With high-throughput whole-genome sequencing technologies, large-scale population genomics studies have become the standard not only for biological scientists in deciphering the code of life, but also for the general public dealing with global crises such as the COVID-19 pandemic. In the first chapter, I present my analysis of the DNA satellite regions of the Korean reference genome. Satellite DNA, which accounts for 5–10 percent of the human genome, is missing from the current Korean reference genome.
Here, in this chapter, I present methods for improving the contiguity of KOREF and building its replicated library from unstacked scaffolds for subsequent updates. A large-scale genomic investigation provides correlations between different mortality rates between ethnic groups and genomic variants. Finally, I present a comprehensive genomic study on variants associated with COVID-19 and an improved Korean reference genome assembly with a library of repetitive sequences.
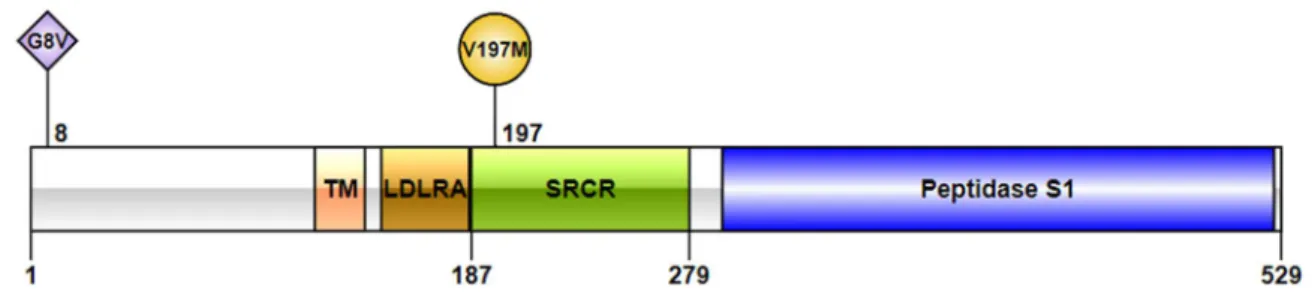
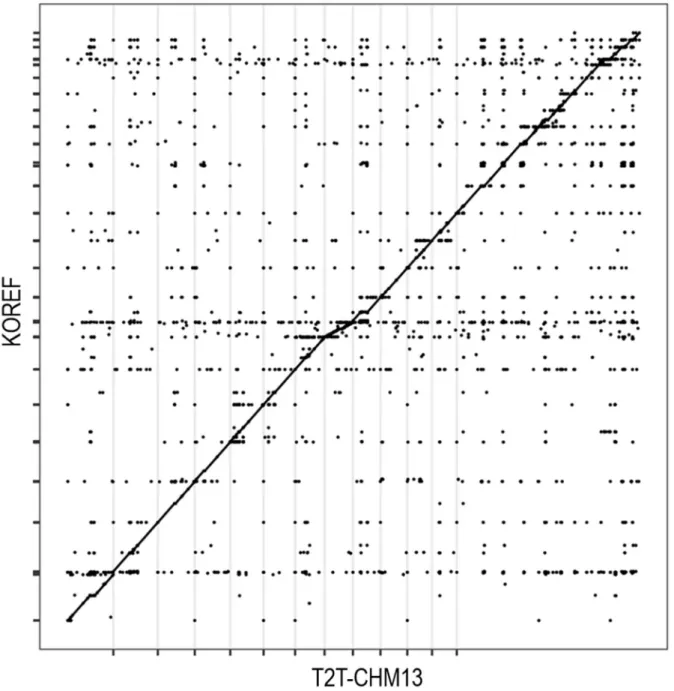
Dotplot presenting alignment between KOREF as a query on the Y-axis and T2T-CHM13 as a reference on the X-axis. The entire table of unplaced scaffolds which are assigned to the chromosomes https://docs.google.com/spreadsheets/d/1SqiwLJ16YEQKPZRz7JgwbyyjnHGj6cIjxVyJKfz0ohM. V197M, a nonsynonymous genomic variant which alters ‘valine’ into ‘methionine’ at the 197th residue of the TMPRSS2 protein.
G8V, a nonsynonymous genomic variant that changes 'glycine' to 'valine' at residue 8 of the TRMSS2 protein.
Chapter 1. The repeat library of the Korean reference genome
- Introduction
- Methods
- Results
- Discussion
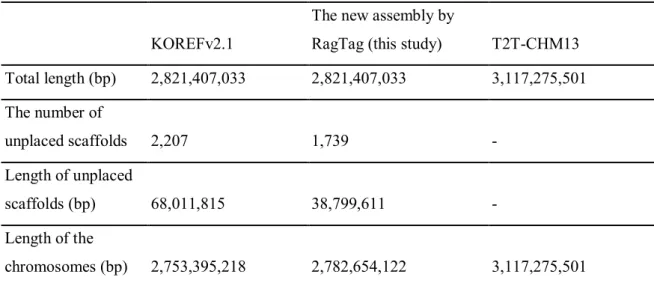
To predict the reliability of KOREF scaffold assignment to the T2T-CHM13 genome, whole genome alignment was performed using Minimap2 version 2.2433 with default options. Alignment of the unpaired scaffolds with the T2T-CHM13 genome shows that the unpaired scaffolds can be assembled as well as the guide reference genome (Figure 1). Centromeric and pericentromeric satellite DNA were extracted from the T2T-CHM13 genome through the bed file of “a centromere/satellite repeat annotation Cen/Sat v2.0 bed file” from https://github.com/marbl/CHM13 using getfasta34 bed tools.
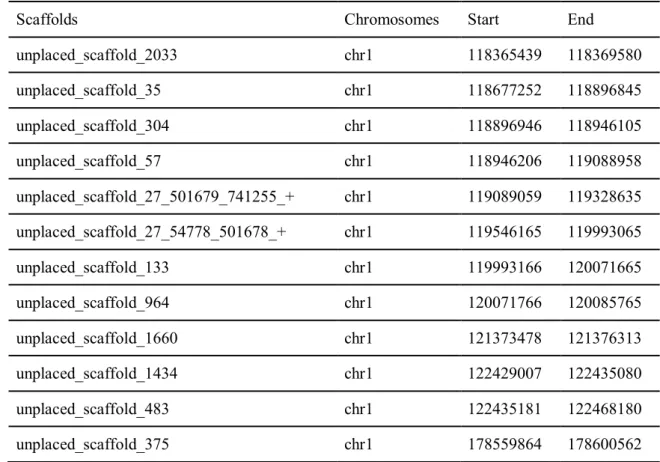
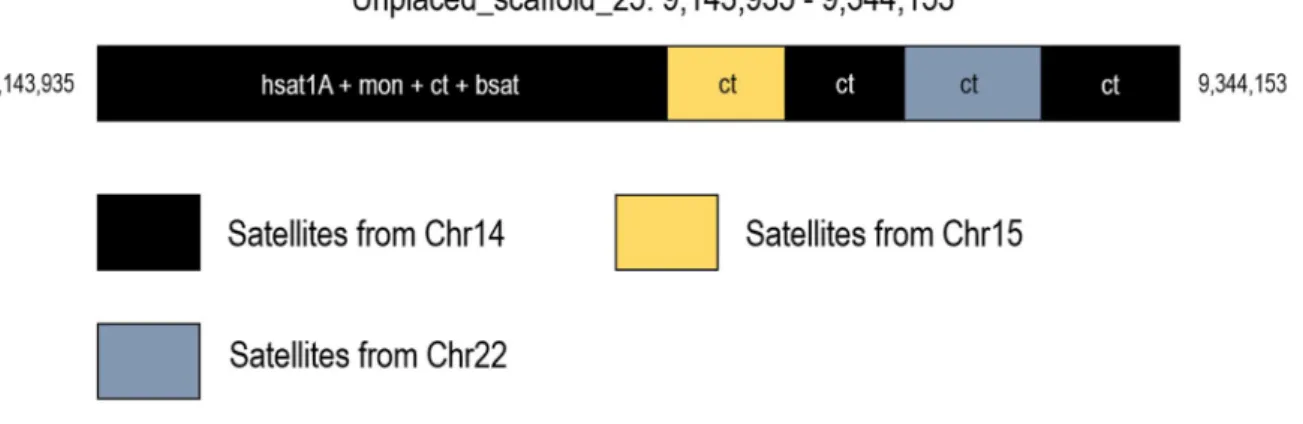
Unstacked scaffolds were aligned to centromeric and pericentromeric satellite DNA from the T2T-CHM13 genome using RepeatMasker version 4.1.335 with RMBlast version 2.11.0. Unplaced scaffolds comprise 68 Mbp, accounting for 2.5 percent of all scaffolds from the paternal KOREF set (Table 1). Excluding complex frameworks that consist of more than two regions from different chromosomes, unstacked frameworks have been shown to be parts of human satellites 1, 2 and 3, beta satellites, gamma satellites, alpha satellites, including higher order repeats (HOR ), highly conserved ribosomal DNA and centromeric transition regions.
The percentages of satellites have not yet been characterized due to the low resolution of RepeatMasker to show satellites and simple repeats and longer unselected scaffolds (>300 Kbp) which were probably assembled incorrectly (Figure 2 and Table 3). KOREF and T2T-CHM13 genome statistics. A total of 468 scaffolds (29 Mbp) were placed and assembled into chromosomes. The shortest scaffolds were shown to have ribosomal DNA (rDNA), human satellite repeat 2 (HSAT2), and high-order alpha repeats (HOR) sequences.
Since the satellite sequences were identified by an alignment to the T2T-CHM13 as a guide genome, potential novel Korean-specific satellites could not be identified and completely excluded from this study. To build the genuine Korean reference satellite DNA database, very precise and contiguous de novoassembly methodologies for KOREF must be applied. Furthermore, the longest scaffolds (>300 Kbp) were excluded from the downstream analysis due to its complex composition of various centromeric satellites, which resulted in a loss of sequences to break down.
Therefore, this study only provides a resource for further studies on the KOREF satellite DNA and improvement of the existing assembly. Although this study has many limitations, it is the first genome-wide research to examine the satellite repertoire of KOREF. Through this study, KOREF has been shown to have complex unlocalized scaffolds containing multiple fragments from different chromosomes.
Chapter 2. COVID-19-related genomic variant analysis
- Introduction
- Methods
- Results
- Discussion
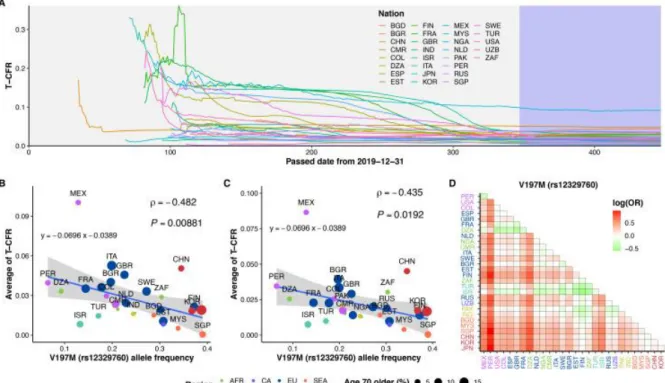
These two variants were found among 20 TMPRSS2 exonic variants with >1% AF globally; of which another 13 were in the three major untranslated regions (3' UTR) and five were synonymous. In contrast, G8V AF was not found to significantly bind to AT-CFR and AT-CFR-10038. The size of the dots indicates the percentage of people older than or equal to 70 in the countries.
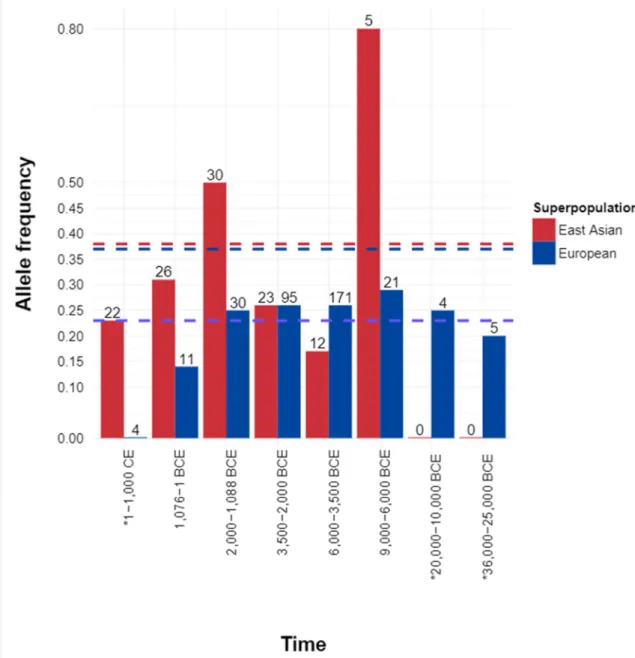
V197M is located in an extremely well-conserved position of the SRCR domain (phastCons17way_primate: 0.958), suggesting that it is under purifying selection38. Interestingly, V197M is absent in great apes55, 56 and in all archaic Denisovan and Neanderthal sequenced genomes38. The variant was observed in humans 40,000 years ago in East Asia from the Tianyuan human genotype38, 61 and in ancient genomes from Mongolia47.
Although small sample sizes precluded statistical analysis, V197M AF turned out to be higher in ancient East Asian populations (33.3%) than in ancient Europeans (16.3%) (Figure 5)38. Dashed lines denote allele frequencies in present day East Asians (red) and Europeans (blue) obtained from gnomAD database. Number on the bar indicates the total number of samples used for the time frame denoted on the x axis.
The study of the variant associated with COVID-19-CFR also has limitations in the application of variants in heterochromatin regions because the strategy is not reliable without worldwide multiethnic satellite databases which are not currently available. We used only public sources for genome databases and variant frequency information not directly associated with COVID-19 patients and AT-CFR. Also, I could not normalize the AT-CFR with environmental factors, such as SARS-CoV-2 test availability, standards of medical care, lockdown measures, mask availability, and proportion of the population smoking.
However, the variant was excluded from this study due to the small amount of genomic data from sex chromosomes. The old genome analysis could not be validated with statistical methods due to the small sample size. Nevertheless, this comprehensive analysis, including contemporary and ancient human genomes, is the first study to examine the history of variants associated with COVID-19 susceptibility.
Conclusion
The Korean Genome Project also played important roles in the process of finding correlations between AF and AT-CFR as a major genomic resource. Genome Holland, C., Whole genome sequence variation, population structure and demographic history of the Dutch population. Kubo, M.; Hirose, N.; Kamatani, Y., Whole-genome deep sequencing reveals recent signatures of selection associated with evolution and disease risk of the Japanese.
Yaegashi, N.; Kinoshita, K.; Kure, S.; Yasuda, J.; Yamamoto, M., Construction of JRG (Japanese reference genome) with single-molecule real-time sequencing. Makino, S.; Okamura, Y.; Kikuchi, A.; Sugimoto, S.; Kawashima, J.; Otsuki, A.; Sakurai- Yageta, M.; Yasuda, J.; Kure, S.; Kinoshita, K.; Yamamoto, M.; Tamiya, G., Construction and integration of three de novo Japanese human genome assemblies toward a population-specific reference. S.; Bhak, J., KoVariome: Korean National Standard Reference Variome database of whole genomes with comprehensive SNV, indel, CNV, and SV analyses.
Manica, A.; Lee, S.; Bhak, J., Comparison of chromosome-level assembly of the Korean KOREF reference genome from PromethION and PacBio with Hi-C mapping information. S.; Jeon, S.; Kim, Y.; Kim, C.; Bhak, J.; Bhak, J., KOREF_S1: a phased, parental trio-binded Korean reference genome using long reads and Hi-C sequencing methods. M.; Mantovani, A.; Duga, S., ACE2 and TMPRSS2 variants and expression as candidates for gender and national differences in the severity of COVID-19 in Italy.
K.; Backman, V., Evidence for possible association of vitamin D status with cytokine storm and unregulated inflammation in COVID-19 patients. P.; Xu, S., PGG.SNV: understanding the evolutionary and medical implications of human single nucleotide variations in diverse populations. F.; Vuckovic, D.; Girotto, G.; Matullo, G.; Polasek, O.; Kolcic, I.; Gasparini, P.; Soranzo, N.; Toniolo, D.; Mezzavilla, M., A bird's-eye view of Italian genomic variation through whole-genome sequencing.
Bosch, E.; Kucinskas, V., Patterns of genetic structure and adaptive positive selection in the Lithuanian population from high-density SNP data. Zhang, M.; Gao, X.; Cui, Y.; Reich, D.; Stoneking, M.; Fu, Q., Ancient DNA indicates human population shifts and intermingling in northern and southern China. Strauss, A.; Fehren-Schmitz, L.; Krause, J.; Reich, D., Reconstructing the deep population history of Central and South America.
Villapalos-Garcia, G.; Zubiaur, P.; Rivas-Duran, R.; Camps-Nord, P.; Arevalo-Roman, C.; Fernandez-Rico, M.; Garcia-Frare Frare, L.; Fernandez-Campos, P.; Soria-Chacartegui, P.; Fernandez de Cordoba-Onate, S.; Delgado-Wicke, P.; Fernandez-Ruiz, E.;. Gonzalez-Alvaro, I.; Sanz, J.; Abad-Santos, F.; dels Sants, I., Transmembrane protease serine 2 (TMPRSS2) rs75603675, comorbiditeit en seks is die primêre voorspellers van COVID-19-erns.