Sewell is a senior lecturer in pharmacology at the Welsh School of Pharmacy, Cardiff University, United Kingdom and also director of the postgraduate M.Sc. It has also raised ideas, hypotheses and theories that represent a significant step forward in the knowledge of the molecular basis of protein aggregation and aggregate toxicity.
INTRODUCTION
The other is that in the microbial world (but examples have also been reported in mammals) amyloids can perform specific biological functions, further highlighting the general importance of protein aggregation in biology. In particular, an increasing wealth of information is accumulating on the cell biology of the complex quality control mechanisms of protein folding either in the cytosol or in the endoplasmic reticulum (ER).
ESSENTIALS OF PROTEIN FOLDING
For example, different modules or domains in the same polypeptide chain have been shown to fold in a rather independent manner (Dinner et al., 2000), and native interactions between residues are established independently in different regions of the polypeptide. Contact order is calculated as the average separation in the amino acid sequence of the contacting residues in the folded state.
KNOWLEDGE OF PROTEIN FOLDING IS FUNDAMENTAL TO UNDERSTAND THE MOLECULAR BASIS OF PROTEIN
The primary physiological function of the molecular chaperones both in the cytosol (heat shock proteins, crystallins, prefoldin, Hsc70) and in the ER (Bip, Grp94, calnexin) is to favor folding of nascent polypeptide chains or refolding of inappropriately folded molecules, to guide later stages of the folding process and to avoid inappropriate interactions of misfolded or incompletely folded polypeptides (Bukau and Horwich, 1998; Hartl and Hayer-Hartl, 2002). Indeed, protein aggregation is well established as one of the major difficulties associated with the production and handling of proteins in the biotechnological and pharmaceutical industries (Clark, 1998).
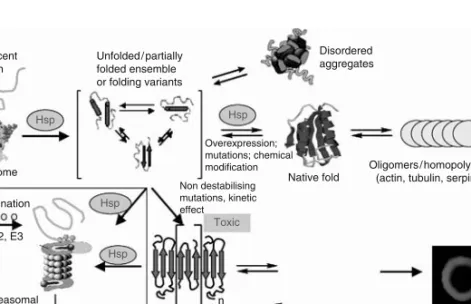
PROTEIN MISFOLDING DISEASES
Recent data suggest that other proteins or peptides are able to propagate a toxic conformation to the natively folded counterparts (Lundmark et al., 2002; Xing et al., 2002; Giasson et al., 2003) or even to different low propensity proteins. to merge (heterogeneous seed) (Xing et al., 2002). The proposed factors associated with increased risk for sporadic amyloidosis include specific genotypes (such as the ApoE4 variant in Alzheimer's disease) (Myers and Goate, 2001) or incorrect lifestyle leading to hypercholesterolemia or to excessive caloric intake (Austen et al., 2002).
SERPINOPATHIES
Support for this mechanism of pathogenesis has been provided by in vitro experiments showing that the same α1-antitrypsin mutants associated with disease are also able to form polymers when incubated in vitro under physiological conditions (Dafforn et al ., 1999).α1-antitrypsin polymers have also been shown in inclusion bodies from the cirrhotic liver of patients carrying homozygous mutations of the protein (Lomas et al., 1992). The subsequent conformational destabilization of neuroserpin leads to the appearance of intracellular protein aggregates that are sequestered in inclusion bodies (Collins'bodies) found in cells in the deeper layer of the cerebral cortex and in the substantia nigra (Davis et al., 2002).
ER STORAGE DISEASES
The tools in the ER that aim to perform the quality control of protein folding normally lead to degradation of a significant part of polypeptide chains before they have reached their native conformations; ER protein degradation is accomplished by the ubiquitin-proteasome pathway after retrograde translocation to the cytosol (Reits et al., 2000; Schubert et al., 2000). However, under ER stress conditions with accumulating unfolded proteins in the ER lumen, GRP78 binds to the unfolded proteins to promote their refolding.
AMYLOID DISEASES
Finally, protein aggregation into amyloid is favored under conditions that result in impairment or overload of the molecular machinery that targets performance. The latter includes cytosolic molecular chaperones, ATP-dependent proteolytic complexes in mitochondria, and components of the ubiquitin-proteasome pathway in the cytosol and in the nucleus (Goldberg, 2003).
STRUCTURE OF AMYLOID FIBRILS AND THEIR PRECURSORS Despite the differences in size, amino-acid composition, sequence and structure of
Self-propagating, molecular-level polymorphism in Abfibrils that is associated with different toxicity and that results from subtle differences in the conditions of fibril growth has also been highlighted (Petkova et al., 2005). A key issue in the investigation of the amyloid structures is the description of either the mechanism by which they are formed from their soluble precursors and the structural features of their intermediates.

STRUCTURAL FEATURES FAVORING PROTEIN FIBRILLIZATION
Such an effect is also confirmed by the increase in the rate of aggregation due to the presence of preformed oligomers in the solution of aggregating monomers. This is the case of the heterogeneous grafting, a phenomenon described for a number of proteins (Xing et al., 2002), which confirms the common basic structural features of the inner ordered core of amyloid fibrils.
STRUCTURAL RELATIONS BETWEEN MISFOLDED PROTEINS AND THEIR TOXIC EFFECTS
In elderly people, medin amyloid aggregates are deposited in the aortic mid-vessels, mostly localized in the upper body, a condition without well-established clinical significance (Peng et al., 2005). The maturation of Pmel17 into fibrils is strikingly similar to the mechanism of amyloidogenesis of the gelsolin variant in Finnish amyloidosis.
MECHANISMS OF AMYLOID TOXICITY
Recently, a new hypothesis on AD pathogenesis underlined the importance of the association with a pathogen as a trigger for the disease (reviewed in Robinson et al., 2004). Infectious agents such as Chlamydia pneumoniae and spirochaetes have also been suggested to be associated with AD at least in mice (Miklossy, 1993; Little et al., 2004).
BIOLOGICAL SURFACES AS PRIMARY SITES OF AMYLOID ASSEMBLY AND TOXICITY
Indeed, anionic membrane phospholipids such as PS and PG have been shown to interact with amyloid aggregates, possibly by recognizing a shared fold (Zhao et al., 2004). It has also been reported that alteration in cholesterol homeostasis could be a shared primary cause of several neurodegenerative diseases (Kakio et al., 2003).
ION CHANNEL HYPOTHESIS
Similarly, more information is needed about the cellular site(s) where these interactions occur; for example, whether there are specific membrane receptors for common structural features of prefibrillar aggregates. This behavior is reminiscent of the action of several bacterial pore-forming toxins such as perfringolysin (Hotze et al., 2002) (see Section 1.3), although eukaryotic examples of this mechanism have also been described.

TWO FACES OF PROTEIN BIOLOGY
In general, it is assumed that the interaction of the aggregates or their precursors with the cell membranes is itself able to destabilize the phospholipid bilayer, creating disordered regions, allowing the reduction or destruction of the ion gradients (Kayed et al. Demuro et al., 2005). Abnormal levels of molecular chaperones have recently been found in the brains of AD patients (Yoo et al., 2001), suggesting the possibility of searching for the presence of abnormalities in the regulation of the corresponding genes in protein deposition diseases (Macario and de Macario, 2002).

DEGENERATIVE CONDITIONS WITH PROTEIN AGGREGATES MAY BE MUCH MORE COMMON
The key role in the molecular pathogenesis of a number of degenerative retinal diseases, played by specialized opsin chaperones, has also recently come to light (reviewed in Chappie et al., 2001). Recent findings highlight the possible amyloid basis of scalp hypotrichosis simplex (HSS), an autosomal dominant form of isolated alopecia caused by mutations in the CDSN gene encoding corneodesmosin (Jonca et al., 2002; Levy-Nissenbaum et al. , 2003). .
AMYLOIDS MAY PERFORM BIOLOGICAL FUNCTIONS
This finding suggests that the silkmoth chorion is a natural amyloid with protective properties necessary for survival and development of the oocyte and embryo (Iconomidou et al., 2000a, 2000b). It has been suggested that the toxic effects of the SAA and AA aggregates are a disadvantage of the proposed physiological function of the hexameric assemblies to protect against infection by forming toxic channels in the cell membranes of the invading bacteria (Hirakura et al. , 2002). .
FINAL CONSIDERATIONS
Implication for pathogenesis and therapy.Proceedings of the National Academy of Sciences of the United States of America. Proceedings of the National Academy of Sciences of the United States of America Mitochondrial involvement in Parkinson's disease.
INTRODUCTION
EPIDEMIOLOGY OF AD
The increase in the number of patients with AD will not only lead to an increase in the emotional burden for more and more patients and their social environment, but also to an increase in the economic costs for society in general. For example, in the Netherlands, the annual cost for an AD patient living at home is estimated at e5000–6000.
GENETIC RISK FACTORS
Prior to the discovery that ApoE plays a role in the etiology of AD, its function in lipid metabolism was extensively studied. When comparing homozygous fore4 with homozygous fore3, a ninefold increase in the incidence of cardiac ischemia on ECG was found in the former.
ASSESSMENT
D IAGNOSTIC R OUTE
Laboratory evaluation: The American Academy of Neurology recommends screening for B12 deficiency and hypothyroidism in patients with dementia (Knopman et al., 2001). Effects on mortality are exacerbated when cognitive dysfunction and depression coexist (Mehta et al., 2003).
PATHOLOGY
O XIDATIVE S TRESS
Other known mediators of inflammation have also been found in plaques, such as interleukin-1b (IL-1b), interleukin-6 (IL-6), and tumor necrosis factor (TNF)-a (Griffin et al., 1995; Gonzalez-Scarano and Baltuch, 1999). Furthermore, it was shown that use of cholesterol synthesis-lowering drugs was associated with a decreased incidence of AD (Wolozin et al., 2000b).
ANIMAL MODELS OF AD
M ICE
- PDAPP
- APP23
- HPS1
This model contains the Swedish (K670N and M671L) mutations in addition to the Indiana (V717F) mutation of APP, under the control of a Hamster prion protein promoter in C3H=B6 strains (Chishti et al., 2001). These mice contain the same mutations as the TgCRND8 mice, but are under the control of the PDGF minigene in a C57=Bl63DBA2J background (Hsia et al., 1999).
BIOMARKERS OF AD
Elevated serum or plasma levels of homocysteine have been shown to be a risk factor for AD (Ravaglia et al., 2005), although Furthermore, CSF levels of specific isoprostanes have been shown to be increased in MCI (Pratico et al., 2002).
THERAPIES
1999) Increased DNA oxidation and decreased levels of repair products in Alzheimer's disease ventricular CSF.Journal of Neurochemistry. 2004) Genetic testing has no place as a routine diagnostic test in sporadic and familial cases of Alzheimer's disease. Journal of the American Geriatrics Society.
CHOLINERGIC TRANSMISSION AND ACETYLCHOLINE RELEASE ENHANCERSRELEASE ENHANCERS
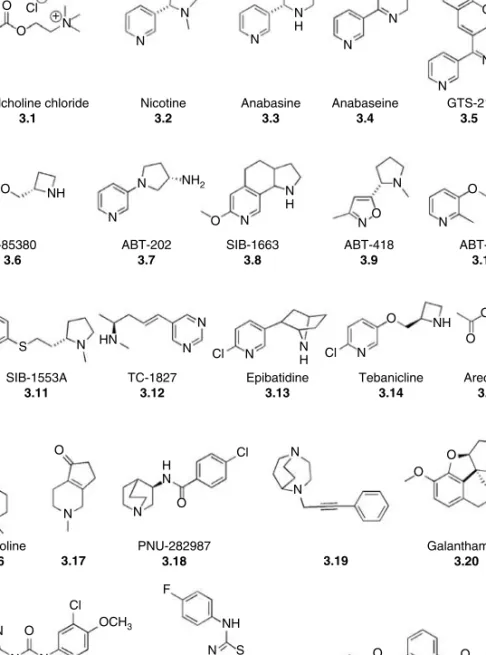
AC H R ELEASE E NHANCERS : N ICOTINIC S TIMULATION
However, this quaternary structure forms even when all appropriate cysteines have been replaced (Bon et al., 2003). Further research led to the isolation of the active component, namely ursolic acid (3.70) (Chung et al., 2001).
CONCLUSION ON THE CLINICAL USE
Characterization in vivo.Journal of Pharmacology and Experimental Therapeutics Rivastigmine for Alzheimer's disease.Expert Review of Neurotherapeutics. A randomized, double-blind, placebo-controlled study of the efficacy and safety of donepezil in patients with Alzheimer's disease in the nursing home setting. Journal of the American Geriatrics Association.
SECRETASE AND APP PROCESSING
The 50th end of the BACE1 mRNA represses effective translation of the protein (Lammich et al., 2004). Mutation of either of the two Asp residues abolishes g-secretase activity (Wolfe et al., 1999).

NONSTEROIDAL ANTI-INFLAMMATORY DRUGS Bruno P. Imbimbo and Francesca Speroni
COX-1 AND COX-2 I NHIBITORS
The interaction between 4-methylsulfonylphenyl and 4-methylsulfamoylphenyl substituents and Arg513 appears to be necessary for the time-dependent inhibition of COX-2 by these inhibitors (Kurumbail et al., 1996). All COX-2 selective inhibitors cause time-dependent inhibition of the COX-2 isoenzyme, but are time-independent competitive inhibitors of COX-1 (DeWitt, 1999; Smith et al., 2000).

NSAID S AND A LZHEIMER ’ S D ISEASE
The mechanism by which COX-2 inhibitors may increase the risk of cardiovascular events is not clear, but their depressant effect on prostaglandin I2 formation has been suggested as a cause of blood pressure elevation, acceleration of atherogenesis, and exaggerated thrombotic response to rupture of an atherosclerotic plaque ( Fitzgerald, 2004). Indeed, ADAPT was prematurely suspended due to an apparent increase in cardiovascular and cerebrovascular events in the naproxen arm compared with placebo.