From discovering my talent to applying my skills in the right areas of work, she guided me with patience and enthusiasm and helped me throughout my project and writing this assignment. Furthermore, the phonon dispersion curves of the investigated compounds showed the flat nature of the phonon bands, and we also found that the low-lying optical and acoustic modes intersect at a lower frequency range, indicating that the investigated compounds may have reasonably low thermal conductivity. Typically, materials with small electronegativity differences have high mobility and low effective mass, while materials with narrow bandgaps can offer low mobility and high effective mass[5] due to the ratio of the density of states to the overall dispersion ratio in motion space.
This property of zintl materials allows control of the carrier concentration through precise doping without disturbing the mobility of the carrier. This thesis is structured as follows: chapter 2 describes the theoretical background, and chapter 3 explains the computational aspects of the current research.
Born-Oppenheimer Approximation
The Hartree approximation
Hartree-Fock Method
The limitations of this approach are that it does not include correlation, the wave function is not antisymmetric and it does not remove the (n,l)-random degeneracy of the hydrogenoid atom. The Hartree-Fock equations describe non-interacting electrons under the influence of a mean field potential consisting of the classical Coulomb potential and a non-local exchange potential. In addition, regardless of this method's success in describing various systems, it failed due to the weak exchange and correlation limits of electrons.
Density Functional Theory
- Thomas-Fermi Equation
- Hohenberg - Kohn Theorem
- Khon-Sham Method
- Local Density Approximation (LDA)
- Generalised Gradient Approximation (GGA)
- Tran-Blaha modified Becke-Johnson potential (TB-mBJ)
The correlation energy is the difference between the full ground state energy and the exchange correlation energy. A practical implementation can be done using the Kohn-Sham method, which is discussed in the next section. If good approximations for these functionalities could be found, direct minimization of the energy would be possible.
These Kohn-Sham equations have the same structure as the Hartree-Fock equations with the non-local exchange potential replaced by the local exchange-correlation potential VXC =δEδρ(~XCr)(~r). Since the ground state electron density is the fundamental variable, this gives us an advantage by reducing the 3N dimensional problem to a 3 dimensional problem. LDA approach is a key contribution by Kohn-Sham, which for calculations of the quantum ground state of many-particle systems proved to be superior to both Thomas-Fermi and Hartree-Fock theories, the basis of the local density approximation for the exchange and correlation -energy functional is the theory of the homogeneous electron gas.
Because the LDA approximates the energy of the true density with the energy of a local constant density, it fails in situations where the density undergoes rapid changes, such as in molecules. An improvement of this situation can be made by taking into account the so-called electron density gradient. Tran-Blaha modified Becke-Johnson potential (TB-mBJ) can be one of the solutions to this problem as this exchange correlation function can capture the exact band gap.
This is used in this thesis to accurately determine the band gaps of the compounds examined.
Introduction to Thermoelectric Materials
The direction and magnitude of the Seebeck voltage (V) depend on the temperature difference between the two junctions of the thermocouple and on the materials composing the thermocouple, i.e. on the Seebeck coefficient [57]. The rate of heating or cooling at an intersection is found to be proportional to the strength of the current and changes sign when reversing the direction of the current. The Peltier coefficient (π) is determined by the ratio between the heating rate (Q) and the current (I).
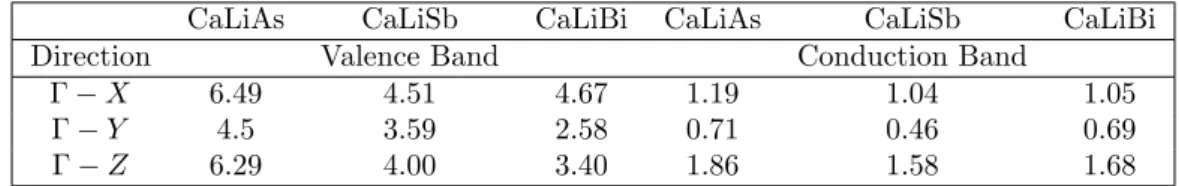
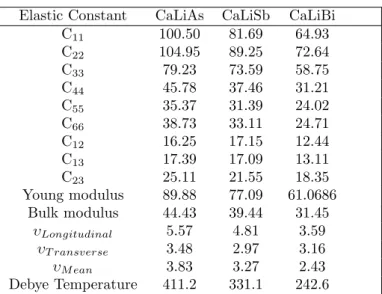
The investigated compounds CaLiP n(P n= As, Sb, Bi) crystallize in orthorhombic structure with space groupPnma(62), and the crystal structure is given in figure. Elastic constants are the fundamental material parameters that determine the material's resistance to applied mechanical deformation. ΘD is a fundamental quantity that correlates various physical properties, such as specific heat, thermal conductivity and melting point of the crystal, with elastic constants.
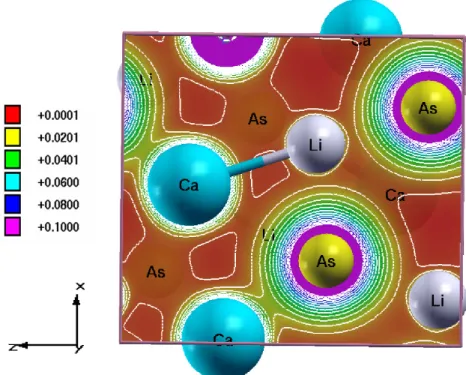
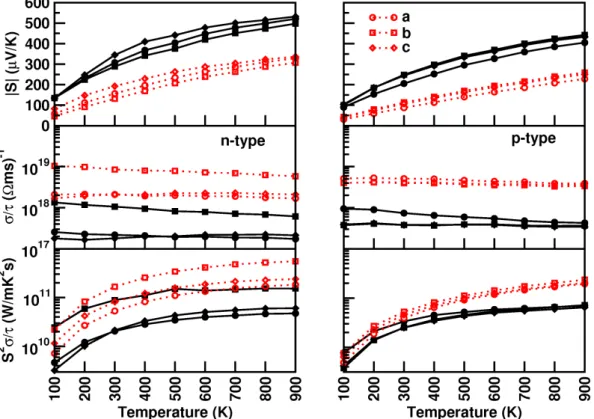
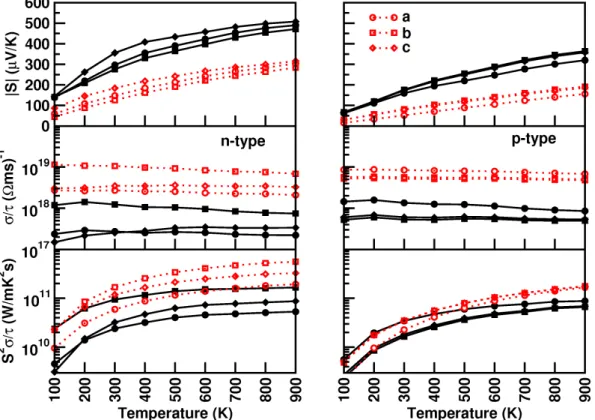
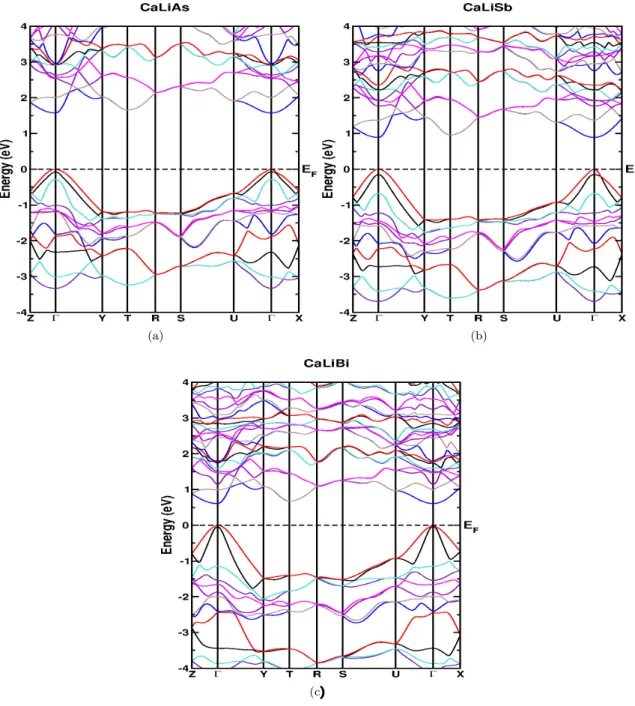
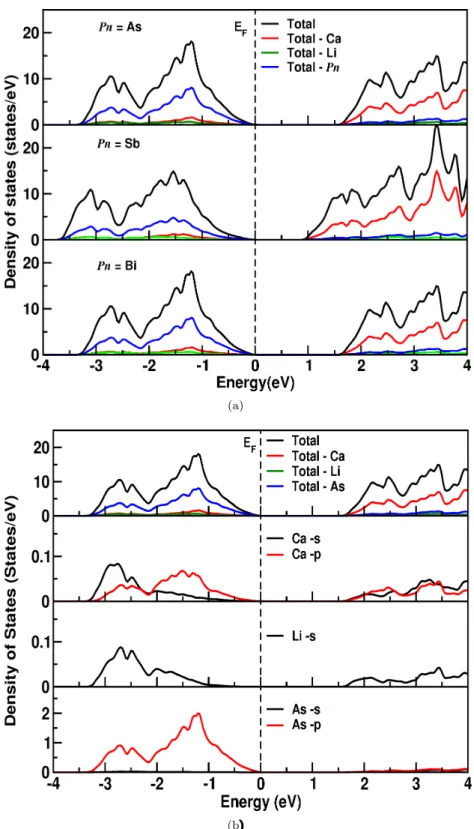
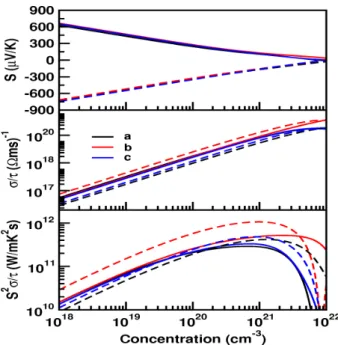
However, we tried to compare the coefficients of the elastic constant of the currently investigated compounds with the prototype family of SrLiAs material [17]. It is found that in SrLiAsC11>C22 This indicates that the studied compounds may also have low thermal conductivity compared to SrLiAs. Further analysis of the electronic properties requires the total and partial density of states of these compounds, and the same is represented in Fig 4.3. In the valence band, anion P nstates predominate and in the conduction band Ca states predominate for all three compounds. From CaLiAs to CaLiBi, the contribution of Li states near the Fermi level is found to be increased. The heavy band found just below the valence band maximum arises from the P n-p states, and the light band found just below is mainly due to Ca-p and Li-s states. Furthermore, we also calculated the charge density of CaLiAs to analyze the bonding nature of the investigated compounds and the same is shown in figure. In general, the weak electro-negativity of the 'Li' atom will form the ionic bond in most cases, but in the present case it makes a covalent bond similar to SrLiAs [17]. Covalent bonding between Li-As reduces the mass of carriers at the band edge resulting in light carriers. Heavy bands usually contribute to high thermopower as it is proportional to the effective mass (m∗)[13], while lighter bands contribute to high mobility (mobility, μ=τ e/m∗, where e is the charge) . For CaLiAs, the change in the density of states near the Fermi level is almost similar for the valence band and the conduction band, while in the case of the other two compounds the change in the density of states near the Fermi level is more in the conduction band than in the of valence. This behavior indicates that for CaLiAs the TE properties of holes and electrons may be similar, whereas the electron doping of the other two will be more favorable. The higher values in the case of electrons as carriers may be due to the increased number of minima in the conduction band region compared to the valence band [49]. In the case of thermopower, almost isotropic behavior is observed along different crystallographic directions and this may be due to the similar band structure nature along different crystallographic directions. Almost a similar type of variation is observed in the case of electrical conductivity scaled by relaxation time in all the compounds. A higher value of electrical conductivity is noted in the case of electron concentration compared to holes, which is very similar to that of the thermopower of the investigated compounds. Furthermore, we find a significant anisotropic character for electrons along the b-axis compared to the a- and c-axes. This may be due to the lower value of effective mass along the Γ−Y direction compared to other two directions in the case of conduction band. In the case of holes, nearly isotropic behavior inσ/τ along different crystallographic directions is observed. In the case of hole doping, the power factor values are found to be nearly isotropic along different crystallographic directions. As previously mentioned, electron doping is more favorable compared to holes in CaLiAs, and a similar situation is also seen in the case of SrLiAs. Further slightly higher value of the thermopower in the case of CaLiAs resulted in a higher value of the power factor compared to SrLiAs. In general we can say that electronic doping is more favorable for TE properties for all investigated compounds. To analyze how better the TE properties of the investigated compounds are, we have compared them with SrLiAs. Next, we tried to evaluate the order of thermal conductivity of the investigated compounds. In this case, we also tried to predict the minimum thermal conductivity for all investigated compounds and it is shown in table 4.4. From Table 4.4, it is clear that the κmin of the investigated compounds is below unity. It is also found that the calculated value of κmin for the investigated compounds is in the same order as other Ca5Al2Sb6 zinc phase compounds based on Ca [6]. This further confirms that the investigated Ca-based pnictide compounds are good TE candidates compared to SrLiAs. Further analysis of the electronic structure indicates that the investigated compounds possess mixed heavy and light bands, indicating that they have good thermoelectric properties. We also calculated the thermoelectric properties of all investigated compounds for both carrier concentrations at different temperatures. Among the compounds examined, we find that CaLiBi has a bipolar conductivity at high temperatures, which may be due to the lower band gap of this material compared to the rest of the compounds. Furthermore, the phonon dispersion curves of the investigated compounds show a flat phonon band, which will absorb more heat, resulting in low thermal conductivity. We also observed the intersection of lower optical and acoustic branches, further indicating that the investigated compounds may have reasonably low thermal conductivity.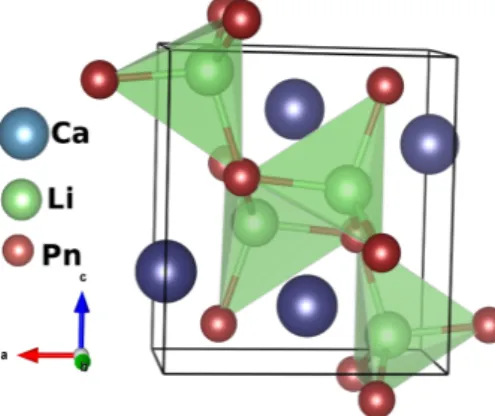
Electronic properties
Thermoelectric properties
Lattice dynamics
![Table 4.1: Calculated structural parameters of CaLiP n (P n = AS, Sb, Bi) along with the experi- experi-mental [16] parameters](https://thumb-ap.123doks.com/thumbv2/azpdfnet/10468934.0/23.892.235.685.158.255/table-calculated-structural-parameters-calip-experi-experi-parameters.webp)