Effect of cerulenin and its analogues at a concentration of 10 µM on the cell viability of RAW 264.7 cells. Inhibition of fatty acid synthase activity in RAW 264.7 cells by cerulenin and its analogues at a concentration of 10 µM.
Introduction
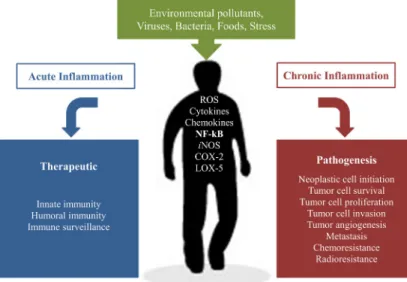
Diseases associated with chronic inflammation
COPD is now the fourth leading cause of death in the world, but will rise to third place by 2030. IBD is a chronic inflammatory bowel disease caused by a dysregulated immune response resulting from increased immune system activity in the intestinal microflora, which subsequently leads to tissue damage.
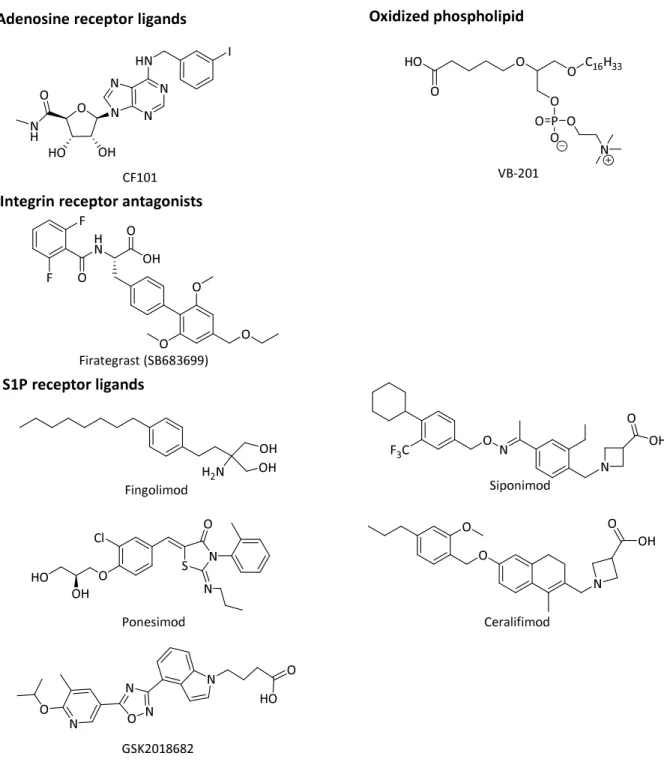
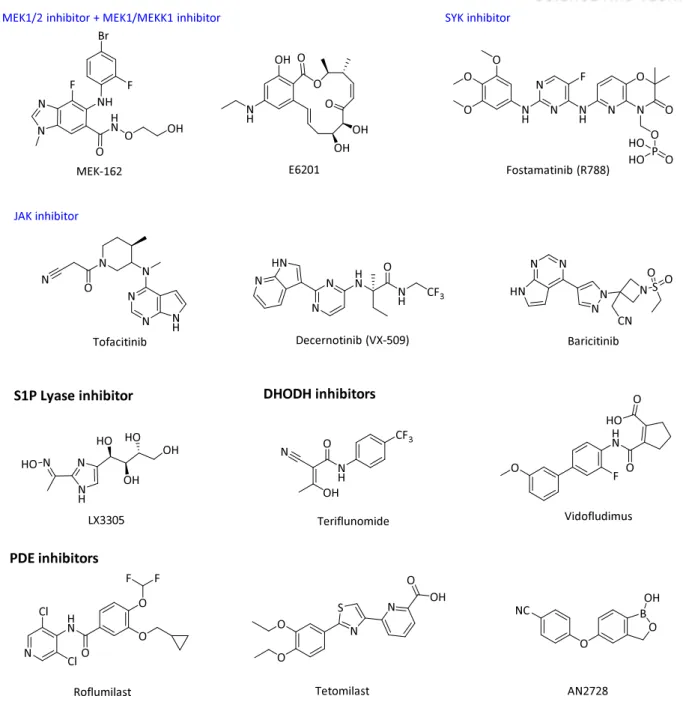
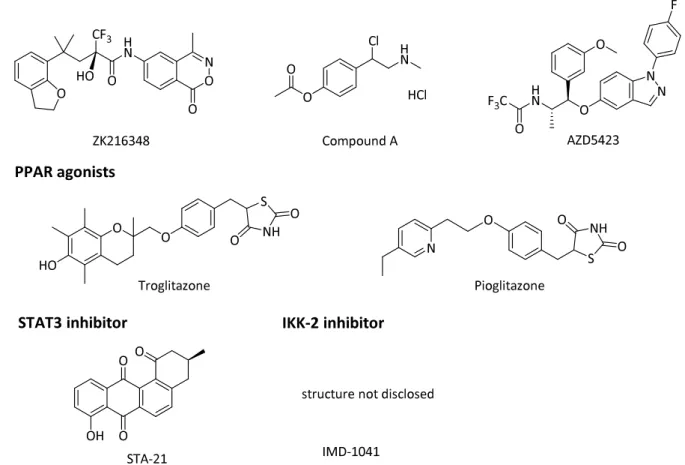
Variable small molecules to target inflammatory mediators
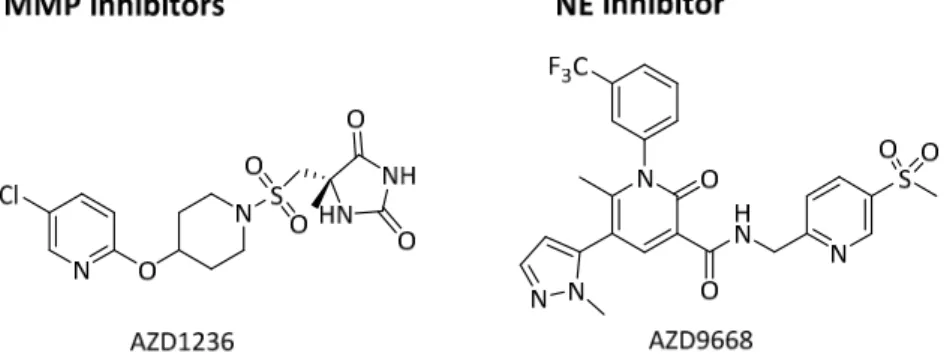
Other CXCR2 antagonist AZD5069 showed safety and tolerability in a phase I and was also tested in a phase II trial for COPD and asthma. The IKK-2 inhibitor IMD-1041 is currently being evaluated in a phase II clinical trial for COPD, but no results have yet been confirmed and the structure is not being disclosed.
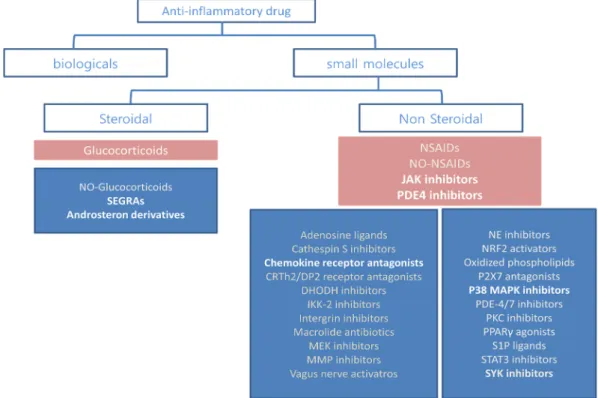
A promising therapeutic target: NF-κB
Inhibition of NF-κB signaling
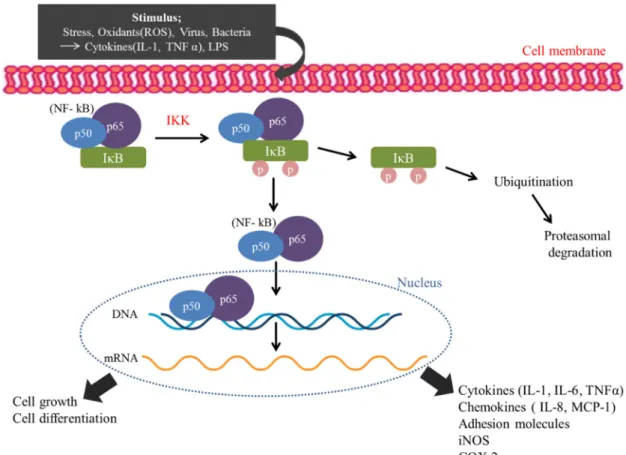
Antioxidants are also potential therapeutic inhibitors of NF-κB.51 However, the exact mechanism for antioxidants to block NF-κB activation is unknown. IKK is a prime target for the development of inhibitors of NF-κB signaling and also for kinase inhibitors for therapeutic applications because it plays an important role in NF-κB activation. Glucocorticoids (GC) are the molecules that are applicable at this level by inhibiting NF-κB by different methods.59 Dexamethasone increases the level of IκBα, causing NF-κB (p50,p65) to remain in the cytoplasm.60 In addition, hydrocortisone induces the expression of IκBα and inhibits the translocation of NF-κB to the nucleus in human cells.61.
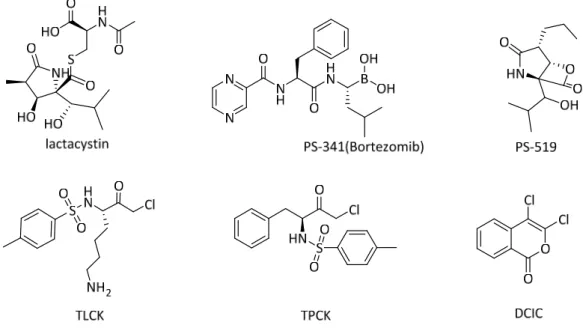
NSAIDs, sulfasalazine and leflunomide block nuclear translocation of NF-κB through inhibition of IκB-α degradation, which may be caused by a direct effect on IKK or upstream signals. Sulfasalazine suppresses nuclear translocation of NF-κB by phosphorylation and degradation of IκBα.62 Likewise, natural product gliotoxin from Aspergillus inhibits the phosphorylation and degradation of IκB-α, inhibiting activation of NF-κB-α induced by LPS.63. MG132 inhibits the chymotrypsin-specific activity of the proteasome complex that blocks IκBα degradation and prevents radiation-induced activation of NF-κB.65. In addition, dipeptidyl boronate analogs also serve as proteasome activity inhibitor.
The final step of the NF-κB activation pathway involves nuclear translocation, DNA binding, and transcriptional activation, and by blocking these steps, we can successfully inhibit NF-κB activation. The natural product maealamine, mushroom-derived wortmannin, and LY294, 002 block NF-κB transactivation induced by specific genes. 74 Furthermore, specific proteins Bcl-2 and antithrombin are reported to inhibit transactivation by interfering with interactions with RelA. 75. Consequently, NF-κB inhibitors should be considered when developing anti-inflammatory pharmaceuticals.
TonEBP, potential target for inflammation
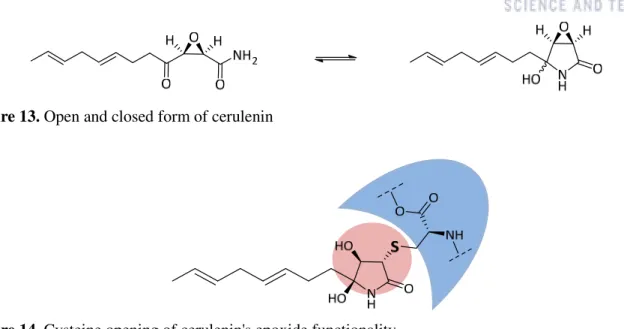
Cerulenin, also remarkable inhibitor for inflammation
Similarly, we are interested in cerulenin analogs as a potential tool to elucidate the mechanism of protein complexes derived from those of FASN. The development of chemically more stable synthetic derivatives is therefore also important due to the cytotoxicity of cerulenin which has pharmacological limitations.
Results and Discussion
Preparation of the click compounds
Parikh-Doering oxidation (SO3 pyr, DMSO) of the alcohol 4 yielded aldehyde 5,104. Aldehyde 5 was reacted with vinyl magnesium bromide to give secondary alcohol 6. Reaction of the acryloyl chloride with the racemic alcohol 6 in the presence of Hunig's base gave the acryloyl ester 7. Highly efficient sodium hypochlorite-olefin epoxidation of 10 yielded epoxy anhydride 11, which was converted to amide 12 by reacting with ammonia.
Bromocarboxylic acid 14 was treated with lithium (trimethylsilyl)acetylide, which was prepared by deprotection of trimethylsilylacetylene with butyllithium, to give compound 15 in 56% yield. After reduction with lithium aluminum hydride and deprotection of the trimethylsilyl group (TMS), the desired alkynol 16 was obtained. Scheme 2) Ester-amide 20 was prepared starting from the known epoxy maleic anhydride. Sodium tungstate catalyzed the epoxidation of maleic acid or its anhydride gave the disodium salt in 99% yield on a molar scale, which was converted to the carboxylic acid 17 via the corresponding barium salt in 75–80% overall yield.107 The anhydride 18108 heating 17 with 2 equivalents of TFAA in dichloromethane formed.
Ring opening of anhydride 18 was achieved by reaction with the alkynol 16 to give the monosubstituted ester acid 19 in good yield. Attempts to prepare amide 20 using coupling reactions such as DCI and DMAP and oxalyl chloride did not give the desired compound. Finally, the carboxamide 20 was prepared by activating the ester acid with PC15 and then quenching the intermediate acid chlorides with NH3 gas to give the ester amide 20.
Preparation of the cerulenin analogues
In addition, the ester alcohol 25 was readily prepared by esterification of the known meso-epoxydiol 24 prepared by m-CPBA epoxidation of cis-butene-1,4-diol. (Scheme 5).109 Ester acid 26 was prepared by oxidation. of the epoxy alcohol 25 with RuCl3 to the acid and subsequent amide formation using gaseous ammonia to afford 27. We first tried to make amide acid by reacting anhydride 18 directly with octylamine to prepare diamide, but this did not work out. Next, we prepared acid amide to react with octylamine to make diamide, but the acid amide failed to react with octylamine.
Due to the difficulties encountered with the synthesis of structure 30, we tried an alternative route to prepare 30 which is discussed below. The third series of target molecules were carboxamides that have thioester and sulfone on the opposite side. Ring opening of the anhydride 18 with thiol and DMAP in acetonitrile/pyridine solution successfully affords the acid thioester 31 in moderate yield.
Copper-catalyzed conjugate addition of thiol to methyl propiolate 33, which proceeds through an allenolate intermediate, gave a mixture of stereoselective isomers 34 and 35 in 42% and 20% yields (Z/E Oxidation of the Z-vinyl sulfide 34 with excess sodium sulfonate gave sulfonate 36 in moderate yield Stereoselective oxidation to the epoxide 37 was carried out following the procedure described by Hegedus et al.chloride and DMF as a catalyst afforded ester-amide 42.
Structure activity relationship
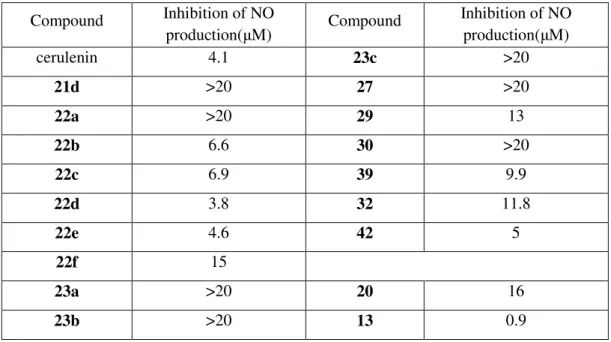
In general, we cannot ensure which analog is the strongest in inhibiting NO production, but most of the epoxycarboxamide analogs we synthesized successfully inhibit the inflammatory process. Interestingly, from the NO production results of some analogs, it seems likely that the open straight chain form of the cerulenin analogs can be considered as active conformation when binding with cysteine residues of the protein, which was an undisclosed issue. Inhibition of NO production in RAW 264.7 cells by cerulenin and its analogs Compound inhibition of NO.
The results obtained with the MTS assay (Figure 2) show that all the synthesized aliphatic analogues 20-42 have similar cytotoxicity with cerulenin. Furthermore, the cytotoxicity of the analogues appears to be independent of chain length and other functionalities around the epoxide moiety. Because cerulenin and certain of its analogs are known to inhibit FASN, the inhibitory activity of the synthesized compounds against FASN has been determined.
However, cerulenin analogs showing satisfactory inhibition of NO production had no significant inhibitory effects on fatty acid synthesis. Likewise, these compounds are essentially inactive against fatty acid synthase but inhibit the inflammatory response, meaning that the target for the inhibitory activities of cerulenin analogs is clearly distinct from that of cerulenin. Therefore, the analogs we synthesized are more closely related to the inhibition of other proteins associated with inflammation than to the suppression of fatty acid synthesis.
Conclusion
Experimental
After completion of the reaction, the reaction mixture was quenched with ammonium chloride (2 mL) and the aqueous layer was extracted with ethyl acetate (2 x 10 mL). The reaction mixture was stirred at room temperature until the reaction was complete as monitored by TLC. After completion of the reaction monitored by TLC, the reaction mixture was cooled to 0 °C and H2O (50 µL) and 15% NaOH solution (50 µL) were added slowly.
After completion of the reaction, monitored by TLC, the reaction mixture was filtered through a pad of celite and washed with dichloromethane and concentrated to dryness. After completion of the reaction, monitored by TLC, the reaction mixture was concentrated by evaporation under reduced pressure. After completion, the reaction mixture was cooled to room temperature and washed with 1 M hydrochloric acid solution (10 mL) and the aqueous phase was re-extracted with ethyl acetate (2x15 mL).
The reaction mixture was stirred at room temperature for 3 h, then 2 M hydrochloric acid (5 mL) was added. The mixture was extracted ethyl acetate (2 x 20 mL), dried over anhydrous Na 2 SO 4 , filtered, concentrated under reduced pressure. After slowly adding triethylamine (0.21 mL, 1.5 mmol) to the mixture, the mixture was stirred at room temperature for 1 hour.
A catalytic amount of DMF (2.3 µL, 0.03 mmol) was added and the mixture was stirred at room temperature for 1 h. The reaction mixture was evaporated and diluted with EtOAc (10 mL), washed with brine (2 x 5 mL), dried over anhydrous MgSO 4 , filtered and concentrated in vacuo.
Fishman-Furman, S.; Farbstein, M.; Bar Yehuda, B.; Fishman, P., Clinical evidence for the use of the A3 adenosine receptor as a target for the treatment of rheumatoid arthritis: data from a phase II clinical trial. A.; Fernández-Martíneź, A.; Castrillo, A.; Bosca, L.; Martı́n-Sanz, P., Selective inhibitors of cyclooxygenase-2 delay the activation of nuclear factor KB and attenuate the expression of inflammatory genes in mouse macrophages treated with lipopolysaccharide. Pahan, K.; Schmid, M., Activation of nuclear factor-kB in the spinal cord of experimental allergic encephalomyelitis.
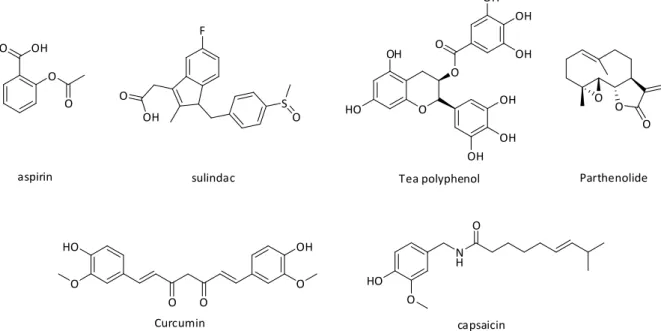
Ghosh, S., Inhibition of NF-kappa B by sodium salicylate and aspirin. B., Sulindac inhibits the activation of the NF-κB pathway. A.; Cancelas, P.; Gómez-Gerique, J.; Millan, J.; Egido, J., Red wine intake prevents activation of nuclear factor-κB in peripheral blood mononuclear cells of healthy volunteers during postprandial lipemia. De Bosscher, K.; Vanden Berghe, W.; Haegeman, G., Crosstalk between nuclear receptors and nuclear factor [kappa]B. S., Role of transcriptional activation of IκBα in mediation of immunosuppression by glucocorticoids.
In Methods in Enzymology, Academic Press: 1999; Vol. J., Radiation-induced activation of nuclear factor-κB involves selective degradation of plasma membrane-bound IκBα. L., Lactacystin, Proteasome Function and Cell Fate. Sawa, Y.; Kaneda, Y.; Higaki, J.; Ogihara, T., In vivo transfection of the cis element [ldquo]decoy[rdquo]. Anti-nuclear factor-[kappa]B binding site prevents myocardial infarction. Proceedings of the National Academy of Sciences of the United States of America b) Hiltunen, M.; Söderhäll, K., Inhibition of polyketide synthesis in Alternaria alternata by the fatty acid synthesis inhibitor cerulenin.