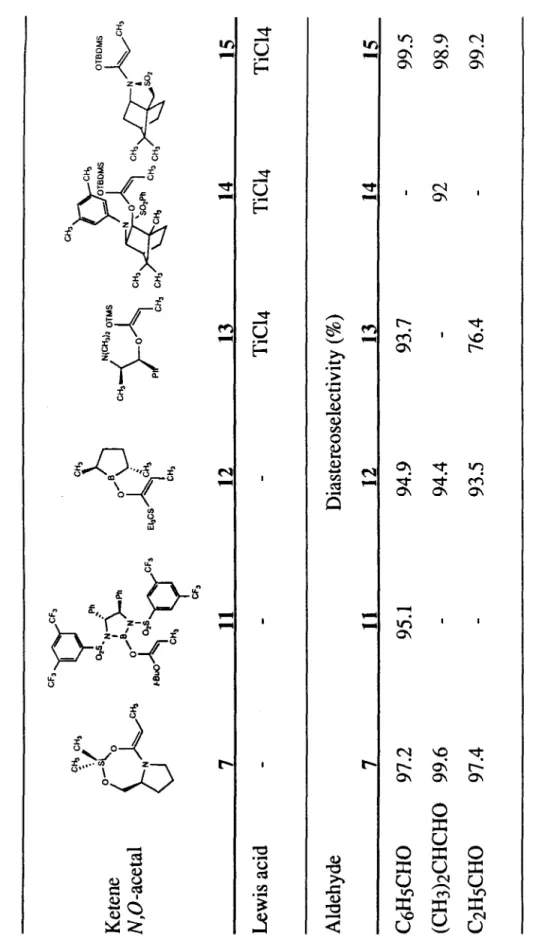
O -silyl ketene N,O-acetals have been shown to react with aldehydes without catalysis to form aldol-type products with high diastereoselectivity. For example, the prolinol-derived O-silyl ketene N,O-acetal 7 reacts with benzaldehyde to form the antialdol product 9 with >98% diastereoselectivity. Mechanistic studies are presented that support a scheme involving the formation of trigonal bipyrarnide (tbp) G, pseudorotation to form tbp.
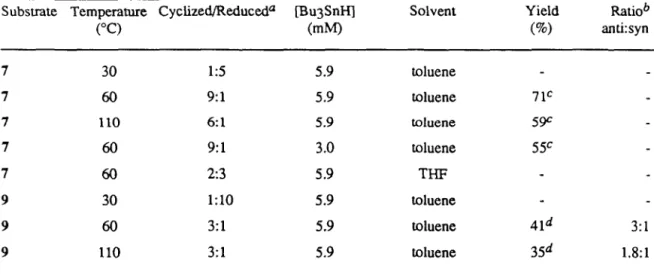
Aldehydes have been shown to react with pyridine, benzeneselenol, dichlorodimethylsilane, and an allylic alcohol to form O-allyloxy dimethylsilyl hemiselenoacetals in high yield. These derivatives undergo efficient free radical-mediated cyclization upon treatment with BU3SnH; as illustrated by the transformation of 7~8. Together these steps provide an efficient method for forming the carbon-carbon bond between the carbonyl carbon of an aldehyde and the end of an allylic alcohol.
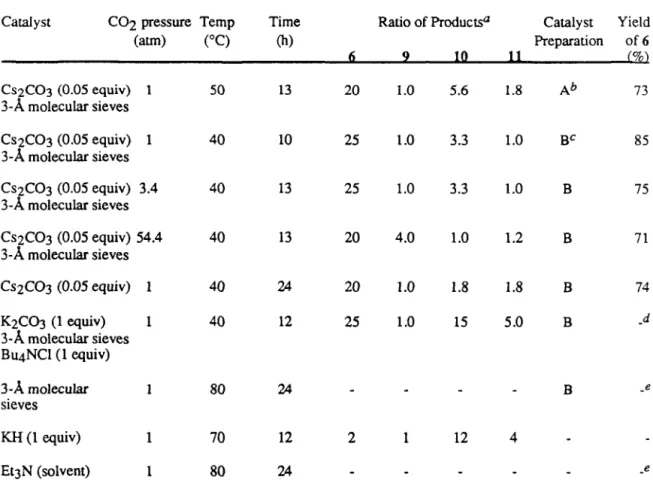
2,3-Epoxy alcohols are converted to C2-inverted cyclic carbonates upon treatment with cesium carbonate under an atmosphere of carbon dioxide (e.g., 5-6). The reaction is completely stereospecific, within experimental error, and forms only 5-ring cyclic carbonates.
CHAPTER I
Aldol reactions of O-silylketene N,O-acetals are accelerated dramatically compared to reactions of the corresponding O-silylketene acetals. Anti-selectivity is unusual for Z-enolates.2 The syn-selectivity observed in most Z-enolates is often explained by the evocation of a chair transition state. A second route involving condensation of the dianion (prepared from prolinol propionamide and 2 equiv LDA at -78°C) with dichlorodimethylsilane was investigated.
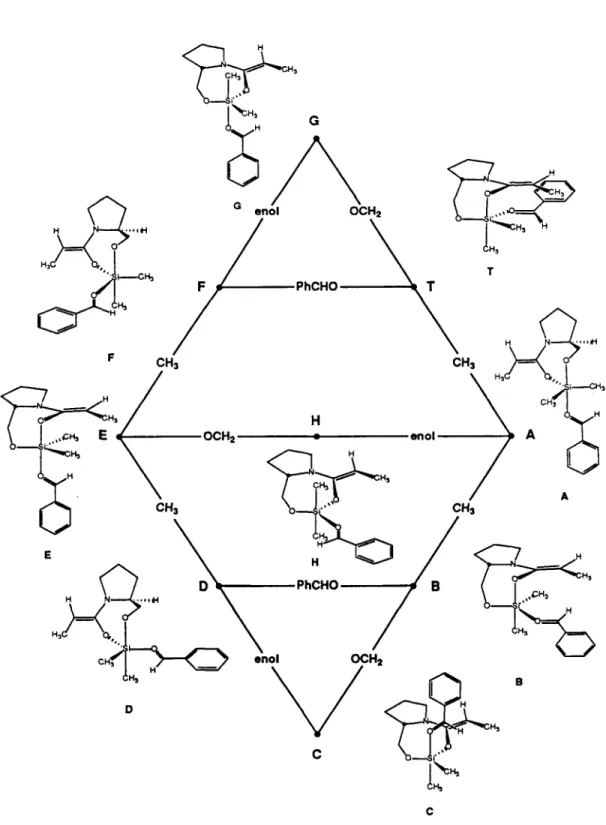
The reaction proceeds readily in noncoordinating solvents (CH2Ch, hexanes, benzene) but is slow in coordinating solvents (THF, DMF), an observation that supports coordination of the aldehyde to silicon. First, the boat transition state provides better overlap between the 1t aldehyde and O -silyl ketene N,O -acetal systems. Examination of four hypothetical transition states provides a possible explanation for the observed selectivity.
The models suggest that it is difficult for the nitrogen lone pair to fully conjugate to the enol double bond when attack occurs at the Re face of the prolinol-derived O -silyl ketene N,O -acetal 7 . probably not formed directly by attack of the aldehyde on the O-silyl ketene N,O-acetal 7. Analysis of an aliquot of the reaction mixture by 1H NMR indicated that the reaction was complete.
Part of the reaction mixture was further purified by thin-layer preparative chromatography (50% ethyl acetate-hexanes) to separately obtain pure (±)-3 and (±)-4.
CHAPTER II
Control of absolute stereochemistry of the cyclization products can then be achieved using optically active allylic alcohols. Initial efforts to access intermediate 1 by generation of radical anion 2 in the presence of silyl chloride 3 led to the exclusive formation of dimeric pinacol products. The hemiselenoacetal 6 appeared to be an almost ideal precursor in this regard, since the selenium-carbon bond is easily cleaved by trialkyltin radicals. This cleavage should be particularly easy in the present case given the stability of the product radical.
This mode of addition may prevent the formation of the less reactive silyl selenide.6 The order of addition of 4 and allylic alcohols to dichlorodimethylsilane is also important. Addition of allylic alcohol before the putative benzeneselenol-aldehyde adduct (4) results in about 15% of the symmetrically substituted siloxane «CH3hSi(ORh). In addition, small amounts (-5%) of hydrocinnamyl alcohol were formed, apparently as a result of competitive blocking of the radical intermediate with tributyltin hydride.
The radical is derived from 7 attacks at the less hindered terminus of the double bond to form a more stable secondary radical. The regioselectivity is different for 9, where both sides of the double bond are substituted, and the more stable benzyl radical is produced by 6-exocyclization. It cannot be inferred that this radical cycle is reversible; since the thermodynamic stabilities of the product radicals can be well reflected in the relative stabilities of the respective transition states.
Similar results were recently observed by Koreeda7 during his investigation of radical cyclizations of silyl ethers 12 and 14. Treatment of the product selenides with m-CPBA and triethylamine led to olefins 17 and 18 in moderate yields. 10 The presence of olefin E 18 among the products is almost certainly due to cleavage of the siloxane functional group prior to elimination to form a double bond.
The silica pad was washed with ethyl acetate (110 mL) to ensure complete elution of the diols. The reaction mixture was partitioned between saturated aqueous sodium bicarbonate solution (30 mL) and ethyl acetate (40 mL). The reaction mixture was partitioned between saturated aqueous sodium bicarbonate (30 mL) and ethyl acetate (40 mL).
CHAPTER III
The relative amounts of Payne rearrangement products could also be reduced by increasing the pressure of carbon dioxide, although this had little effect on the reaction rate. The exact role of molecular sieves in the reaction is uncertain, but their incorporation causes a twofold increase in the reaction rate. The highest selectivity occurred with DMF as solvent, although the reaction also occurred in non-polar (toluene) and protic (isopropanol) solvents.
Mixtures of potassium carbonate and tetrabutylammonium chloride were less effective than cesium carbonate as catalysts, and triethylamine did not accelerate the reaction. However, only the hydroxycarbonates 15 and 16 were observed, although not surprisingly higher reaction temperatures were required for the reaction to proceed. In order to determine the reaction mechanism, the transformation of 14 into 15 and 16 was monitored by NMR spectroscopy.
After an additional 40 min, the reaction mixture was warmed to 0 °e and quenched with saturated aqueous ammonium chloride solution (50 mL). The solution was partitioned between 1:1 ethyl-octetate-hexanes (100 mL) and brine (100 mL). The aqueous layer was extracted with 1:1 ethyl acetate-hexanes (2 x 100 mL) and the combined organic phases were dried over sodium sulfate. The reaction mixture was heated to 72°C and heated for 16 hours under an atmosphere of carbon dioxide.
The organic layer was separated and the aqueous layer was washed with a 40 ml portion of ethyl acetate. The reaction mixture was partitioned between saturated ammonium chloride solution (50 mL), saturated brine (50 mL), and ethyl acetate (100 mL). The reaction mixture was partitioned between saturated ammonium chloride solution (20 mL), saturated brine (20 mL), and ethyl acetate (30 mL). The aqueous layer was further extracted with two 30 mL portions of ethyl acetate.
The reaction mixture was partitioned between saturated ammonium chloride solution (20 mL), saturated brine (20 mL), and ethyl acetate (30 mL). The reaction mixture was concentrated under vacuum (0.1 Torr) and used without further purification in the following reactions. The reaction was quenched with saturated aqueous citric acid solution (5 mL) and the aqueous layer was extracted with two 20 mL portions of ethyl acetate.
The reaction was quenched with saturated aqueous citric acid solution (5 mL) and the aqueous layer was extracted with two 20 mL portions of methylene chloride. The reaction was quenched with saturated aqueous citric acid (5 mL) and the aqueous layer was extracted with two 20 mL portions of methylene chloride.
Appendix I