1358-1364.
[34] B.B. Sarma, R. Neumann, Nat. Chem., 5 (2014) 1-6.
[35] A. Wang, T. Zhang, Acc.Chem. Res., 46 (2013) 1377-1386.
ACCEPTED MANUSCRIPT
23
[36] J. Zhang, B. Hou, A. Wang, Z. Li, H. Wang, T. Zhang, AIChE Journal, 60 (2014) 3804- 3813.
[37] M. Sasaki, K. Goto, K. Tajima, T. Adschiri, K. Arai, Green Chem., 4 (2002) 285-287.
[38] T.P. Mawhinney, M.A. Madson, M.S. Feather, Carbohyd. Res., 86 (1980) 147-150.
[39] M. Orazov, M.E. Davis, PNAS, 112 (2015) 11777-11782.
[40] M. Bicker, S. Endres, L. Ott, H. Vogel, J. Mol. Catal. A: Chemical, 239 (2005) 151-157.
[41] C.B. Rasrendra, I.G.B.N. Makertiharta, S. Adisasmito, H.J. Heeres, Top. Catal., 53 (2010) 1241-1247.
[42] K. Nemoto, Y. Hirano, K.-i. Hirata, T. Takahashi, H. Tsuneki, K.-i. Tominaga, K. Sato, Appl. Catal. B, 183 (2016) 8-17.
[43] M.L. Hayes, n.J. Pennings, A.S. Serianni, R. Barker, J. Am. Chem. Soc., 104 (1982) 6764-6769.
[44] R. Ooms, M. Dusselier, J.A. Geboers, B. Op de Beeck, R. Verhaeven, E. Gobechiya, J.A.
Martens, A. Redl, B.F. Sels, Green Chem., 16 (2014) 695-707.
[45] M. Tao, D. Zhang, H. Guan, G. Huang, X. Wang, Sci. Rep., 6 (2016) 1-13.
[46] H. Niiyama, Y. Saito, S. Yoshida, E. Echigoya, Nippon Kagaku Kaishi, 1982 (1982) 569- 573.
ACCEPTED MANUSCRIPT
24
[47] L. Petrus, M. Petrusova, Z. Hricoviniova, in: A.E. Stutz (Ed.) Glycoscience:
Epimerization, Isomerization and Rearrangement Reactions of Carbohydrates, Springer Berlin Heidelberg, 2001.
[48] V. Bilik, Chem. Zvesti, 26 (1972) 183-186.
ACCEPTED MANUSCRIPT
25 Figure Captions
Figure 1. Product distributions in the autogenous (A) and in the presence of 0.5 MPa of O2 (B).
Reactions: 1 wt% of cellulose in water; 11 mM of catalyst; 175 oC; 1 h.H3PMo and H4SiMo are the abbreviation of H3PMo12O40 and H4SiMo12O40.
Figure 2. Effect of oxygen pressure on the activity of H3PMo12O40. Reactions: 1 wt% of cellulose in water; 11 mM of catalyst; 175 oC; 1 h.
Figure 3. The selectivity of reaction at different reaction temperatures. Reactions: 1 wt% of cellulose in water; 11 mM of H3PMo12O40 catalyst; 1 h; 0.6 MPa.
Figure 4. Effects of reaction time (A) and catalyst loading amount (B) on catalytic conversion of cellulose in water at 175 oC, 0.6 MPa O2. A was performed with 0.08 mmol of catalyst while 1 h of reaction time was selected for B.
Figure 5. Selective conversion of cellulose into glycolic acid by using Mo-based HPAs catalyst with molecular oxygen.
Scheme captions
Scheme 1. Chemo-catalytic reaction pathways on transformation of cellulose into some high- value added oxygenated chemicals over acid catalysts.
Scheme 1
ACCEPTED MANUSCRIPT
26 Figure 1
ACCEPTED MANUSCRIPT
27 Figure 2
ACCEPTED MANUSCRIPT
28 Figure 3
ACCEPTED MANUSCRIPT
29 Figure 4
ACCEPTED MANUSCRIPT
30 Figure 5
ACCEPTED MANUSCRIPT
31
ACCEPTED MANUSCRIPT
32 Table captions
Table 1. Catalytic conversion of sugars with H3PMo12O40 catalyst Table 1
Table 1. Catalytic conversion of sugars with H3PMo12O40 catalysta
Ent
ry Substrate
Reaction
Condition X Selectivity (%) (oC
)
(min )
(mol
%)
Glucos e
Mannos e
Fructos e
GlycAl d
Erythro se
Glyc A
Lac
A FA
1 Glucose
autogenous
85 60 29.5 97.4 - - - - -
2 Glucose 12
5 60 55.7 67.5 7.7 - -
16.6 - 2.9
3b Glucose 12
5 60 56.6 75.6 3.7 3.9 4.2
- - -
4 Glucose
O2(0.6 MPa)
85 60 30.8 95.1 - - - - - -
5 Glucose 12
5 60 76.8 36.9 7.2 - 2.9
30.1 4.8 8.9
6c Glucose 12
5 60 84.8 24.5 3.2 5.3 2.5
- - -
7 Mannose 12
5 60 72.2 29.2 8.5 - 2.5
33.8 10.7 10.
3
8 Fructose 12
5 60 91.1 5.5 - - -
14.1 16.9 11.
6
9 Sorbose 12
5 60 91.5 4.1 8.1 - -
16.1 17.9 13.
8
10 DHA 12
5 60 >95 - - - - -
17.3 34.9 19.
8 11 Glyceraldeh
yde
12
5 60 >95 - - - - -
14.0 30.0 18.
0 12 Glycolic
acid
12
5 60 5.5 - - - - -
- 11.
3
aReactions were performed with 5:1 molar ratio of substrate to catalyst; bwith 50:1 molar ratio; cusing MoO3 (an equimolar of Mo with entry 5) as a catalyst instead of H3PMo12O40).