BACKGROUND AND RESEARCH OBJECTIVES
This is followed by specific binding of a TCR to T cells in the tumor-draining lymph nodes to the cognate peptide-major histocompatibility (MHC) complex displayed on an antigen-presenting cell (APC), such as macrophages or dendritic cells. Because DC can activate antigen-specific T cells, the DC vaccine aims to restore immune suppression by administering 'trained' DCs and triggering adaptive immune responses in the patient. Oxidizing the whole tumor lysate with hypochlorous acid or infecting the tumor cell with Newcastle disease significantly improved DC vaccination results in the preclinical setting (84–86).
Chemotherapy can selectively reduce the number of Tregs in the tumor bed, tumor-draining lymph nodes, and peripheral blood. The vaccine peaked with mature antigen-presenting cells in the draining lymph nodes (dLN) and increased local and systemic helper and cytotoxic T cells. Potential vaccine neoepitopes are then predicted based on their binding affinity to the HLA subtype determined in the patient.
It is noteworthy that based on recent developments, several personalized vaccines are now underway or in the pipeline, awaiting their evaluation in a clinical setting. Also, the endothelium of tumor blood vessels often regulates the expression of leukocyte adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) or increases the level of Endothelin B receptor in the tumor microvasculature. . Such molecular mechanisms may explain the robust desmoplastic response in pancreatic cancer, which can capture T cells in the stroma and prevent migration into the tumor cell-rich areas.
ISGF3 translocates to the nucleus to induce genes regulated by IFN-stimulated response elements (ISREs), resulting in the expression of several immunoregulatory cytokines, cell death factors, and proteins associated with the antiviral response.
MUSYC DOSING OF ADJUVANTED CANCER VACCINES OPTIMIZES ANTITUMOR RESPONSES
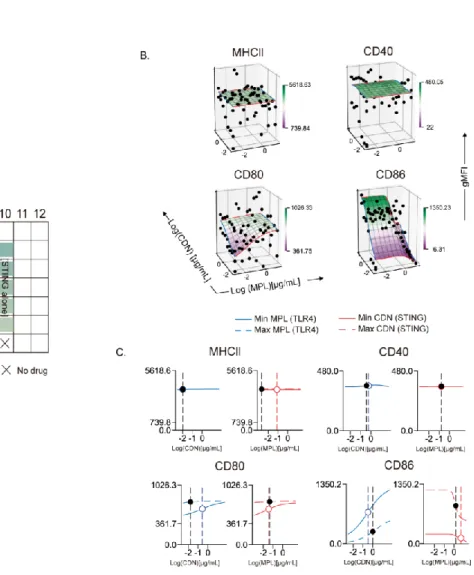
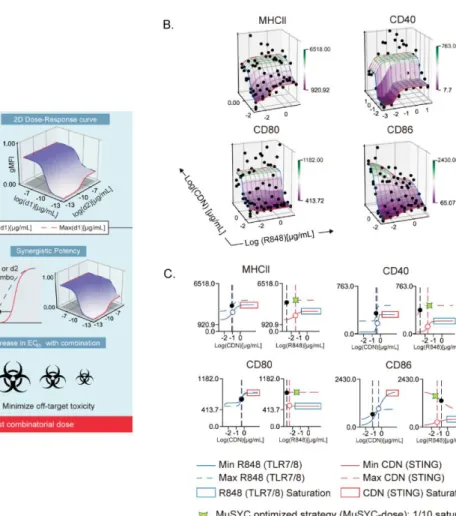
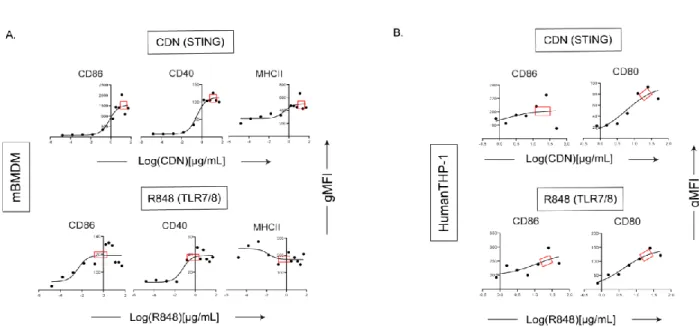
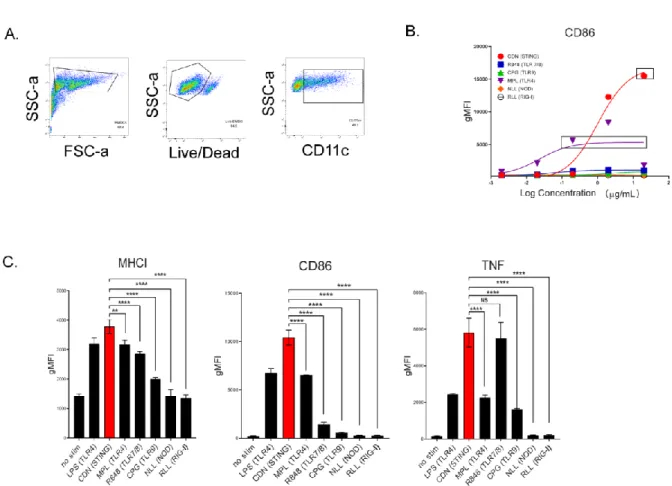
Optimization and derivation of MuSyC dose for the combination [1/10 saturated dose of R848 and saturated dose of CDN]. Therefore, we can validate the MuSyC dose without performing the checkerboard method with the MuSyC assay (50 samples) and instead use the single agent dose responses with the MuSyC dose strategy (10 samples). We next wanted to determine whether the dose of MuSyC would translate to other types of APCs.
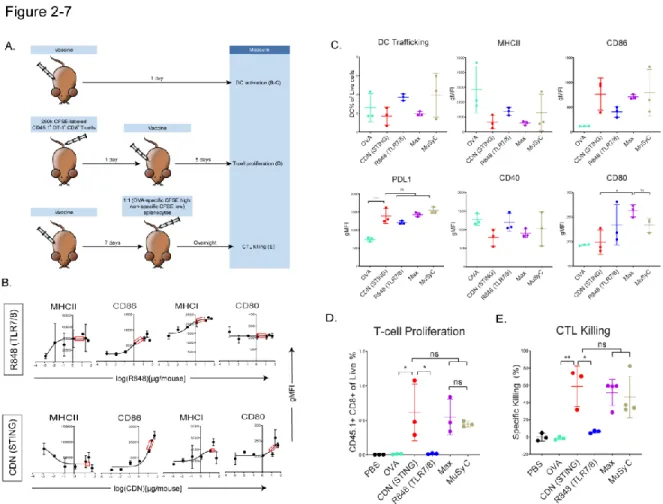
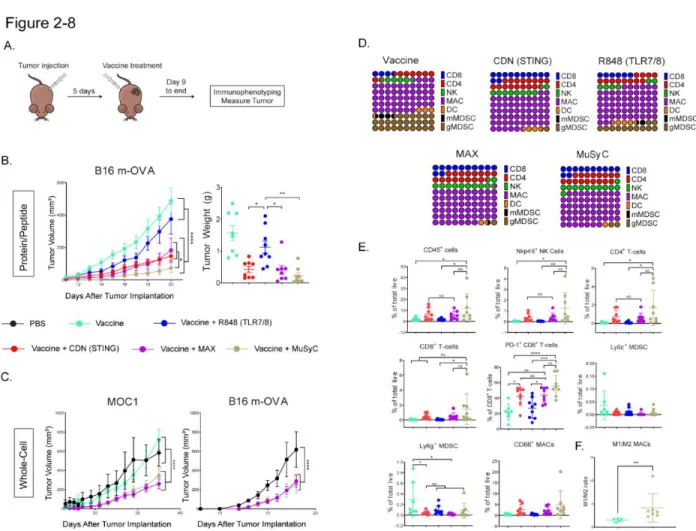
After showing that MuSyC-generated combination dose (MuSyC dose) of CDN and R848 is equivalent/comparable to the Max dose across several APC models in vitro, we tested whether the MuSyC dose also leads to synergistic T cell activation in vivo (Fig. .2-7 A). This tumor reduction trended with the tumor weights, but the MuSyC dose tumor weight was not significant compared to Max dose and CDN (Fig.2-8 B). In both cases, MuSyC dose or Max dose significantly decreases the tumor volume compared to PBS or GVAX alone (Fig.2-8 C).
However, there was no significant difference in the maximum dose and dose response of MuSyC (Figure 2-8 C). These higher CD45+ immune infiltrates induced by MuSyC dose treatment resulted in an increased percentage of CD8+ T-cells, CD4+ T-cells, NK cells, and CD68+ MACs in the tumor compared to other groups (Fig. 2-8 E). MuSyC-dose vaccine does not cause additional weight loss and reduces IL-6 plasma levels compared with CDN vaccine.
The MuSyC dose-vaccinated mice recovered significantly more than the CDN and Max dose groups than the R848 vaccine forty-eight hours after injection. However, MuSyC dose does not significantly affect weight loss compared to CDN or Max dose vaccines for the 24 and 48 hour time points. IL-6 is significantly lower in the MuSyC dose treatment group than in the CDN group (Fig. 2-10 C).
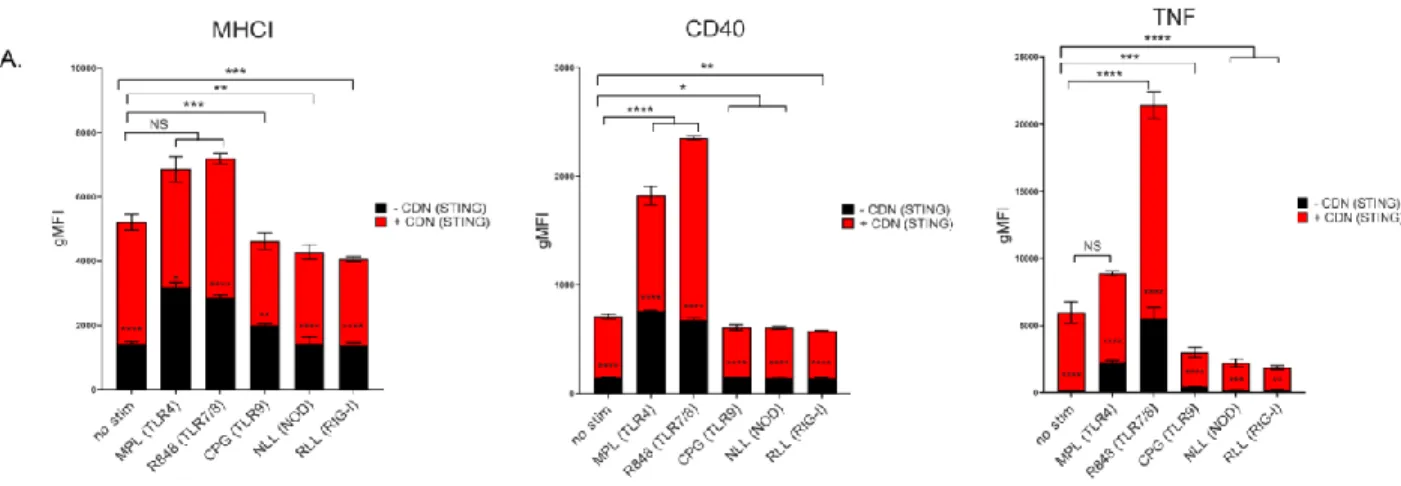
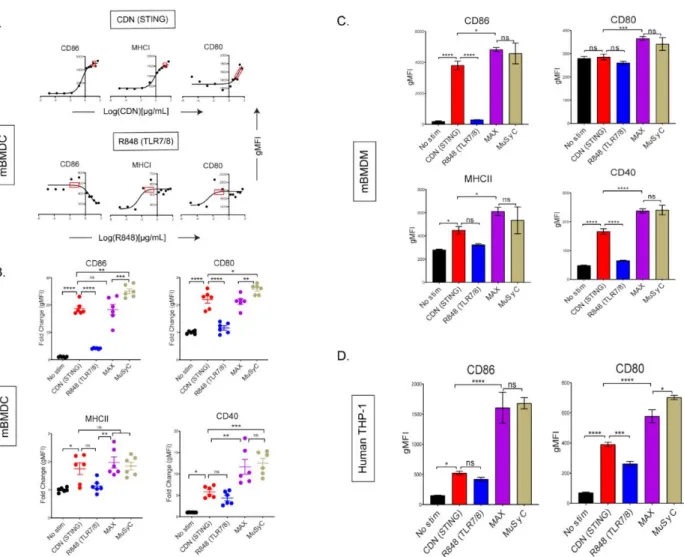
Surprisingly, vaccines with the highest dose or the MuSyC dose do not significantly affect the remaining cytokines with CDN treatment. The MuSyC algorithm classified the combination of CDN and R848 as synergistically effective and potent for APC activation. Regardless, our MuSyC dose derivation for CDN and R848 resulted in similar or better APC activation effects compared to the maximum in vitro dose for several models.
Specifically, MuSyC dose-adjuvanted vaccines optimized the antitumor response and induced novel changes in the tumor microenvironment. In addition to increased tumor-infiltrating lymphocytes, the MuSyC CD45+ dosed vaccine population contained a high proportion of macrophages, suggesting a myeloid-based mechanism for the antitumor response.
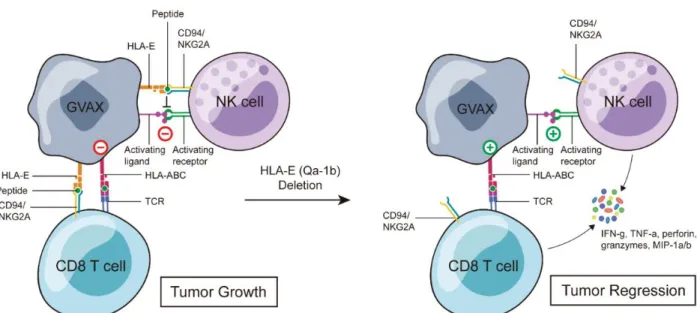
ABROGATION OF HLA-E/QA-1B EXPRESSION ON TUMOR CELLS SIGNIFICANTLY INCREASES
To circumvent this, we generated the mouse homologue of HLA-E, Qa-1b, knocked out tumor cells and used them to generate GVAX without HLA-E/Qa-1b (QVAX). Mice were inoculated with B16-mOVA or MOC2 on the flank and then administered GVAX on day five and NKG2A blockade two days later. However, by using a whole-cell vaccine instead of a peptide, NKG2A blockade may also work at the vaccination site, since the tumor cells in GVAX also express HLA-E/Qa-1b.
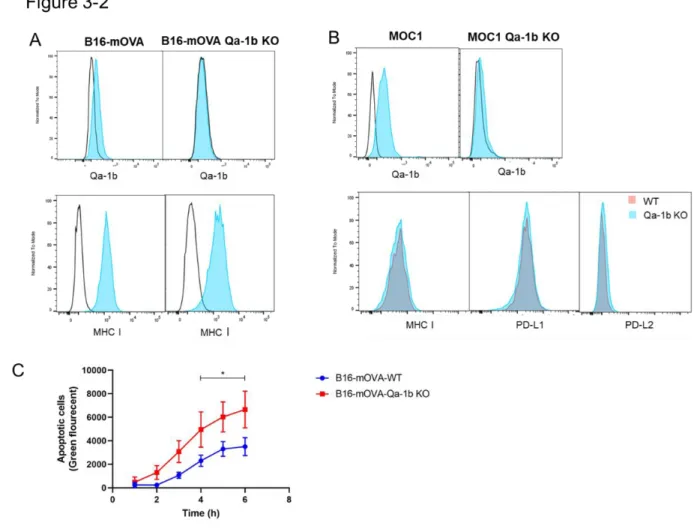
Loss of Qa-1b does not alter MHC I or PD-L1 expression on mouse tumor cells. To separate the effect of NKG2A blockade on the primary tumor and the vaccination site, we generated Qa-1b-deficient B16-mOVA and MOC1 tumor cells to use as a vaccine source. In addition, we performed an NK killing assay to determine whether there was a functional loss of Qa-1b. NK cells express high levels of NKG2A, and it was previously shown that NK cell-mediated killing is inhibited by the expression of HLA-E/Qa-1b ( 375 ).
Furthermore, B16-mOVA Qa-1b KO cells were significantly more sensitive to NK cell-mediated killing than WT B16-mOVA cells (Fig. 3-2 C), suggesting a functional loss of Qa-1b. Figure 3-2: MOC1 and B16-mOVA Qa-1b KO cells have normal expression of MHCI, PD-L1 and increased sensitivity to NK cell-mediated killing. B16-mOVA-WT and B16-mOVA-Qa-1b-KO cells were co-cultured with IL-2-activated NK cells at an effector-to-target ratio of 25:1. We next determined whether GVAX generated with tumor cells lacking Qa-1b (QVAX) is superior to GVAX in controlling WT tumor growth.
1e6 CD45.1 CFSE labeled OT-1 CD8 T cells were adoptively transferred into B6 mice and then vaccinated with B16-mOVA GVAX or QVAX the following day. In this study, we showed that the NKG2A ligand HLA-E/Qa-1b inhibits the anti-tumor immune response elicited by GVAX. We have shown that murine and human tumor cells comprising clinical GVAX express HLA-E/Qa-1b after interferon-gamma stimulation.
We reasoned that expression of HLA-E/Qa-1b at the site of vaccination is likely to have the same immune-suppressive effects as we and others have observed in primary tumors ( 373 , 374 ). To determine whether HLA-E/Qa-1b was suppressing immune activation at the site of vaccination, we generated tumor cells lacking Qa-1b, which had normal MHC I and PD-L1 expression. When Qa-1b-deficient tumor cells were used in GVAX, which we called QVAX, we observed a remarkable reduction in tumor volume compared to GVAX.
CONCLUSION AND FUTURE PERSPECTIVES