저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게
l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다:
l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다.
l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다.
저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다.
Disclaimer
저작자표시. 귀하는 원저작자를 표시하여야 합니다.
비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다.
변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
Doctoral Thesis
TonEBP in DNA repair and the pathogenesis of rheumatoid arthritis
Byeong Jin Ye
Department of Biological Sciences
Ulsan National Institute of Science and Technology
2021
Byeong Jin Ye
Department of Biological Sciences
Ulsan National Institute of Science and Technology
TonEBP in DNA repair and the pathogenesis of
rheumatoid arthritis
1
Overall Abstract
Tonicity-responsive enhancer binding protein (TonEBP), also called NFAT5, was initially identified as a stress protein responding to hypertonicity. Recent studies have revealed that TonEBP might be a more general stress protein responding to a variety of stresses including genotoxic and autoimmune stresses. In this thesis, I have explored the role of TonEBP in response to genotoxic and autoimmune stress.
TonEBP is recruited to DNA damage sites where it mediates a variety of repair processes. A major source of DNA damage is the accumulation of R-loops, which consist of a transcriptionally generated RNA-DNA hybrid and the displaced single-stranded DNA. A recent study has shown that TonEBP is specifically recruited to R-loops where it initiates a signaling pathway for resolution of R-loops. In this study, I found that TonEBP was associated with poly (ADP-ribose) polymerase 1 (PARP1). TonEBP was poly ADP-ribosylated (parylated) in response to DNA damages, and the parylation was required for the recruitment of TonEBP to R-loops. Pharmacological inhibition of PARP1 resulted in the elevated levels of R-loops and DNA damages in cancer cells. Thus, TonEBP contributes to resistance to DNA damage by inhibiting R-loop accumulation in oncogenic cells.
Rheumatoid arthritis (RA) is a chronic and systemic inflammatory autoimmune disease accompanied by inflammation of the synovium of the peripheral joint, cartilage destruction, bone erosion, and a loss of function. Although complex interactions of genetic and environmental factors are recognized, the exact cause of the disease is unknown. Patients with RA require lifelong treatment. Even with effective drug treatment, more than 60% of patients develop drug resistance and side effects within two years. Therefore, it is necessary to develop therapeutic agents targeting newly discovered pathological pathways.
Almost complete suppression of RA has been demonstrated in TonEBP haplo-deficient mice demonstrating critical role of TonEBP. Here, I used myeloid-specific TonEBP deficient mice to investigate the TonEBP’s role in myeloid cells including macrophages and dendritic cells (DCs).
2
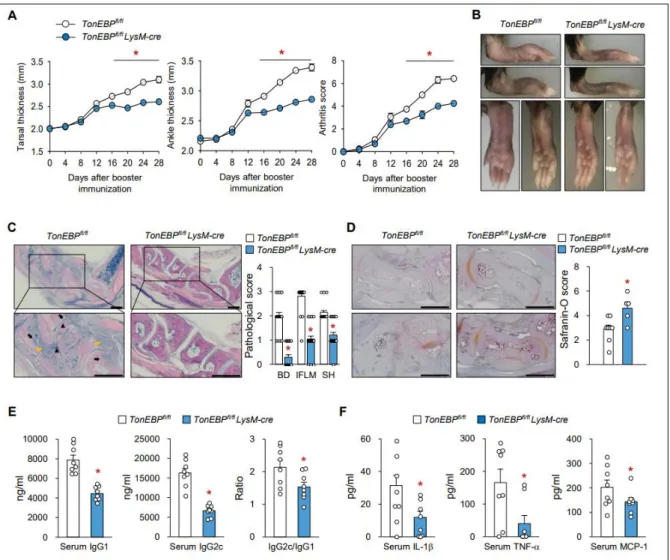
Myeloid cell-specific TonEBP deletion reduced disease severity in a mouse model of RA - collagen- induced arthritis - in association with reduced incidence of bone destruction, infiltration of inflammatory cell, and cartilage damage. It has been reported that TonEBP was the rate-limiting component of the pro-inflammatory enhanceosome which drives inflammatory genes including TNF- α, IL-1β, IL-6, iNOS, and COX2. Here, I found that TonEBP was essential in the maturation of DCs as it was required for the surface expression of MHC II and costimulatory molecules leading to pathological differentiation of Th1 and Th17 cells from naïve CD4 T cells. Thus, TonEBP mediates the autoimmune inflammatory pathways in macrophages and DCs.
In order to develop therapeutic agents for RA, I investigated cerulenin which disrupts the assembly of the pro-inflammatory enhanceosome which consists of TonEBP, NF-κB, AP-1, and p300. I found that cerulenin exhibited clear therapeutic efficacy in mouse models of inflammatory arthritis. Using an alkynyl derivative of a cerulenin and click reaction, I found that cerulenin was covalently attached to TonEBP by alkylation but not to other components of the enhanceosome. TonEBP displayed elevated thermal stability by cerulenin, demonstrating that cerulenin alkylation altered the structure of TonEBP.
These data suggest that cerulenin disrupts the pro-inflammatory enhanceosome by covalent modification of TonEBP.
In summary, I have found that TonEBP induced by genotoxic and autoimmune stress contributes to oncogenesis and RA, respectively. Cerulenin inhibits inflammatory arthritis by targeting TonEBP.
Cerulenin might prove to be a useful therapeutic agent for inflammatory diseases.
3
Contents
Overall Abstract --- 1
Contents --- 3
List of figures --- 5
Abbreviations --- 7
Chapter 1. Background --- 9
1-1. Rheumatoid arthritis --- 9
1-2. Treatment of Rheumatoid arthritis --- 10
1-3. Dendritic cell in rheumatoid arthritis --- 11
1-4. Tonicity -responsive enhancer binding protein (TonEBP) --- 12
1-5. References --- 16
Chapter 2. PARP1-mediated PARylation of TonEBP prevents R-loop–associated DNA damage 2-1. Abstract --- 20
2-2. Introduction --- 21
2-3. Materials and methods --- 23
2-4. Results --- 27
2-5. Discussion --- 37
2-6. Supplementary information --- 40
2-7. References --- 41
4
Chapter 3. TonEBP in dendritic cells mediates proinflammatory maturation and Th1/Th17 responses
3-1. Abstract --- 45
3-2. Introduction --- 46
3-3. Materials and methods --- 48
3-4. Results --- 53
3-5. Discussion --- 65
3-6. Supplementary information --- 68
3-7. References --- 76
Chapter 4. Cerulenin as a novel anti-inflammatory agent targets TonEBP for treatment of chronic inflammatory diseases 4-1. Abstract --- 82
4-2. Introduction --- 83
4-3. Materials and methods --- 84
4-4. Results --- 88
4-5. Discussion --- 98
4-6. Supplementary information --- 100
4-7. References --- 102
Conclusion --- 105
Acknowledgement --- 107
5
List of Figure
Chapter 1. Background
Figure 1.1. Effects of RA on the body
Figure 1.2. Clinical features of RA and major targets for treatment of RA Figure 1.3. Dendritic cells play crucial roles in RA
Figure 1.4. TonEBP in chronic inflammatory diseases
Figure 1.5. TonEBP is a rate-limiting component of pro-inflammatory enhanceosome Figure 1.6. TonEBP as an immunometabolic stress protein
Chapter 2. PARP1-mediated PARylation of TonEBP prevents R-loop–associated DNA damage Figure 2.1. TonEBP interacts with PARP1 through the Rel-homology domain
Figure 2.2. PARP1 mediates PARylation of TonEBP in response to camptothecin
Figure 2.3. PARylation of TonEBP mediated by PARP1 is required for recruitment of TonEBP to sites of R-loop-associated DNA damage
Figure 2.4. PARP1-mediated PARylation of TonEBP prevents R-loop–associated DNA damage Figure 2.5. RHD of TonEBP is required for prevention of R-loop–associated DNA damage and cell
survival in response to CPT
Figure 2.6. TonEBP prevents R-loop–mediated DNA damage in HepG2 human hepatoma cells Figure S2.1. TonEBP interactomes, related to R-loop–associated DNA damage
Chapter 3. TonEBP in dendritic cells mediates proinflammatory maturation and Th1/Th17 responses
Figure 3.1. Myeloid TonEBP deficiency reduces the severity of collagen-induced arthritis
Figure 3.2. Myeloid TonEBP deficiency reduces immune responses in the paw tissues of CIA mice Figure 3.3. Myeloid TonEBP deficiency inhibits T-cell differentiation and maturation of DCs in CIA
mice
Figure 3.4. TonEBP is required for maturation and inflammatory responses of BMDCs in response to LPS
Figure 3.5. TonEBP in DCs promotes proliferation of CD4+ T-cells and subsequent differentiation into Th1/Th17 cells
Figure 3.6. TonEBP in DCs controls LPS-mediated DC maturation via p38 MAPK
Figure 3.7. TonEBP expression by DCs promotes maturation and contributes to development of rheumatoid arthritis
Figure S3.1. Myeloid TonEBP deficiency reduces the severity of arthritis.
6
Figure S3.2. Increased immune responses in paw tissue of CIA mice
Figure S3.3. Flow cytometry gating strategies and the expression of DC migration-related genes in inguinal lymph nodes of CIA mice
Figure S3.4. Generation and characterization of BMDCs Figure S3.5. Expression of DC maturation markers in BMDCs
Figure S3.6. Expression of MHC II-related genes in BMDCs and BMDMs Table S3.1. Primers used for real time PCR
Chapter 4. Cerulenin as a novel anti-inflammatory agent targets TonEBP for treatment of chronic inflammatory diseases
Figure 4.1. Cerulenin reduces the severity of ajduvant-induced arthritis
Figure 4.2. Cerulenin inhibits RANKL-induced osteoclast differentiation and MMPs production in BMDMs
Figure 4.3. Clickable probe of cerulenin to identify target molecule Figure 4.4. Cerulenin or click probe bind to TonEBP
Figure 4.5. TonEBP is a specific molecular target of cerulenin in pro-inflammatory enhanceosome Figure 4.6. Cerulenin is colocalized with TonEBP
Figure 4.7. Anti-inflammatory effects of cerulenin was mediated by TonEBP
Figure 4.8. Cellular thermal shift assay (CETSA) to evaluate cerulenin target interactions Figure 4.9. Cerulenin breaks up assembly of pro-inflammatory enhanceosome specifically by
targeting the TonEBP
Figure S4.1. Local injected cerulenin or oral administrated cerulenin also reduced paw thickness and inflammation
Figure S4.2. Anti-inflammatory effects of cerulenin is not associated with fatty acid synthase (FASN)
7
Abbreviations
anti-TNFα : Anti-tumor necrosis factor antagonist APCs : Antigen-presenting cells
AIA : Adjuvant-induced arthritis ATM : Ataxia-telangiectasia mutant
bDMARDs : biologic disease modifying anti-rheumatic drugs BMDCs : Bone marrow-derived-dendritic cells
BMDMs : Bone marrow-derived macrophages CatD : Catalytic domain
ChIP : Chromatin immunoprecipitation CPT : Camptothecin
CIA : Collagen-induced arthritis CII : Collagen type II
CIITA : Coactivator class II transactivator co-IP : Co-immunoprecipitation
DCs : Dendritic cells
DMARDs : Disease modifying anti-rheumatic drugs DMEM : Dulbecco’s modified Eagle’s medium EdU : 5-ethynyl-2′-deoxyuridine
FAM : Azide conjugated fluorescein FASN : Fatty acid synthase
FBS : Fetal bovine serum
FLS : Fibroblast-like synoviocytes
GM-CSF : Granulocyte/macrophage colony-stimulating factor HCC : Hepatocellular carcinoma
MAPK : Mitogen-activated protein kinase
8 MKO : Myeloid-specific TonEBP knockout
MHC II : Major histocompatibility complex class II MMP : Matrix metalloproteinase
MTX : Methotraxate
NFAT5 : Nuclear factor of activated T cells
NFκB : nuclear factor kappa-light-chain-enhancer of activated B cells PARP1 : Poly (ADP-ribose) polymerase 1
PAR : Poly(ADP-ribose) PBS : Phosphate-buffered saline
PCNA : proliferating cell nuclear antigen PLCγ1 : Phospholipase Cγ1
RANKL : Receptor activator of nuclear factor κB ligand RHD : Rel-homology domain
RA : Rheumatoid arthritis RF : Rheumatoid factor
SHPRH : SNF2 histone linker phd ring Helicase
TonEBP : Tonicity-responsive enhancer binding protein TLR4 : Toll-like receptor 4
TRAP : Tartrate-resistant acid phosphatase UV : Ultraviolet
WT : Wildtype YY1 : Yin and yang 1
9
Chapter 1. Background
1-1. Rheumatoid arthritis (RA)
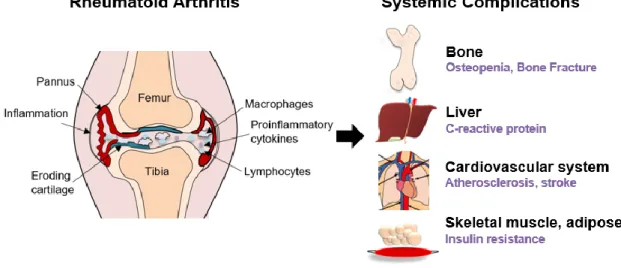
Rheumatoid arthritis (RA) is a chronic inflammatory disease that starts with synovial inflammation of the peripheral joints, destroys cartilage and bone, deforms joint structure, and causes loss of function (1). RA is a systemic autoimmune disease, accompanied by autoantibodies such as rheumatoid factor (RF) or anti-cyclic citrullined protein antibody (anti-CCP Ab), caused by a combination of genetic and environmental factors (2). RA invades not only the joints but also other tissues and organs, which can lead to complications like osteoporosis, cardiovascular disease, and metabolic disease. Patients with RA carry a higher mortality rate than the general population (Figure 1-1) (3).
Figure 1-1. Effects of RA on the body.
Rheumatoid arthritis is a representative chronic disease that affects approximately 1% of the world's population. As society ages, the number of patients with rheumatoid arthritis is expected to continue to increase (4). The prevalence of rheumatoid arthritis shows large regional deviation. Therefore, genetic and environmental factors seem to have a significant influence (5). RA patients show various clinical symptoms with varied response to drugs and a high recurrence rate. Immunity, the function of blocking invading bacteria, is supported by lymphocytes (6). The interaction of genetic and environmental factors
10
promotes loss of tolerance to self-proteins results in a cascade of autoimmune reactions (7). When immune cells misrecognize a part of our body as bacteria, autoimmune diseases occur. Autoimmune reactions increase expression of inflammatory cytokines which cause chronic inflammation (Figure 1- 2) (8).
Figure 1-2. Clinical features of RA and major targets for treatment of RA
Resulting in proliferation of abnormal layer of fibrovascular tissue and inflammation, Pannus formation is promoted. Pannus invades cartilage and bone surface and lymphocytes attack the synovial membrane, leading to joint destruction (9).
1-2. Treatment of Rheumatoid arthritis
RA treatments include non-steroidal anti-inflammatory drugs, steroids, immunosuppressive drugs (disease modifying anti-rheumatic drugs: DMARDs) and biologic drugs (biologic DMARDs, bDMARDs) that are prescribed when symptoms persist or worsen even after use of existing DMARDs (10). Treatment guidelines of RA are well established. Among the existing DMARDs, methotraxate (MTX) is widely used as the basis for rheumatoid arthritis treatment (11). Patients with insufficient therapeutic response to MTX are usually changed or added to other existing DMARDs or use biological
11
agents. Anti-tumor necrosis factor antagonist (anti-TNFα) show excellent therapeutic effects in patients who do not respond to existing anti-rheumatic drugs or have insufficient therapeutic effects (12).
Existing rheumatoid arthritis treatments have limitations, however. First, more than 30% of patients with rheumatoid arthritis have adverse reactions to the drug from the beginning, so the biggest problem is the drug-non-reactive patient group who cannot change the drug or even use the replacement drug.
Second, biological agents are expensive, inconvenient, and have various side effects such as infection problems and resistance. Third, 60% of patients develop drug resistance and side effects within 1-2 years (13). Therefore, development of new target-based treatments that is effective for drug-non- reactive patient is needed (14).
1-3. Dendritic cell in rheumatoid arthritis
Dendritic cells (DCs), professional antigen-presenting cells (APCs) that link the innate and adaptive immune responses, play critical roles in initiation and progression of rheumatoid arthritis (RA) (15).
Accordingly, DCs which is APCs, mediate differentiation of pro-inflammatory Th1 and Th17 cells.
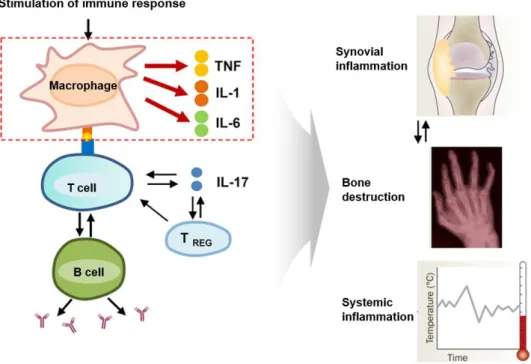
DCs are generated from blood mononuclear cells and localized in lymphoid and non-lymphoid tissues (16). Upon exposure to antigen, immature DCs mature and migrate to lymphoid tissues, where they up- regulate surface MHC expression, costimulatory molecules, and expression of inflammatory cytokines, down-regulate their antigen uptake capacity, and increase their T cell stimulatory capacity. DCs present processed antigens to naïve T cells, and induce immunity or tolerance (17). Maturation of DCs activates naïve T cells by presentation of antigenic peptide on MHC molecules, co-stimulatory molecules (CD80/86) and immunomodulatory cytokines. The IL-12 from DCs induce the differentiation of Th1 cells from naïve T cells. Th1 cells produce IFN γ and IL-2, which induce inflammation. Th1 cells activate B cells to differentiate into plasma cells. The pro-inflammatory cytokines, such as IL-6, IL-1, and IL-23, induce the differentiation of Th17 cells from naïve T cells. Th17 cells produce IL-17, IL21, and IL22, the pro-inflammatory cytokine that drives joint damage and inflammation. Dendritic cells play critical roles in rheumatoid arthritis by activating Th1, Th17 and B cells in response to autoantigens (Figure 1-3) (18).
12 Figure 1-3. Dendritic cells play crucial roles in RA
DC numbers dramatically increase in the synovial joint tissues of RA patients compared to normal synovium. Highly expressed inflammatory cytokines, such as IL-6, IL-1, IL-8, IL-10, and TNFα in the rheumatoid synovium, are associated with the migration of dendritic cells (19). DCs are important in the development of RA and are an attractive therapeutic target. Aspirin inhibits maturation of DCs.
Aspirin-treated DC captured antigen well, but expressed lower levels of Class II MHC and co- stimulatory molecules and were weak T cell stimulators (20). B cells, type 1 and type 17 T helper cells are regulated by DC are also considered additional targets for immune-based therapy.
1-4. Tonicity-responsive enhancer binding protein (TonEBP)
Tonicity-responsive enhancer binding protein (TonEBP) was originally discovered as a mediator of transcription promoted by hypertonicity (21). The Rel homology domain (RHD) of TonEBP forms a homodimer and binds to DNA; dimerization is required for DNA binding (22). The RHD is shared by other Rel proteins, such as NFκB proteins and NFAT1–NFAT4. Because the crystal structure of the
13
DNA binding domain is surprisingly similar to NFκB (23-24), TonEBP is considered to belong to the Rel family alongside NFAT and NF-κB.
TonEBP has been reported to interact with multiple proteins, such as protein kinases ABL and ATM, phospholipase Cγ1 (PLCγ1), the p65 subunit of NFκB, the DNA-binding transcription factor yin and yang 1 (YY1), the histone acetyltransferase p300, the E3 ubiquitin ligase SHPRH, the deubiquitinase USP1, RNA helicase A and Poly (ADP-ribose) polymerase 1 (PARP1) (25-27). 5 functions to regulate PCNA polyubiquitination in response to DNA damage inhibiting DNA replication (25). TonEBP is induced by DNA-damaging agents. Interestingly, TonEBP forms foci at sites of DNA damage with bulky adducts. In addition, according to our interactomic data, TonEBP associates with proteins that resolve R-loop structures, such as RNA helicases (DHX9) and inhibitors of R-loop accumulation (PARP1, TOP1) (25, 28).
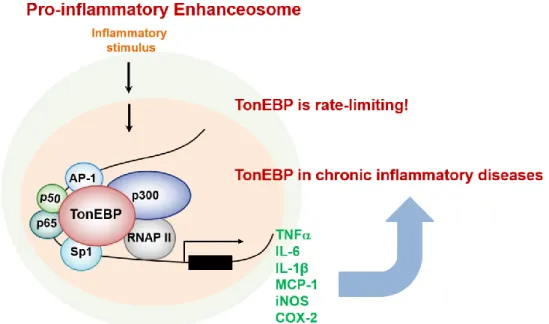
Figure 1-4. TonEBP in chronic inflammatory diseases
14
Recent studies show TonEBP is critically involved in the initiation and progression of chronic inflammatory diseases such as rheumatoid arthritis, atherosclerosis, diabetic nephropathy, acute kidney injury, hyperlipidemia and insulin resistance, autoimmune diseases (including type 1 diabetes mellitus and multiple sclerosis), salt-sensitive hypertension, and hepatocellular carcinoma (Figure. 1-4) (29-32).
Diverse pathological conditions and agents stimulate the activity of TonEBP. These signals increase the levels of TonEBP protein together with an increase in TonEBP mRNA abundance. Stimulation of TonEBP by inflammatory stimuli affects macrophage and microglia polarization and hepatocyte and podocytes activation (26, 29).
As TonEBP was found to be an important modulator of NFκB (26), the effect of TonEBP on inflammation was investigated. When inflammation begins, the expression and activity of TonEBP increases. This means that TonEBP is activated by inflammatory signals like NFκB. Even if the activity of TonEBP is reduced by half, the progression of inflammatory disease is sharply reduced (33).
Therefore, TonEBP is considered to be an attractive target for anti-inflammatory agents (Figure. 1-5).
Figure 1-5. TonEBP is a rate-limiting component of pro-inflammatory enhanceosome
TonEBP is highly expressed in the synovium and synovial macrophages of patients with rheumatoid arthritis (33). Pro-inflammatory cytokines contribute to the autoimmune stress that leads to increased TonEBP production. The anti-type II collagen antibody (CAIA) model in mice is completely suppressed in TonEBP-haplodeficient mice, demonstrating the complete dependence of this inflammatory response
15
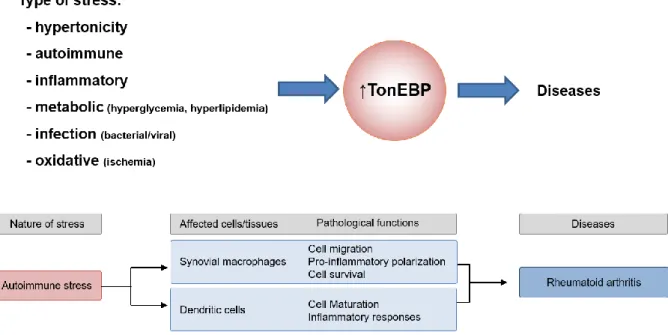
on TonEBP function (33). This surprising effect due to the slight reduction in TonEBP expression suggests multiple functions of TonEBP, including maturation of dendritic cells, activation of cell migration in fibroblast-like synoviocytes, and macrophages activation of the NFκB enhanceosome that drives pro-inflammatory gene expression. TonEBP expression in macrophages contributes to chronic, unrelenting arthritis by making apoptotic resistance to activated macrophages. Synovial fluid of patients with rheumatoid arthritis includes pro-inflammatory cytokines (1). Assembly of pro-inflammatory enhanceosome is related to chronic inflammation in the synovial membrane of patients with chronic arthritis (26). TonEBP is involved in controlling the differentiation and activity of different subsets of CD4+ T cells, thereby contributing to autoimmunity. TonEBP is a pleiotropic stress protein involved in the cellular response to hypertonicity, autoimmune reactions, inflammation, metabolic and genotoxic stress (34). Autoimmune stress and inflammatory stress both contribute to the high expression of TonEBP in rheumatoid arthritis. TonEBP contributes to unrelenting rheumatoid arthritis by cell migration, pro-inflammatory polarization, cell survival in synovial macrophages and cell maturation and inflammatory responses in dendritic cells (Figure. 1-6).
Figure 1-6. TonEBP as an immunometabolic stress protein
16 1-6. Reference
1. Gibofsky, A. Overview of epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis.
Am. J. Manag. Care 18, S295–S302. (2012).
2. Kang, E.H., Ha, Y.J., Lee, Y.J. Autoantibody Biomarkers in Rheumatic Diseases. Int J Mol Sci.
21, 1382. (2020)
3. Kim, J.W., Suh, C.H. Systemic Manifestations and Complications in Patients with Rheumatoid Arthritis. J Clin Med. 9, 2008. (2020)
4. Løppenthin, K., Esbensen, B.A., Østergaard M., Ibsen, R., Kjellberg, J., Jennum, Poul. Morbidity and mortality in patients with rheumatoid arthritis compared with an age- and sex-matched control population: A nationwide register study. J Comorb. 9, doi: 10.1177/2235042X19853484. (2019) 5. Edwards, C.J., Cooper, C. Early environmental factors and rheumatoid arthritis. Clin Exp Immunol.
143, 1-5. (2006)
6. Koyasu, S., Moro, K. Role of innate lymphocytes in infection and inflammation. Front. Immunol.
https://doi.org/10.3389/fimmu.2012.00101. (2012)
7. Vojdani, A. A Potential Link between Environmental Triggers and Autoimmunity. Autoimmune Dis.
2014, 437231. (2014)
8. Moudgil, K.D., Choubey, D. Cytokines in Autoimmunity: Role in Induction, Regulation, and Treatment. J Interferon Cytokine Res. 31, 695-703. (2011)
9. Ainola, M.M., Mandelin, J.A., Liljeström, M.P., Li, T.F., Hukkanen, M.V.J., Konttinen, Y.T. Pannus invasion and cartilage degradation in rheumatoid arthritis: involvement of MMP-3 and interleukin- 1beta. Clin Exp Rheumatol. 23, 644-650. (2005)
10. Singh, J.A., Saag, K.G., Bridges S.L.J, Akl, E.A., Bannuru, R.R., Sullivan, M.C., Vaysbrot, E., McNaughton, C., Osani, M., et al. 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care Res (Hoboken), 68, 1-25. (2016)
11. Braun, J. Methotrexate: Optimizing the Efficacy in Rheumatoid Arthritis. Ther Adv Musculoskelet Dis. 3, 151-158. (2011)
17
12. Radner. H., Aletaha, D. Anti-TNF in rheumatoid arthritis: an overview. Wien Med Wochenschr.
165, 3-9. (2015)
13. Pope, J., Combe, B. Unmet Needs in the Treatment of Rheumatoid Arthritis Journal of Rheumatology and Autoimmune Diseases. 3, 65-78. (2013)
14. Smolen, J. S., Aletaha, D., McInnes, I. B. Rheumatoid arthritis. Lancet 388, 2023–2038 (2016) 15. Ganguly, D., Haak, S., Sisirak, V. & Reizis, B. The role of dendritic cells in auto-immunity. Nat.
Rev. Immunol. 13, 566–577 (2013)
16. ) Shortman, K. & Naik, S. H. Steady-state and inflammatory dendritic-cell development. Nat. Rev.
Immunol. 7, 19–30 (2007)
17. Dalod, M., Chelbi, R., Malissen, B. & Lawrence, T. Dendritic cell maturation: functional specialization through signaling specificity and transcriptional programming. EMBO 33, 1104–
1116 (2014)
18. Khan, S., Greenberg, J.D., Bhardwaj, N. Dendritic cells as targets for therapy in rheumatoid arthritis. Nature Reviews Rheumatology. 5, 556-571. (2009)
19. Yu, M.B., Langridge, W.H.R. The function of myeloid dendritic cells in rheumatoid arthritis.
Rheumatol Int. 37, 1043-1051. (2017)
20. Hackstein, H., Morelli, A.E., Larregina, A.T., Ganster, R.W., Papworth, G.D., Logar, A.J., Watkins, S.C., Falo, L.D., Thomson, A.W. Aspirin inhibits in vitro maturation and in vivo immunostimulatory function of murine myeloid dendritic cells. J Immunol. 166, 7053-7062. (2001) 21. Miyakawa, H., Woo, S.K., Dahl, S.C., Handler, J.S., Kwon, H.M. Tonicity responsive enhancer binding protein, a rel-like protein that stimulates transcription in response to hypertonicity. Proc.
Natl Acad. Sci. USA 96, 2538–2542. (1999)
22. Lee, S.D., Woo, S.K., Kwon, H.M. Dimerization is required for phosphorylation and DNA binding of TonEBP/NFAT5. Biochem Biophys Res Commun. 28, 968-975. (2002)
23. Lopez-Rodriguez, C., Aramburu, J., Rakeman, A.S., Rao, A. NFAT5, a constitutively nuclear NFAT protein that does not cooperate with Fos and Jun. Proc. Natl Acad. Sci. USA 96, 7214–7219. (1999)
18
24. Stroud, J.C., Lopez-Rodriguez, C., Rao, A., Chen, L. Structure of a TonEBP-DNA complex reveals DNA encircled by a transcription factor. Nat Struct Biol. 9, 90-94. (2002)
25. Kang, H.J., Park, H., Yoo, E.J., Lee, J.H., Choi, S.Y., Kwon, W.L., Lee, K.Y., Hur, J.H. et al.
TonEBP regulates PCNA polyubiquitination in response to DNA damage through interaction with SHPRH and USP1. iScience. 19, 177–190. (2019)
26. Lee, H.H., Sanada, S., An, S.M., Ye, B.J., Lee, J.H., Seo, Y.K., Lee, C.W. et al. LPS-induced NF- κB enhanceosome requires TonEBP/NFAT5 without DNA binding. Sci. Rep. 6, 24921. (2016) 27. Irarrazabal, C. E., Gallazzini, M., Schnetz, M.P., Kunin, M., Simons, B.L, Williams, C.K, Burg,
M.B., Ferraris, J.D. Phospholipase C-γ1 is involved in signaling the activation by high NaCl of the osmoprotective transcription factor TonEBP/OREBP. Proc. Natl Acad. Sci. USA 107, 906–911.
(2010)
28. Kang, H.J., Cheon, N.Y., Park, H., Jeong, G.W., Ye, B.J., Yoo, E.J., Lee, J.H., Hur, J.H., Lee, E.A., Kim, H.T et al. TonEBP recognizes R-loops and initiates m6A RNA methylation for R-loop resolution. Nucleic Acids Res. 11, 269-284. (2021)
29. Lee, J.H., Suh, J.H., Choi, S.Y., Kang, H.J., Lee, H.H., Ye, B.J., Lee, G.R., Jung, S.W., Kim, C.J., Lee-Kwon, W., et al. Tonicity-responsive enhancer-binding protein promotes hepatocellular carcinogenesis, recurrence and metastasis. Gut. 68, 347–358. (2019)
30. Choi, S., You, S., Kim, D., Choi, S.Y., Kwon, H.M., Kim, H.S., Hwang, D., Park, Y.J., Cho, C.S., Kim, W.U. Transcription factor NFAT5 promotes macrophage survival in rheumatoid arthritis. J.
Clin. Investig. 127, 954–969. (2017)
31. Ye, B.J., Lee, H.H., Yoo, E.J., Lee, C.Y., Lee, J.H., Kang, H.J., Jung, G.W., Park, H., Lee-Kwon, W., Choi, S.Y., et al. TonEBP in dendritic cells mediates pro-inflammatory maturation and Th1/Th17 responses. Cell Death Dis. 11, 421. (2020)
32. Halterman, J.A., Kwon, H.M., Leitinger, N., Wamho, B.R. NFAT5 expression in bone marrow- derived cells enhances atherosclerosis and drives macrophage migration. Front. Physiol. 3, 313.
(2012)
19
33. Yoon, H. J., You, S.Y., Yoo, S.A., Kim, N.H., Kwon, H.M., Yoon, C.H., Cho, C.S., Hwang, D.H., Kim, W.U. NFAT5 is a critical regulator of inflammatory arthritis. Arthritis Rheum. 63, 1843–1852 (2011)
34. Choi, S.Y., Lee-Kwon, W.S., Kwon, H.M. The evolving role of TonEBP as an immunometabolic stress protein. Nat Rev Nephrol. 16, 352-364. (2020)
20
Chapter 2. PARP1-mediated PARylation of TonEBP prevents R-loop–associated DNA damage
2-1. Abstract
Lack of coordination between the DNA replication and transcription machineries can increase the frequency of transcription–replication conflicts, leading ultimately to DNA damage and genomic instability. A major source of these conflicts is the formation of R-loops, which consist of a transcriptionally generated RNA–DNA hybrid and the displaced single-stranded DNA. R-loops play important physiological roles and have been implicated in human diseases. Although these structures have been extensively studied, many aspects of R-loop biology and R-loop–mediated genome instability remain unclear. We found that in cancer cells, tonicity-responsive enhancer-binding protein (TonEBP, also called NFAT5) interacted with PARP1 and localized to R-loops in response to DNA- damaging agent camptothecin (CPT), which is associated with R-loop formation. PARP1-mediated PARylation was required for recruitment of TonEBP to the sites of R-loop–associated DNA damage.
Loss of TonEBP increased levels of R-loop accumulation and DNA damage, and promoted cell death in response to CPT. These findings suggest that TonEBP mediates resistance to CPT-induced cell death by preventing R-loop accumulation in cancer cells.
21 2-2. Introduction
Maintenance of genome integrity depends on spatiotemporal coordination between the DNA replication and transcription machineries. Lack of such coordination can lead to DNA damage and genomic instability resulting from an elevated frequency of transcription–replication conflicts (1, 2). A major source of these conflicts is the formation of R-loops, which consist of a co-transcriptionally generated RNA–DNA hybrid and the displaced single-stranded DNA (3). R-loops, which arise naturally during transcription in organisms from bacteria to mammals, play multiple roles in cellular processes, including regulation of gene expression (4), transcription termination (5), and DNA replication (6). In addition to these physiological functions, R-loops also threaten genome integrity, resulting in deleterious effects on the cell (7-9). Abnormal accumulation of R-loops and collisions with replication forks can increase the rate of DNA damage and genome instability (2, 10). R-loops have been implicated in many human diseases, including neurological disorders, cancer, and autoimmunity (11, 12). Accordingly, R-loops must be tightly regulated in living cells.
R-loop homeostasis is regulated by the factors and cellular processes that control the formation and resolution of these structures. In humans, proteins involved in chromatin modification and the DNA damage response play important roles in maintaining the proper R-loop balance (13-15), and aberrant expression or function of these factors can contribute to disease (16, 17). Moreover, R-loops can regulate gene expression by recruiting protein factors (13). However, the molecular mechanisms linking these factors to R-loop biology remain unclear, and we require a clearer understanding of their identities and the mechanisms by which they are recruited.
Tonicity-responsive enhancer-binding protein (TonEBP), also known as nuclear factor of activated T-cells 5 (NFAT5), is a pleiotropic stress protein involved in the response to various types of stress (18).
TonEBP can promote physiological or pathological consequences depending on the context: for example, TonEBP-mediated responses to osmotic stress (19-22) and bacterial infection (23) have protective functions, whereas the responses to autoimmune and metabolic stresses contribute to the pathogenesis of human diseases such as rheumatoid arthritis (24, 25), atherosclerosis (26), hepatocellular carcinoma (HCC) (27, 28), obesity (29), and diabetes mellitus (29, 30). Although
22
TonEBP has well-established functions in the responses to a range of cellular stresses, its role in the cellular response to DNA damage remains to be elucidated. To systematically explore the cellular functions of TonEBP, we analyzed proteins that interacted with TonEBP using tandem affinity purification-mass spectrometry. The results of that study revealed that TonEBP interacts with DNA repair proteins; in addition, TonEBP is recruited to DNA damage sites and protects cells from genotoxic stress (31). TonEBP mediates DNA damage tolerance through ubiquitination of proliferating cell nuclear antigen (PCNA), a DNA clamp required for replication and repair. Furthermore, TonEBP is involved in the dynamic association of DNA repair proteins and their recruitment to DNA damage sites.
Our interactomic data also revealed that TonEBP associates with proteins that resolve R-loop structures, such as RNA helicases and inhibitors of R-loop accumulation (31, Supplementary Figure 2-1), raising the possibility that it plays a role in R-loop biology.
In this study, we explored the role of TonEBP in R-loop–associated DNA damage. We found that in cancer cells, TonEBP interacted with poly (ADP-ribose) polymerase 1 (PARP1), localized to R-loops, and prevented R-loop accumulation and DNA damage in response to an R-loop–associated DNA- damaging agent. Together, these findings suggest that TonEBP mediates resistance to DNA damage by preventing R-loop accumulation in oncogenic cells. These findings provide new insights into the molecular mechanisms underlying DNA damage and repair responses, and reveal novel functions of TonEBP in R-loop biology.
23 2-3. MATERIALS AND METHODS
Cells and reagents
HEK293T (ATCC CRL-3216), U2OS (ATCC HTB-96) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS; ThermoFisher Scientific, Waltham, MA, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (GE Healthcare, Salt Lake City, UT, USA). Hep G2 (ATCC HB-8065) cells were maintained in modified Eagle's medium (Hyclone, South Logan, UT, USA) supplemented with 10% FBS and penicillin–streptomycin. All cells were maintained at 37°C in an incubator in an atmosphere containing 5% CO2. Cells were transfected with the same concentrations of scrambled or gene-targeted siRNAs for 24 h using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA). All siRNA duplexes were purchased from Integrated DNA Technologies. All plasmids were purified using an endotoxin-free purification system (Qiagen, Hilden, North Rhine-Westphalia, Germany) and transfected into cells using Lipofectamine 2000 (Invitrogen) for 24 h. Transfected cells were then cultured in fresh complete medium and analyzed as indicated in the figure legends.
Immunofluorescence and EdU staining, and image analysis
Cells were plated on LabTek chamber slides (Thermo Fisher Scientific), cultured for 1 day, and then fixed with 100% methanol at 20°C for 30 min. For chromatin-bound proteins, cells were pretreated with 0.5% Triton X-100 for 2 min before fixation. For UV micro-irradiation, UVA laser (55 mW) irradiation was performed by means of a Palm MicroBeam laser microdissection workstation. The fixed cells were stained with the indicated primary antibodies overnight at 4°C. After washes with 0.05% Triton X-100, Alexa Fluor 488–, 568–, and 633–conjugated secondary antibodies (Invitrogen) were added and incubated for 1 h. The stained cells were mounted. EdU staining was performed by using Click-iT EdU assay kit (Invitrogen). Images were acquired on an Olympus FV1000 confocal fluorescence microscope and processed using the Olympus Fluoview or ImageJ software.
Immunoprecipitation
24
To prepare the total cell lysates, cells were washed three times with ice-cold PBS, and lysed and incubated with RIPA buffer on ice (37). The lysates were incubated with the indicated antibody overnight at 4°C under rotary agitation, followed by incubation with Protein A/G Sepharose beads (GE Healthcare Sciences) for 2 h. Bead–antibody–antigen complexes were pelleted by centrifugation at 4°C for 1 min, and the supernatant was removed. The beads were washed three times for 10 min at 4°C in RIPA buffer, resuspended in sample buffer, and boiled at 95°C for 5 min. The complexes were analyzed by immunoblotting.
Immunoblotting
Cells were washed twice with cold PBS and lysed in RIPA buffer (0.01 M Tris pH 7.4, 0.15 M NaCl, 0.001 M EDTA, 0.001 M EGTA, 1% Triton X 100; all from Sigma-Aldrich, Saint Louis, MO, USA) containing 2 M PMSF (Sigma-Aldrich) and protease inhibitors (Roche, Rotkreuz, Switzerland). After centrifugation of the lysate, the supernatant was used for immunoblot analysis. Protein concentration was measured using the BCA protein assay system (Pierce Biotechnology, Rockford, IL, USA). Proteins were denatured in Laemmli buffer. Equal amounts of protein from each sample were separated on SDS- PAGE gels and transferred to PVDF membranes. Blocking, incubation with primary antibody, and washing of the membrane were performed in PBS supplemented with 0.05% Tween-20 (v/v) and 5%
(w/v) non-fat dry milk. The primary antibodies used for immunoblotting were anti-TonEBP (16; 1:3000), anti-Myc-tag (#2778), anti-PAR (#83732) (all from Cell Signaling Technology, Danvers, MA, USA), anti-FLAG tag (#F1804, Sigma-Aldrich), anti-S9.6 (#ENH001, Kerafast, Boston, MA, USA), and anti- PARP1 (#ab227244, Abcam, Cambridge, UK). All antibodies were used at a 1:1000 dilution unless stated otherwise. Horseradish peroxidase-conjugated goat anti-mouse (62-6520; Thermo Fisher Scientific) or anti-rabbit (65-6120; Thermo Fisher Scientific) secondary antibodies were diluted 1:5000.
Reactive bands were detected by chemiluminescence on an ImageQuant LAS 4000 imaging system (GE Healthcare).
Proximity ligation assay
25
For the PLA, cells were pre-extracted with cold 0.5% NP-40 for 3 min on ice. The cells were then fixed with 4% PFA/PBS for 15 min, washed three times with PBS, and blocked for 1 h at RT with 2%
BSA/PBS. The fixed and blocked cells were incubated with primary antibody overnight at 4°C. The next day, the cells were washed three times in PBS and incubated in a pre-mixed solution of PLA probe anti-mouse minus and PLA probe anti-rabbit plus (Sigma-Aldrich) for 1 h at 37˚C. The Duolink In Situ Detection Reagents (Green) were used to perform the PLA reaction. Slides were mounted in Duolink In Situ Mounting Medium with DAPI and imaged on a Zeiss Axioscope at 40X. PLA foci were quantified using the ImageJ software.
Cell survival analysis
U2OS and HepG2 cells were plated in triplicate in 96-well plates and subjected to the MTT assay.
Absorbance at 490 nm was measured using a multi-well plate reader. LDH release was calculated as a percentage using the following formula: percentage = (sample–spontaneous release/maximum release − spontaneous release) × 100.
S9.6 IP
Cells were trypsinized, washed with PBS, and resuspended in 25 mL PBS. The cells were then crosslinked in 1% formaldehyde (Pierce Biotechnology, Rockford, IL, USA), quenched with 0.125 M glycine, and washed twice in PBS containing protease inhibitor (PI; Roche). The fixed cells were lysed with lysis buffer (50 mM HEPES pH 7.9, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100) with proteinase inhibitor cocktail, and chromatin was sonicated in shearing buffer (0.1%SDS, 1 mM EDTA, 10 mM Tris pH 8.1) on a ultrasonicator (Covaris, Woburn, MA, USA) to an average size of 1 kb. Washed Protein A/G Sepharose beads (Pierce Biotechnology) were used to pre- clear chromatin for 2 h. Ten micrograms of the chromatin fraction was mixed with either 20 μg S9.6 antibody or 20 μg mouse IgG, and incubated overnight at 4°C. Pre-washed Protein A/G Sepharose beads were then added to the chromatin/antibody mixture for 2 h. After washing three times with binding buffer (10 mM NaPO4 pH 7, 140 mM NaCl, 0.05% Triton X-100), bound beads were boiled in 30 μL
26
5X sample buffer (10% SDS, 500 mM DTT, 50% glycerol, 250 mM Tris-HCL and 0.5% bromophenol blue dye, pH 6.8) and loaded on a 4–20% gradient gel (Bio-Rad,Hercules, CA, USA).
Statistical analysis
Data are expressed as the mean ± standard deviation or standard error of the mean. The statistical significance of differences between two conditions was estimated using the unpaired t-test.
Comparisons between more than two conditions were evaluated by one-way ANOVA with Tukey’s post-hoc test. A p-value < 0.05 was deemed significant. All statistical analyses were performed using the GraphPad Prism 8.2 software (GraphPad, San Jose, CA, USA).
27 2-4. Results
TonEBP interacts with, and is PARylated by, PARP1
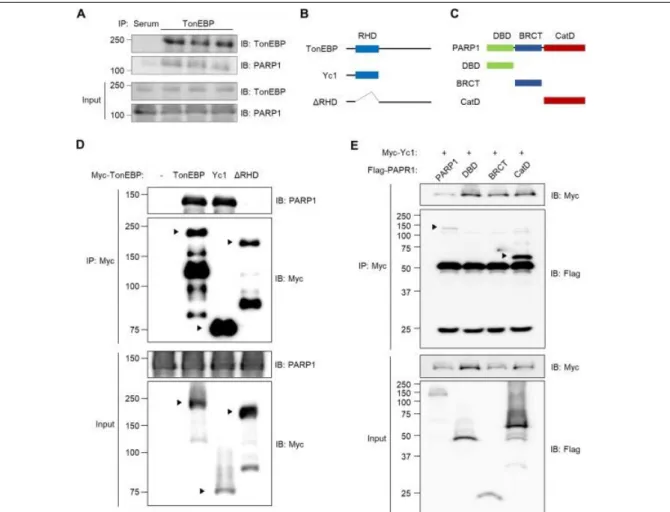
Our interactomic data from a previous study revealed that TonEBP interacts with proteins involved in R-loop resolution, including PARP1 and RNA helicases (31, Supplementary Figure 1). Given the pivotal role of PARP1 in resolving R-loop–associated DNA damage (13), we decided to investigate the interaction between TonEBP and PARP1, and its role in R-loop biology. To this end, we first sought to determine whether TonEBP physically binds to PARP1. Co-immunoprecipitation (co-IP) experiments in HEK293T cells revealed that endogenous TonEBP and PARP1 proteins interact (Figure 2-1A). Next, we defined the molecular basis of the TonEBP–PARP1 interaction using constructs expressing several recombinant variants of TonEBP (Figure 2-1B) and PARP1 (Figure 2-1C). Constructs containing the Rel-homology domain (RHD), including full-length TonEBP and Yc1 (N terminus of TonEBP), interacted with PARP1, whereas a mutant lacking the RHD (∆RHD) did not (Figure 2-1D), indicating that the RHD of TonEBP is essential for its interaction with PARP1. To determine which structural elements of PARP1 are required for the interaction with TonEBP, we transfected cells with constructs expressing Yc1 and full-length PARP1 or a deletion mutant. Full-length PARP1 and the catalytic domain (CatD) both bound to Yc1, whereas the DNA-binding domain and BRCA1 C-terminal domain did not (Figure 2-1E). These data indicate that the RHD of TonEBP and the CatD of PARP1 mediate the interaction between the two proteins.
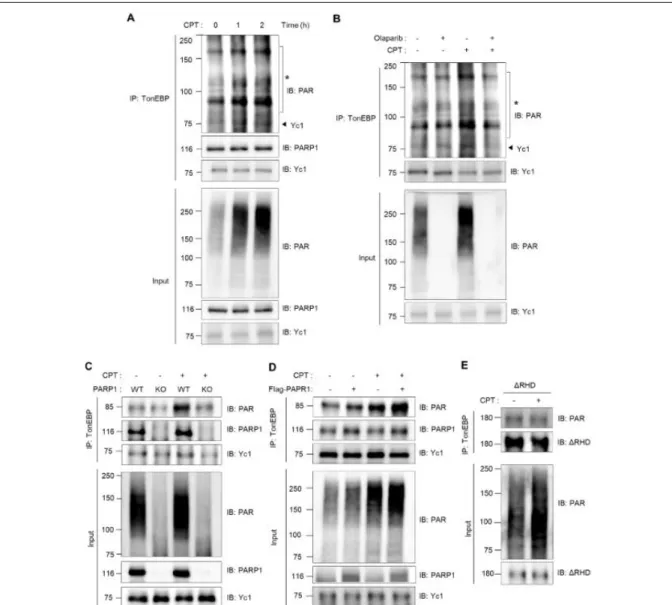
In response to DNA damage, PARP1 is activated and promotes the formation of poly (ADP-ribose) polymer on its substrates (32). In light of the observation that TonEBP interacts with PARP1, we asked whether TonEBP was poly-ADP-ribosylated (PARylated) by PARP1 in Yc1-overexpressing cells. For these studies, we used the U2OS human osteosarcoma cell line, which has a robust and well- characterized response to DNA damage, and camptothecin (CPT), which promotes R-loop–associated DNA damage. The content of PARylated protein increased in a time-dependent manner in response to CPT (Figure 2-2A, bottom panel). Notably, co-IP experiments revealed that CPT induced increased PARylation intensity and up-shifting smears of Yc1 (Figure 2-2A, top panel), and this effect was abrogated by the PARP1 inhibitor olaparib (Figure 2-2B). The interaction between TonEBP and PARP1
28
remained even after PARylation of TonEBP (Figure 2-2A, top panel). Next, we examined whether PARylation of TonEBP in response to CPT was mediated by the binding. PARylation of TonEBP was dramatically reduced in the PARP1 knockout cells (Figure 2-2C), whereas PARP1 overexpression induced a clear increase both in basal and CPT-induced PARylation of TonEBP (Figure 2-2D). In addition, the ∆RHD mutant was not PARylated (Figure 2-2E). Together, these data demonstrate that TonEBP interacts with PARP1, and that PARP1 mediates the PARylation of TonEBP in response to CPT.
Figure 2-1. TonEBP interacts with PARP1 through the Rel-homology domain. (A) HEK293T cells were immunoprecipitated (IP) with normal serum (Serum) or anti-TonEBP antibody (TonEBP).
Cell lysates (input) and precipitated proteins were immunoblotted for TonEBP and PARP1. (B) Domain structures of human TonEBP and deletion constructs Yc1 and RHD. (C) Domain structures of human PARP1 and deletion constructs containing the DNA-binding domain (DBD), BRCA1 C- terminal domain (BRCT), or catalytic domain (CatD). (D) U2OS cells were transfected with a plasmid expressing Myc-tagged TonEBP, Yc1, or ΔRHD for 24 h. Cell lysates were immunoprecipitated with anti-Myc antibody. Cell lysates and precipitated proteins were immunoblotted for PARP1 and Myc. (E) U2OS cells were transfected for 24 h with plasmids expressing Myc-Yc1 in combination with FLAG tagged PARP1, DBD, BRCT, or CatD. Cell lysates were immunoprecipitated with anti-Myc antibody. Cell lysates and precipitated proteins were immunoblotted for FLAG and Myc.
29
Figure 2-2. PARP1 mediates PARylation of TonEBP in response to camptothecin. (A) U2OS cells transfected with Yc1 were treated with 10 M camptothecin (CPT) for the indicated times (0 - 2 h). Proteins were immunoprecipitated (IP) with anti-TonEBP antibody (TonEBP). Precipitates and cell lysates were immunoblotted for PAR (poly (ADP-ribose)), PARP1 and TonEBP. *, PARylated proteins. (B) The cells were pretreated with 1 M olaparib for 1 h, followed by treatment with 10
M CPT for 2 h as indicated. Cell lysates were prepared and immunoprecipitated with anti-TonEBP antibody. Precipitates and cell lysates were immunoblotted for PAR and TonEBP. (C) PARP1 wild type (WT) or knockout (KO) HEK-293T cells transfected with Yc1 were treated with 10 M CPT for 2 h. Cells were then immunoprecipitated with anti-TonEBP antibody. Precipitates and cell lysates were immunoblotted for PAR, PARP1 and TonEBP. (D) U2OS cells transfected with Yc1 were transfected a second time with a plasmid expressing FLAG-empty (-) or FLAG-PARP1 (+) for 24 h.
The transfected cells were treated with 10 M CPT for 2 h and then immunoprecipitated with anti- TonEBP antibody. Precipitates and cell lysates were immunoblotted for PAR, PARP1 and TonEBP.
(E) U2OS cells were transfected with ΔRHD mutant of TonEBP and treated with 10 M CPT for 2 h. Cells were then immunoprecipitated with anti-TonEBP antibody. Precipitates and cell lysates were immunoblotted for PAR and TonEBP.
30
Figure 2-3. PARylation of TonEBP mediated by PARP1 is required for recruitment of TonEBP to sites of R-loop-associated DNA damage.
(A and B) U2OS cells transfected with scrambled (Scr) or PARP1-targeted siRNA (si-PARP1) were transfected again with GFP-Yc1 for 24 h. The cells were then subjected to laser micro-irradiation after pretreatment with vehicle (-) or 1 M olaparib for 30 min. (A) After irradiation, green fluorescence images were acquired at the indicated times (0 to 60 sec). (B) The fluorescence intensity in the micro-irradiated area at indicated time point was determined from 10 cells. Data (means ± SD) were obtained from three independent experiments (n = 3). * p < 0.01 compared to Scr; -. (C) Cells were transfected with siRNA and treated with olaparib, as in A and B. Cells were then treated with vehicle (-) or 10 M CPT, followed by immunostaining for TonEBP. (Left) Representative images.
(Right) Numbers of TonEBP foci (means ± SD) were determined from at least 50 cells in each
31
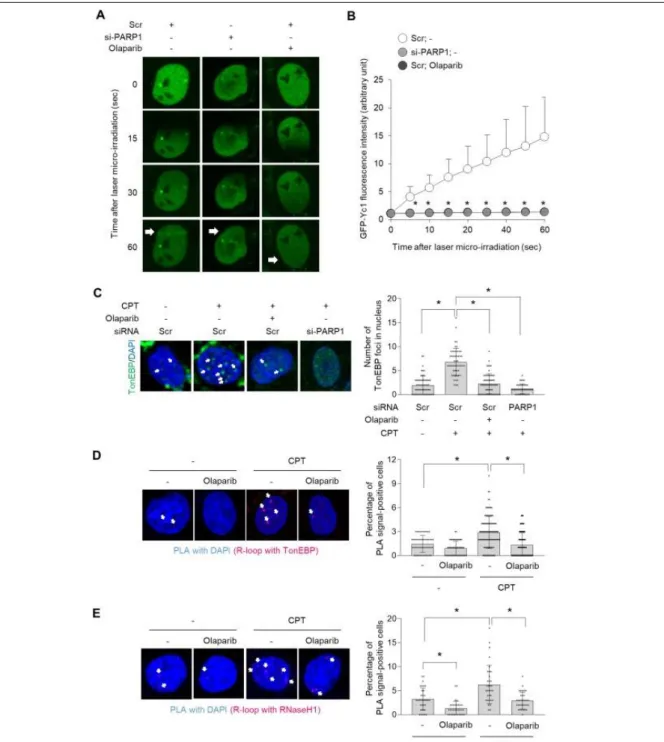
PARylation of TonEBP mediated by PARP1 triggers recruitment of TonEBP to sites of DNA damage PARylation of target proteins by PARP1 is one of the earliest steps in the cellular response to DNA lesions, leading to activation of DNA damage response pathways and facilitating DNA damage repair.
Indeed, many DNA repair factors are PARylated to access DNA damage sites (32). Our previous study provided evidence that TonEBP is recruited to DNA damage sites and promotes the DNA damage bypass pathway (31). Hence, we investigated whether PARP1 is required for recruitment of TonEBP to DNA damage sites. To this end, we performed laser micro-irradiation to induce double-strand breaks in DNA and then examined laser damage-induced recruitment of GFP-tagged Yc1 proteins. Yc1, which contains the RHD of TonEBP, was translocated to the micro-irradiated region of the nucleus, but this translocation was not observed in olaparib- or PARP1-siRNA–treated cells (Figure 2-3A and 2-B).
Furthermore, we examined DNA damage-induced foci formation of TonEBP in response to CPT and found that both pretreatment with olaparib and knockdown of PARP1 significantly inhibited the CPT- mediated formation of TonEBP foci (Figure 2-3C).
CPT-induced DNA Topoisomerase 1 cleavage complex formation is the major source of DNA damage, most likely through replication-induced DNA damage (33, 34) and R-loop is one of the sources of DNA damage following CPT treatment (35-37). Our recent data show that TonEBP is recruited to R- loops in response to CPT and mediates R-loop resolution via RNaseH1 recruitment (38). Thus, we asked whether PARylation of TonEBP is required for R-loop recognition. To answer this question, we performed a proximity ligation assay (PLA) with anti–DNA/RNA hybrid (S9.6) and anti-TonEBP antibodies, enabling us to monitor the interaction between R-loop–enriched foci and TonEBP. We observed PLA signals in the nucleus following CPT treatment but to a lesser extent in cells treated with olaparib (Figure 2-3D). These data indicate that PARP1-dependent PARylation of TonEBP was essential for its recruitment to sites of R-loop–associated DNA damage in response to CPT. Furthermore, olaparib significantly inhibited recruitment of RNase H1 to R-loops at both basal and CPT-treated conditions condition. * p<0.01. (D and E) Cells were pretreated with vehicle (-) or 1 M olaparib followed by treatment with vehicle (-) or 10 M CPT. The cells were then subjected to PLA between TonEBP and R-loops (D), or RNaseH1 and R-loops (E). (Left) Representative images. (Right) Percentages of PLA signal-positive nuclei were calculated from at least 30 cells. Data (means ± SD) are from three independent experiments (n = 3). * p < 0.01.
32
(Figure 2-3E), demonstrating that PARylation was required for recruitment of RNase H1 to sites of R- loops.
PARP1-mediated PARylation of TonEBP prevents R-loop accumulation
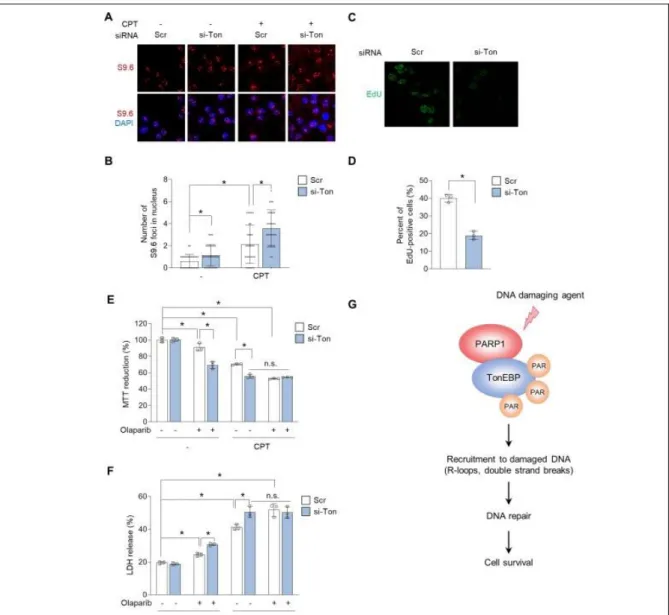
Next, we investigated the potential role of TonEBP and its PARylation in the R-loop response following CPT treatment using cells in which TonEBP had been knocked down with siRNA.
Immunocytochemistry experiments with S9.6 antibodies revealed that the global level of R-loops was Figure 2-4. PARP1-mediated PARylation of TonEBP prevents R-loop–associated DNA damage.
U2OS cells were transfected with scrambled siRNA (-) or TonEBP-targeting siRNA (si-Ton) for 24 h. The cells were then untreated (-) or pretreated (+) with 1 M olaparib for 1 h, followed by treatment with either 10 M CPT or vehicle alone (Veh) for 3 h. (A) R-loops and nuclei were visualized by immunostaining with S9.6 antibody and DAPI staining, respectively. (B) S9.6 foci per nucleus were counted from 30 cells (means ± SD). (C–F) Immunostaining was performed for γH2AX and 53BP1.
Representative images for γH2AX (C) and 53BP1 (E) were shown. Percentages of γH2AX (D) and 53BP1 (F) foci-positive cells were calculated from at least 30 cells. Data (means ± SD) are from three independent experiments (n = 3). * p<0.01, n.s.: not significant.
33
elevated in TonEBP-depleted cells irrespective of CPT treatment (Figure 4A and B). The R-loop signal was also elevated upon PARP inhibition with olaparib, as previously reported (13). However, PARP inhibition did not further increase the R-loop signal in TonEBP-depleted cells, again irrespective of CPT treatment (Figure 2-4A and B), suggesting that TonEBP and PARP1 act in the same pathway to suppress R-loops.
Excessive R-loop accumulation triggers DNA damage and genome instability (3, 7). Hence, we asked whether R-loop accumulation in TonEBP-depleted cells increases the rate of DNA damage. To answer this question, we analyzed H2AX and 53BP1 foci, which are markers of DNA damage. Consistent with the accumulation of R-loops (Figure 2-4A), TonEBP depleted cells had significantly increased levels of H2AX and 53BP1 foci, irrespective of CPT treatment (Figure 2-4C-F). Pretreatment with olaparib also significantly increased the intensities of γH2AX and 53BP1 (Figure 2-4C–F). By contrast, in TonEBP-depleted cells, olaparib did not further increase formation of γH2AX and 53BP1 foci, irrespective of CPT treatment, suggesting that TonEBP and PARP1 are epistatic for signaling or repair of DNA damage induced by CPT.
These data, together with our data in Figures 3 and 4, indicate that PARP1-mediated PARylation of TonEBP is required to prevent R-loop accumulation and DNA damage.
The RHD of TonEBP is required to prevent R-loop–associated DNA damage and cell survival in response to CPT
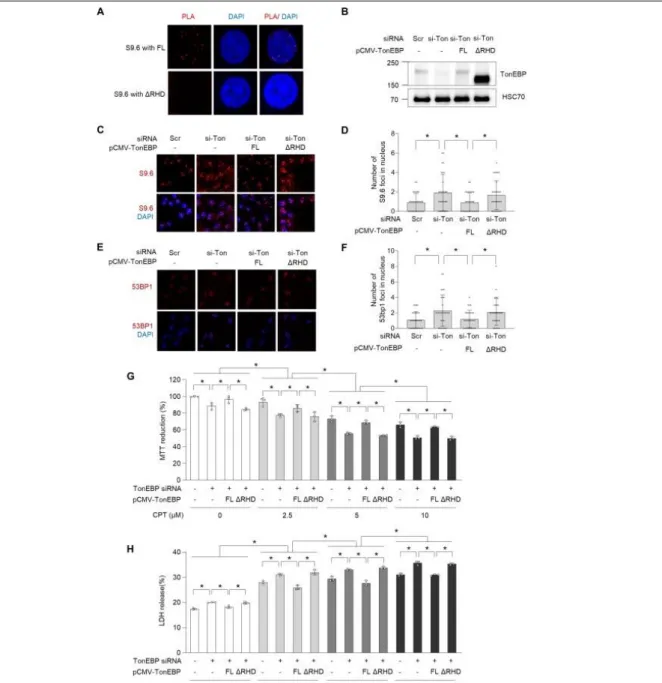
To further explore our observation that RHD of TonEBP was required for PARylation via an interaction with PARP1 (Figures 2-1 and 2), we performed rescue experiments to determine whether the RHD of TonEBP is required for R-loop resolution, maintenance of genome integrity, and cell survival. We first examined whether RHD of TonEBP is critical for R-loop binding, which is required for function of TonEBP at R-loops, using PLA in cells overexpressing intact TonEBP or ∆RHD. The PLA signals were observed in cells overexpressing intact TonEBP but not in ∆RHD mutant (Figure 2-5A), indicating that the RHD of TonEBP is critical for R-loop binding. More importantly, the re-expression of intact TonEBP (Figure 2-5B) rescued the increase in R-loop accumulation caused by TonEBP depletion, whereas re-
34
Figure 2-5. RHD of TonEBP is required for prevention of R-loop–associated DNA damage and cell survival in response to CPT. (A) U2OS cells were transfected with plasmid expressing full- length TonEBP (FL) or ΔRHD for 24 h. The cells were then subjected to PLA between TonEBP and R-loops. Representative images are shown. (B-H) U2OS cells were transfected with scrambled siRNA (Scr or -) or TonEBP-targeting siRNA (si-Ton) for 24h. The siRNA-transfected U2OS cells were then transfected a second time with plasmid expressing full-length TonEBP (FL) or ΔRHD for 24 h. (B) Cell lysates were immunoblotted for TonEBP and HSC70. Representative images are shown. (C) R-loop and nuclei were visualized by immunostaining with S9.6 antibody and DAPI staining, respectively. (D) The number of S9.6 foci per nucleus was determined from at least 30 cells.
(E) Immunostaining was performed for 53BP1. Representative images are shown. (F) Percent of 53BP1 foci-positive cells was calculated from at least 30 cells. (G and H) The transfected cells were treated with vehicle (-) or 2.5, 5, or 10 M of CPT. Cell viability was assessed by MTT reduction (F) and LDH release (G) after 24 h. Data (means ± SD) were from three independent experiments (n = 3). * p < 0.05.
35
expression of the ∆RHD mutant did not (Figure 2-5C-D). Moreover, the ∆RHD mutant did not revert the increase in abundance of 53BP1 foci in TonEBP-depleted cells (Figure 2-5E and F). Consistent with the higher levels of R-loop accumulation and DNA damage, depletion of TonEBP enhanced the dose dependent cell death by treatment with CPT (Figure 2-5G and H). Overexpression of intact TonEBP abolished this increase, whereas overexpression of the ∆RHD mutant did not (Figure 2-5G and H).
Taken together, these data suggest that the RHD of TonEBP prevents R-loop accumulation and DNA damage, and promotes cell survival, following CPT treatment.
Figure 2-6. TonEBP prevents R-loop–mediated DNA damage in HepG2 human hepatoma cells.
(A and B) HepG2 cells were transfected with scrambled siRNA (Scr) or TonEBP-targeting siRNA (si-Ton) for 24 h. The cells were then treated with vehicle (-) or 10 M CPT. R-loop and nuclei were visualized by immunostaining with S9.6 antibody and DAPI staining, respectively (A). The number of S9.6 foci per nucleus was determined from at least 30 cells (B). (C and D) HepG2 cells were
36
TonEBP prevents R-loop–mediated DNA damage in HepG2 human hepatoma cells
The findings described above indicated that PARP1-mediated PARylation of TonEBP promotes DNA repair and cell survival in human osteosarcoma U2OS cells. Given that TonEBP (27, 28) and PARP1 (39) are key drivers of HCC tumorigenesis, and that their inhibition suppresses HCC progression, we investigated the potential role of the PARP1–TonEBP axis in R-loop–associated DNA damage in HepG2 human hepatoma cells. siRNA-mediated depletion of TonEBP in HepG2 cells increased R-loop accumulation in both the presence and absence of CPT (Figure 2-6A and B), and led to a defect in the incorporation of the thymidine analog 5-ethynyl-2′-deoxyuridine (EdU) (Figure 2-6C and D), indicating that TonEBP depletion promotes R-loop accumulation and DNA replication defects. Interestingly, TonEBP depletion exerted synergistic effects on olaparib-induced cell death in CPT-untreated cells (Figure 2-6E and F). TonEBP depletion and olaparib increased sensitivity to cell death in response to CPT treatment (Figure 6E and F). However, olaparib treatment of TonEBP-depleted cells did not further increase cell death in response to CPT (Figure 2-6E and F), as in the case of U2OS cells. Taken together, these data indicate that the PARP1–TonEBP axis suppresses R-loop–associated DNA damage and cell death in cancer cells.
transfected with siRNA as above, and then subjected to Click-iT EdU assay. (C) Representative images of cells in each condition. (D) Percent of EdU positive cells were measured. Mean ± SD, n = 3. *p < 0.01. (E and F) HepG2 cells were transfected with siRNA and then treated with vehicle (-) or 1 M olaparib for 1 h in the absence (-) or presence 10 M CPT as indicated. Cell viability was assessed by MTT assay (E) and LDH release (F) after 24 h. Data (means ± SD) are from three independent experiments (n = 3). * p<0.01. n.s.: not significant. (G) Model: PARP1-mediated PARylation of TonEBP prevents R-loop–associated DNA damage.
37 2-5. Discussion
TonEBP is a well-established transcriptional regulator that serves as both an activator and a repressor (18). TonEBP regulates transcription of a large number of genes, either by direct binding to DNA via a specific motif or indirect binding via an interacting protein. Beyond its role as a transcriptional regulator, TonEBP is recruited to sites of DNA damage and promotes error-prone DNA damage bypass (controlled by PCNA) in response to genotoxic stress (31). Given that one of the hallmarks of cancer is widespread genomic instability, this implies that TonEBP is involved in human cancer. Indeed, TonEBP facilitates DNA repair via recruitment of a prominent DNA repair protein, the ERCC1/XPF complex, and consequently promotes self-renewal and chemoresistance of liver stem cells in response to cisplatin, a DNA-damaging agent that is widely used as an anticancer drug (28). However, it remains to be determined how TonEBP is recruited to sites of DNA damage, and which factors influence its functions in DNA damage and repair response.
Our primary finding of this study was that TonEBP interacts with PARP1, and that PARP1- mediated PARylation is necessary for recruitment of TonEBP to sites of DNA damage. PARP1 associates with TonEBP in cells exposed to high NaCl and suppresses its transcriptional activity (40).
One intriguing possibility is that in addition to promoting recruitment of TonEBP to sites of DNA damage, PARylation of TonEBP is required for regulation of its transcriptional activity. Consistent with this idea, it is already known that transcriptional activity of TonEBP is regulated by post-translational modification, including phosphorylation (11) and sumoylation (41). Future studies should investigate the impact of the TonEBP–PARP1 interaction on other cellular responses, including transcriptional regulation.
PARP1 is an important DNA repair molecule (32), and the successful application of PARP1 inhibitors in treating cancer is a classic example of the rational development of DNA repair-targeted cancer therapy (42). Specifically, olaparib suppresses HCC growth in mouse and HCC patient-derived xenograft models (39). TonEBP also acts as an oncoprotein via multiple mechanisms in HCC (27, 28).
TonEBP plays a critical role in tumorigenesis and tumor progression by activating the NF-κB enhanceosome and upregulating COX2 expression (27). It also promotes stemness of liver cancer and
38
cisplatin resistance via recruitment of the ERCC1/XPF dimer (28). Interestingly, PARP and the ERCC1/XPF complex participate in distinct DNA damage repair pathways (43). This study demonstrated that PARP is involved in a common repair pathway with tyrosyl-DNA phosphodiesterase 1, and that the ERCC1/XPF complex is involved in DNA repair by inducing the H2AX response. Based on these findings, we speculate that TonEBP may be involved in tumorigenesis and tumor progression via both DNA repair pathways involving PARP and XPF-ERCC1 in liver cancer, although additional studies focusing on this issue are still required.
R-loops play important physiological roles and have been implicated in pathogenesis (13-17).
Although the factors and cellular processes associated with R-loops have been extensively studied, many aspects of R-loop biology and R-loop–mediated genome instability remain unclear. Given the multifaceted roles of TonEBP and PARP1 in cancers, we focused on the PARP1-mediated function of TonEBP in R-loop–associated DNA damage in cancer cells, and identified TonEBP as a novel regulator of R-loop–associated DNA damage. We showed that TonEBP is recruited to R-loop–forming loci in response to the R-loop–associated DNA-damaging agent CPT. The loss of TonEBP increased the levels of R-loop accumulation, DNA damage, and DNA replication defect, thereby increasing the rate of CPT- mediated cell death, demonstrating that TonEBP plays an important general role in resolving R-loop–
associated DNA damage in cancer cells. This finding is supported by our recent study showing that the overexpression of catalytically-active RNaseH1, a conserved enzyme that performs R-loop resolution, recovered the increase of DNA damage and the reduction of cell proliferation induced by TonEBP depletion (38).
Similar results were obtained by knocking down or inhibiting of PARP1. However, loss of PARP1 function did not further increase R-loop–associated DNA damage in TonEBP-depleted cells, suggesting that TonEBP and PARP1 act in the same pathway to resolve replication stress, coordinate DNA repair, and promote cell survival (Figure 2-6G). These findings provide new insights into the molecular mechanisms underlying cellular DNA damage and repair response, as well as the novel functions of TonEBP in R-loop biology.
In summary, we have shown that in cancer cells, TonEBP is PARylated via an interaction with
39
PARP1, localizes to R-loops, prevents R-loop accumulation and DNA damage, and protects against cell death induced by an R-loop–associated DNA-damaging agent. These findings suggest that TonEBP mediates resistance to CPT-induced cell death by preventing R-loop accumulation in cancer cells.
Therefore, TonEBP or its protein–protein interactions represent potential therapeutic targets for human disorders associated with dysregulation of R-loop structures. One important issue to be addressed is how the same DNA damage response (DDR) stimulus can lead to markedly different responses, and how complex pathways and events are orchestrated. A better understanding of DDR will open new avenues for rational disease management.
40 2-6. Supplementary information
Supplementary figures
Supplementary Figure 2-1. TonEBP interactomes, related to R-loop–associated DNA damage.
TonEBP interactome, including PARP1.
41 2-7. Reference
1. Hamperl, S.; Cimprich, K.A. Conflict resolution in the genome: how transcription and replication make it work. Cell 2016, 167, 1455–1467.
2. Garcia-Muse, T.; Aguilera, A. Transcription-replication conflicts: how they occur and how they are resolved. Nat. Rev. Mol. Cell Biol 2016, 17, 553–563.
3. Allison, D.F.; Wang, G.G. R-loops: formation, function, and relevance to cell stress. Cell Stress 2019, 3, 38–46.
4. Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Che´ din, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 2012, 45, 814–825.
5. Skourti-Stathaki, K.; Kamieniarz-Gdula, K.; Proudfoot, N.J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 2014, 516, 436–439.
6. Go´mez-Gonza´lez, B.; Garcı´a-Rubio, M.; Bermejo, R.; Gaillard, H.; Shirahige, K.; Marı´n, A.;
Foiani, M.; Aguilera, A. Genome-wide function of THO/TREX in active genes prevents R-loop- dependent replication obstacles. EMBO J. 2011, 30, 3106–3119.
7. Sollier, J.; Cimprich, K.A. Breaking bad: R-loops and genome integrity. Trends Cell Biol. 2015, 25, 514–522.
8. Aguilera, A.; Garcı´a-Muse, T. R loops: from transcription byproducts to threats to genome stability.
Mol. Cell 2012, 46, 115–124.
9. Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396.
10. Aguilera, A.; Gómez-González, B. DNA-RNA hybrids: the risks of DNA breakage during transcription. Nat Struct Mol Biol. 2017. 24, 439–443.
11. Groh, M.; Gromak, N. Out of balance: R-loops in human disease. PLoS Genet. 2014, 18, 10(9).
12. Mackay, R.P.; Xu, Q.; Weinberger, P.M. R-Loop Physiology and Pathology: A Brief Review. DNA Cell Biol. 2020, 39, 1914–1925.
13. Cristini, A.; Groh, M.; Kristiansen, M.S.; Gromak, N. RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage.
Cell reports 2018, 23, 1891–1905.
14. Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat Rev Genet.
2014, 15, 829–845.
15. Wang, I.X.; Grunseich, C.; Fox, J.; Burdick, J.; Zhu, Z.; Ravazian, N.; Hafner, M.; Cheung, V.G.
Human proteins that interact with RNA/DNA hybrids. Genome Res. 2018, 28, 1405–1414.
16. Richard, P.; Manley, J.L. R loops and links to human disease. J Mol Biol. 2017, 429, 3168–3180.