http://iaesjournal.com/online/index.php/TELKOMNIKA/index
Accelerating Computation of DNA Multiple Sequence Alignment in Distributed Environment
DOl: 10.11591/telkomnika .v12il2 .6572: Abstrdct
mオャ|Qーャセ@ NセャGqャjセョ・・@ al1=;inmerrt (MS;\) エセ@ '-' t\'_etmiqul' for firdm.;i セQュゥャj[イゥエL@ "' m<iny ^[アオ・ョ・ャGセN@ Ti-u5 tecM"l.Ub is Yer'{ •n'•IJ01tant to セオーーッイエ@
rn.;nf_ ヲセャIゥGNGヲッョョhャゥ」セ@ tMk <w:h Hr. 1dent;f;•n<;1 Sin•/!6 Nuclaot1de l'olymnrphoHn {S_NI') "n:I ュhセIャセイwGャGャAi@ ヲイ。ァュ・ョエセ@ b1nninq. Th!! セゥイョdQVセエ@
<.!lqcmhrn m M:Sr, 1> $tM f•lgonthm, The cornpll'xity of DN1\ mult1plb セャGアumᄋセセ@ :1l1?nrnent \jセio_@ dtna1nic pG\Gャャセュュゥョ_@ tH.hn1q•Jl' 1> v"rr
!>19h. H.1> イセU・セイセィ@ ャャャョGlセ@ t(· hセ・ャセイGA^エエ@ .:.emputation of Star mセエQーャ・@ Sセᄋセ|Q・ョ」セ@ エ|ィN[ュュセョエ@ •Js•nsi MH5111JO p。ウセQョァ@ lnterfaces HmセiゥN@ The
P"1fonn.anc>J-"1' lt:e ーイセゥ[ッウ・、@ m••thod セᄋセセ@ LLNLLGjャオセエ・、@ by イNャAic|ャャセエ[ョ\j@ セーNNゥ・、オーN@ E1p•mrnent '"as conducted オセュセ@ { .. 1 セLアオ・ョ」Zセ@ of セoセ@ bp
cMャエ」ゥョッMイョ・クM」ゥャイッイイNッセᄋュhᄋM|hh[j@ ヲイ\QアョキQQi[セ@ yiold"'d t·v randomly out from Nf"a-rencx. セッエゥオᄋュ\Z・@ of cMャ[ッゥョ・MイmクM」エゥイッュ。hᄋュセMAャᄋb・エ@ エ。セ・ョ@
lrom N".":51 HセjセエャッイQゥQi@ BNBZ・ャ|エセイ@ for bQエQ|Q」セョNLャッセャG@ !r,forrMt•on). TM 1\'•;••ll's DィoセセGセ、@ lhat エィセ@ ーイッーッセj、@ tochr.ique could \'lbtaio セセャGセ、オセ@ thr<:!.
ャPQQQ・セ@ t,;rng f;ve c-:irnptJt•l" when '.ili;in•n'i' 1,.; セッオ|AoZイエサh@ oi gャQセQイゥッᄋュ\QセM、Qイ\ZMイッャッ\。ュ・MBMbヲ[A@ fra\JITWnt!:. l•!Ort:.t)n·r, Q[Lセ@ 11tcrMWl!J oi 1;1, ..
r"Jrr.b':r of cornpu\<'r-; would セQァッQヲイZセイエャ[G@ ュ」イ・\ゥウセ、@ セーッッ、\jヲNG@ of th., prorion::d
http://iaesjournal.com/online/index.php/TELKOMNIKA/about/editorialTeam
Editorial Team
Advisory Editors
I. Prof. Hamid
A.
Toliyat, Texas A&M University, United States
2. Prof. Patricia Melin, Tijuana Institute of Technology, Mexico
3. Prof. Neil Bergmann, The University of Queensland, Australia
4. Dr. Argyrios Zolotas, University of Lincoln, United Kingdom
5. Prof. Aurelio Piazzi, University of Panna, Italy
6. Prof. Daniel Thalmann, Nanyang Technological University, Singapore
7. Prof. Ajith Abraham, VSB Technical University ofOstrava, Czech Republic
Editor-in-Chief
I. Tole Sutikno, Universitas Ahmad Dahlan, Indonesia
[ USER i
r---··---·---;
i
BBセGBᄚGN[I@
i
AMセセZNセNZZ@
..
セNセ@
.,.I
• vャセキ@
• Sub.crlbe / U11>ub>uob"
QセNセ[Z[NZ[セセ[セZNZ[[ZZZQ@
i
r•;----··
·li'J ·'1-
i
Co-Editor-in-Chief
I.
Prof. Dr. Leo P. Ligthart, Delft University of Technology, Netherlands
2. Prof. Dr. Omar Lengerke, Universidad Aut6noma de Bucaramanga, Colombia
3. Assoc. Prof. Dr. Wanguan Liu, Curtin University of Technology, Australia
4. Dr. Arianna Mencattini, University of Rome "Tor Vergata", Italy
5. Dr. Auzani Jidin, Universiti Teknikal Malaysia Melaka (UTeM), Malaysia
6. Mark S. Hooper, Analog/RF IC Design Engineer (Consultant) at Microsemi, United States
Editors
1. Prof. Dr. Faycal Djeffal, University ofBatna, Batna, Algeria
2. Prof. Dr. Luis Paulo Reis, University ofMinho, Portugal
3. Prof. Dr. Sanjay Kaul, Fitchburg State University, United States
4. Prof. Dr. Sotirios G. Ziavras, University Heights, United States
5. Prof. Dr. Srinivasan Alavandar, CK College of Engineering and Technology, India
6. Prof. Dr. Tarek Bouktir, Ferhat Ab bes University, Setif, Algeria
7. Prof. dr.sc. Maj a Stula, University of Split, Croatia
8. Assoc. Prof. Dr. Nik Rumzi Nik Idris, Universiti Teknologi Malaysia, Malaysia
9. Assoc. Prof. Dr. Lunchakorn Wuttisittikulkii, Chulalongkorn University, Thailand
10. Assoc. Prof. Dr. Jumril YU11as, Universiti Kebangsaan Malaysia, Malaysia
11. Assoc. Prof. Dr. Mochammad Facta, Universitas Diponogoro (UNDIP), Indonesia
12. Asst. Prof. Dr. Ahmet Teke, <;:ukurova University, Turkey
13. Asst. Prof. Dr. Ehsan 0. Sheybani, Virginia State University, United States
14. Asst. Prof. Dr. Supavadee Aramvith, Chulalongkorn University, Thailand
15.
Dr. Ahmad Saudi Samosir, Universitas Lampung (UNILA), Indonesia
16. Dr. Ahn1ed Nabih Zaki Rashed, Menoufia University, Egypt
17. Dr. Deris Stiawan. CCNA, CIEH, CIHFI, Universitas Sriwijaya, Indonesia
18. Dr. Han Yang, University of Electronic Science and Technology of China, China
19. Dr. Jacek Stando, Technical University of Lodz, Poland
20. Dr. MU11awar A Riyadi, Universiti Teknologi Malaysia, Malaysia
21. Dr. Nidhal Bouaynaya, University of Arkansas at Little Rock, Arkansas, United States
22. Dr. Peng Peng, Seagate Technology, Bloomington, MN, United States
23. Dr. Shahrin Md. Ayob, Universiti Teknologi Malaysia, Malaysia
24. Dr. Surinder Singh, SLIET Longowal, India
25. Dr. Tutut Herawan, Universiti Malaysia Pahang, Malaysia
26. Dr. Vassilis S. Kodogiannis, CEng, University of Westminster, United Kingdom
27. Dr. Yin Liu, Symantec Core Research Lab, United States
TELKOMNIKA Indonesian Journal of Electrical Engineering Vol. 12, No. 12, December 2014, pp. 8278 - 8285
DOI: 10.11591/telkomnika.v12i12.6572 • 8278
Accelerating Computation of DNA Multiple Sequence
Alignment in Distributed Environment
Ramdan Satra*1", Wisnu Ananta Kusuma'. Heru Sukoco1
1
Department of Computer Science, Bog or Agricultural University Kampus IPB Dermaga, JI. Meranti, Wing 20 Level 5-6, Boger 16680, Indonesia
Telp./Fax.: +62-251-8625584
'Department of Informatics Engineering, University of Muslim Indonesia, Makassar, Indonesia *Corresponding author, e-mail: [email protected] 1
, [email protected], [email protected]
Abstract
Multiple sequence alignment (MSA) is a technique for finding similarity in many sequences. This technique is very important to support many Bioinformatics task such as identifying Single Nucleotide Polymorphism (SNP), creating phylogenetic tree, and metagenome fragments binning. The simplest algorithm in MSA is Star Algorithm. This algorithm consists of aligning all possible pairs of sequences, finding a sequence Star chosen from sequence that has maximum alignment score, and aligning all sequences refered to the sequence Star. Each of pairwise alignments is conducted using dynamic programming technique. The complexity of DNA multiple sequence alignment using dynamic programming technique is very high. The computation time is increased exponentially due to the increasing of the number and the length of DNA sequences. This research aims to accelerate computation of Star Mutiple Sequence Alignment using Message Passing Interfaces (MP/). The performance of the proposed method was evaluated by calculating speedup. Experiment was conducted using 64 sequences of 800 bp G/ycine-max-chromosome-9-881 fragments yielded by randomly cut from reference sequence of Glycine-max-chromosome-9-881 taken from NCBI (National Center for Biotechnology Information). The results showed that the proposed technique could obtain speed up three times using five computers when aligning 64 sequences of G/ycine-max-chromosome-9-BBI fragments. Moreover, the increasing of the number of computers would significantly increase speed up of the proposed method.
Keywords: DNA multiple sequence alignment, distributed computing, star algorithm, message passing interface
Copyright© 2014 Institute of Advanced Engineering and Science. All rights reserved.
1. Introduction
Multiple Sequence Alignment (MSA) is a technique for aligning multiple biological sequences [1] to find similarities and differences in the sequence [2]. MSA is used in phylogenetic analysis to assessing the origin of species. MSA is also used to search a single nucleotide polymorphism (SNP) for molecular-based plant breeding genetics [3]. The growth of biological sequence databases makes the sequence alignment computation increase significantly [1 ].
The complexity of sequence alignment is 0 (LN), where L is the length of each sequence and N is the number of sequences [4]. The longer and the more sequences to be aligned the longer it's computational time. The optimal time of sequence alignment is increased exponentially with increaseing of the number and length of sequences [5].
Methods or tools are needed to obtain optimal computational process. Previous study to optimize the computational sequence alignment used a dynamic programming algorithm with complexity of 0 (mn) by Zhou and Chen [6]. Other studies used a linear space algorithm for pairwise sequence alignment with the complexity of 0 (n) [7, 8]. Others used parallel computing with MPI [9] and GPU-CUDA [1, 3]. Parallel computing used to propose new clssification algorithm [12] and simulate properties of polymer chain [13] in other case of computational problem.
In this research, parallel computing is utilized on clusters of computers known as Distributed Memory. This research tries to improve a previous work conducted by Sunarto セ@
TELKOMNIKA ISSN: 2302-4046
• 8279
Multiple Sequence Alignment. The new approach could handle more number of sequences and was also easily extended by increasing the number of resources used to compute multiple sequence alignment.
2. Research Method 2.1. Research Data
In this research, we used the sequence of Glycine-max-chromosome-9-881 obtained from NCBI (National Center for Biotechnology Information) [1
OJ.
Detailed research data is shown in Table 1. [image:4.595.94.562.199.279.2]9;\K
S"•tu
セ@
セi@
Table 1 . Research data te.,)"'h
セ@
'.)..!,,..., ("
セセセセセセセセセMGM]BGMBMGMGMGMGNZBBGMZZZNoZBGMB]]ZN⦅⦅Mセセセセセセセセ@ I
Mセセ@
セHセ@
-
·-G/ycine-max-chromosome-9-881 811-850 64
(v(.v..
p
セ@
tu
VI IWi-
ᄉセ@
2.2. Multiple Sequence Alignment (MSA) with Star Algorithm
Algorithms for MSA are actually developed based on probabilistic or heuristic approaches such as the Star method and the Progressive method [3]. This research used the Star method by improving and extending theprevious research conducted by Sunarto AA et al. In the Star alignment algorithm, there are 3 stages, including (i) calculating pairwise alignment scores from all possible sequence pairs, (ii) selecting a Star sequence which has the best alignment score, and (iii) aligning the Star sequence with other sequences [3, 9]. The illustration of multiple sequence alignment using the Star algorithm can be described as follows:
For example, supposed we have DNA sequence data as follows: Seq 1 =ATGG
Seq 2 =ATGC Seq 3 =ATCC Seq 4 =AGCG
The first stage is calculating pairwise similarity scores from all possible sequences pairs. This stage has O(n2) complexity where
n
is number of sequences. This stage begins bycalculating the number of sequence pairs using Equation (1):
(n-1) + (n-2) + .... + 1 or n(n-1)12 (1)
__ ,JWhere
n
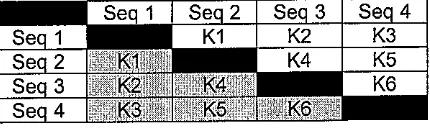
is number of sequences.· The next step is creating identity matrix containing the sequences, symbolized by Kn. Figure 1 shows the identity matrix.
セ@
Figure 1. Pairwise sequence identity matrix
The results of the determination of DNA pairwise sequence are: K1 =Seq 1 (ATGG) and Seq 2 (ATGC)
[image:4.595.180.395.554.618.2]'
.
8280 • ISSN: 2302-4046
Each pair of sequences above is aligned using Needleman Wunsch algorithm. This algorithm uses dynamic programming approach to find global similarity of sequences. Needleman Wunsch algorithm consists of matrix initialization, filling the value of each cell matrix and tracing back. Similarity score of pairwise sequence is calculated using Equation (1) below:
!
Am-1,n-1+
Sm,nA - M Am,n-1
+
W[mnJ -
ax
A Wm-1,n
+
0
(1)
Where:
mis the row and n is the collumn.
A is a matrix of values for each cell is expressed as (A1m,nJ)
S is the score of each cell is expressed as ( S1m,nJ}, match score = 5 and unmatch score= -3
Wexpressed as gap alignment with value = -
4
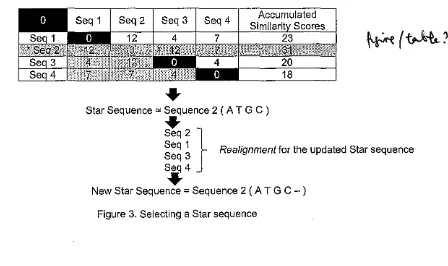
[image:5.600.178.398.322.387.2]The calculation results of similarity scores for all pairs of sequences are shown in Figure 2.
Figure 2. Sequence alignment similarity scores
The second stage of star algorithm is selecting a Star sequence. A Star sequence was selected by comparing the sequence alignment similiartity scores of all sequences. A sequence with the best similarity score was chosen as a Star sequence. Next, the Star sequence was updated by aligning it to the other sequences. The complexity of this stage is O(n), where n is the number of sequences. Illustration of this stage is shown in Figure 3.
Accumulated Similarit Scores
23
...
Star Sequence =Sequence 2 (AT G C )
セRス@
Seq 1 Realignment for the updated Star sequence Seq 3
s・セ@
...
4New Star Sequence= Sequence 2 (AT G C - )
[image:5.600.108.556.504.770.2]TELKOMNIKA ISSN: 2302-4046 • 8281
The third stage of star algorithm is realignment. This stage is realigning the Star sequence to others sequences by using dynamic programming technique. Complexity is O(n), where
n
is the amount of sequences. Illustration for realignment is shown in Figure 4.Seq2(ATGC-) Seq 1 (AT G G )
Seq3(ATCC) Seq4(AGCG)
...
ATGC-ATG-G AT-CC A-GCG
} The results of the sequence alignment
Figure 4. Realignment
2.3 Parallelization MSA with Message Passsing Interfaces (MPI)
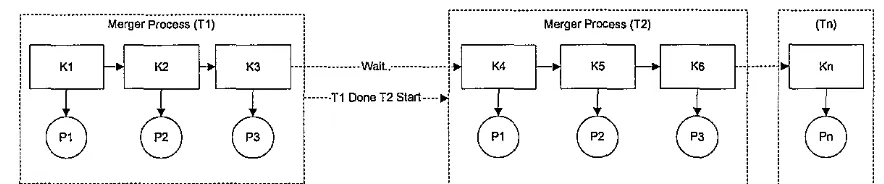
In this research, we used Foster's Methodology for implementing parallel programming. This methodology consists of partitioning, communication, agglomeration and mapping stage [8]. Parallelization was conducted for computing pairwise similarity scores. The calculation began by dividing the sequence pairs into multiple processes (threads) symbolized by K1, K2
... Kn. In the case when the amount of computers is equal to the amount of processes, each process will be allocated to each computer. If Pk is a computer for k
=
1,2,. .. n this allocation can be defined as P1 =Kt, P2=
K2 ... Pn =Kn. Unfortunately the number of sequence pairs is often larger than the number of computers. Sequence pairs which has not been allocated yet must wait until the previous process has been completed. Allocation of processes (threads) when the number of sequence pairs are more than the number of computers or when Kn > Pn can be described in Figure 5.ZQセ@
hGzhセセMMMゥᄋセᄋᄋᄋᄋᄋセᄋMᄋᄋᄋᄋᄋ[[[セ[LᄋMセ@
MZN|ᄋセ@
11
,i i i : I
i
1
r-···--T1 Done T2 sエ。イエMMMMセ@
l :
i
] P 1 P2 P 3 i \ p 1 P2
P 3 i ! P n !
i ...
.J
i_ ··· -... --- ·- ··· ... --.... _ _Il ...
···-
__
_J Figure 5. The distribution of processes when the number of sequence pairs are more than thenumber of computers or Kn > Pn
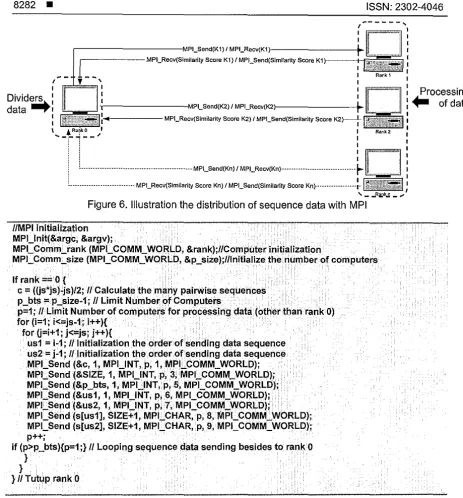
[image:6.602.222.499.128.263.2] [image:6.602.64.506.463.555.2]8282 • ISSN: 2302-4046
,--
セMML@
---,
I I
I I
I I
r - - - I M P l _ S e n d ( K 1 ) / M P l _ R e c v ( K 1 ) - - - ' f - 1 MPl_Recv{Similarity Score K1) f MPl_Send(Simi!arity Score K1) I ェゥヲゥjゥャャヲFイョZセゥャ@ :
I MセGBセ U Qhャャャエj@ I
I Rank 1 I I IF""'=wi I
Dividersl : '
I I
: , Processing
+-
ofdatadata I セMMMMMMMmpャ⦅s・ョ、HkRI@ I
MPl_Recv(K2)---+-: ャPエセᄆqゥセjャUQZMMMMMMmpャ⦅r・」カHsャュゥャ。イゥエケ@ Score K2)/ MP1_Send(Similarlty Score K2)--t
\ .. r。セォo@ I I
'-,--l- - " I
i I
Rank2
i
l ...
MPl_Seod(Ko) /mpャ⦅r・ッカHkッIMᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋᄋセ@
D
L---·----·--··
MPl_Recv(Similarity Score Kn) I MPl_Send(Slmllar!ty Score Kn)···0· - - - : -
ャ[hゥhャセセQL@
... - _,RjWl\Jl - ...
Figure 6. Illustration the distribution of sequence data with MPI
llMPI initialization MPl_lnit(&argc, &argv);
MPl_Comm_rank (MPl_COMM_WOR.LD, &rank);//Computer init_ialization
MPl_Comm_size (MPl_COMM_WORLD, &p_size);//lnitialize the.number of computers If rank== 0 {
c = ((js*js)'js)/2; II Calculate the many pairwise sequences p_bts =.p_size-1; II Limit Number of Computers
p=1; II Limit Number of computers for processing data (other than rank 0) for (i=1; i<=js-1; i++){
for (j=i+1; j<=js; j++){
us1 = i:1; // Initialization the order of sending data sequence us2 = j-1; //Initialization the order.of sending data sequence MPl_Send (&c, 1, MPl_INT, p, 1, MPl_COMM_WORLD); MPl_Send (&SIZE, 1, MPl_INT, p, 3; MPl_COMM_WORLD); MPl_Send (&p_bts, 1, MPl_INT, p,.5, mpャ⦅comm⦅woセl「I[@
MPl_Send (&us1, 1, MPl_INT, p, 6, MPl_COMM_WORLD); MPl;_Send (&us2, 1, MPl_INT, p;7, MPl_COMM_WORLD);
MPl_Send (s[us1], SIZE+1, MPl_CHAR, p, 8, MPl_COMM ... WORLD); MPl_Send (s[us2], SIZE+1, MPl_CHAR, p, 9, MPl_COMM_WORLD); p++;
if (p>p_bts){p=1 ;} II Looping sequence data sending besides to rank O
} }
} II tオエセー@ rank 0
Figure 7. Source code data divider
If rank<> O {
MPl_Recv(&c, 1, MPl_INT, 0, 1, mpャセcomm⦅worldL@ &status); MPl_Recv(&SIZE, 1, MPl_INT, 0, 3, MPl_COMM_WORLD, &status); MPl_Recv(&p_bts, 1, MPl_INT, 0, 5, MPl_COMM_WORLD, &status); p=1;
for (h=O; h<=c-1; h++){ if (rank="p){
}
MPl_Recv(&us1, 1, MPl_INT, 0, 6, MPl_COMM_WORLD, &status); MPl_Recv(&us2, 1, MPl_INT, O; 7, MPl_COMM_WORLD, &status);
MPl_Recv(s[us1], SIZE+1, MPl_CHAR, 0, 8, mpャ⦅commセworldL@ &status); MPl_Recv(s[us2], SIZE+1, MPl_CHAR, 0, 9, MPl_COMM_WORLD; &status); } lle_nd_ rank <> O
[image:7.600.61.524.57.555.2]TELKOMNIKA ISSN: 2302-4046 • 8283
3. Results and Analysis
This research used 5 computers with spesification Intel Core CPU [email protected] with 4 CPU cores, 8 GB of RAM memory. A whole genome data with FASTA format of Glycine-max-chromosome-9-881 was divided randomly into some numbers of sequences such as 3, 4, 8, 12, 14, 16, 20, 24, 28, 32, 36, 40, 44, 48, 52, 56, 60 and 64 sequence. Testing evaluation was conducted in 3, 4 and 5 computers.
3.1. Results
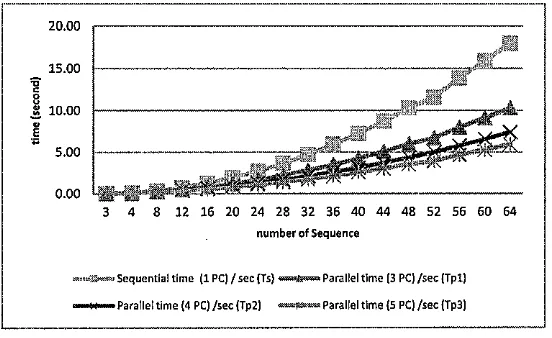
The evaluation results was described in Figure 9-11. Figure g showed the performance of our proposed method in term of execution time.
number of Sequence
BGBセs・アオ・ョエゥ。ャ@ time {1 PC)/ sec (Ts)...,.... Parallel time (3 PC) /sec {Tpl)
_..,_Parallel time (4 PC) /sec (Tp2) セp。イ。ャャ・ャ@ time (5 PC) /sec {Tp3)
Figure 9. the execution time in various number of sequences
The result of sequence alignment computation showed that the parallel computation time was faster than the sequential computation time. Figure 9 showed the difference in computation time of data FASTA Glycine-max-chromosome-9-BBI with the length of 811-850 basepair. Using 3, 4 and 8 sequences the computational time was only slightly different. However, with the incresing of the number of sequences the computational time was significantly different.
3.2. Analysis
The performance analysis or evaluation of this proposed method was conducted by calculating speedup [11 ]. Speedup can be obtained by using Equation (2).
Ts
Speedup = - (2)
Tp
Description:
Ts =Sequential execution time Tp = Parallel execution time
[image:8.595.146.422.219.388.2]•
8284 • ISSN: 2302-4046
3.50 セMMMMMMMMMMM
セsー・・、オー@ 3 PC -speedup4PC
1.00
--w--speedups PC
o.so
+----0.00 MᄋセMMMMMMMMMMM
STXuセRPセセSRSVセᆱTXURセVPセ@
[image:9.595.125.500.64.234.2]number of sequence
Figure 10. The speedup of parallel computing in various number of sequences
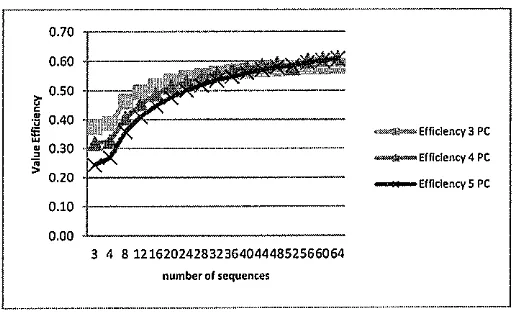
The other metric for showing the parallel performance is efficiency. Increasing the number of processors decreased the value of efficiency, and conversely an increase in the size of data will increase the efficiency [14]. The constant efficiency value indicates that the performance of a parallel system is scalable to the size of the data. Scalable parallel systems means the system is able to maintain performance with increasing number of processors [14] to process the data in a certain size. Efficiency is calculated using Equation (4). The results of efficiency calcucaltion in this study can be seen in Figure 11.
Efficiency = ウセョI@
Limitation efficiency value is 1 Ip <efficiency <1
Description:
S (n) =Speedup of parallel computing p
=
Many processors0.70 · · · · ·
-セ@ a.so
--·-ᄋセ@ 0.40
iii
セ@ 0.30 .
I: 0.20
+.-:---0.10 ·
-o.oo セMMMMMMMMMMM
3 4 8 1216202428323640444852566064
number of sequem;es
セeヲヲゥ」ゥ・ョ」ケ@ 3 PC
セeヲヲゥ」A・ョ」ケTpc@
[image:9.595.155.415.469.625.2]_..._EfflclencyS PC
Figure 11. Efficiency of parallel computing in various number of sequences
(4)
•
'
.
TELKOMNIKA ISSN: 2302-4046 • 8285
4. Conclusion
Parallel computing using MPI is recommended for MSA. This study show that the speed up of using MP! in distributed environment for conducting MSA was ゥョ」イ・セ@ by increasing the size and the number of DNA sequences aligned. The trend of efficiency was almost constant when using 56 of sequences or more. This tendency indicated that this method was scalable to the size of data.
Future research can be conducted by using the larger size of data sequence and increase the number of processors to improve the speedup and efficiency of parallel computing for MSA. In this research, pairwise sequence alignment was conducted in sequential computation. In future, we could conduct hybrid computing by conducting the paiwise sequence alignment in parallel using MP! and CUDA GPU to increase speed up significatly.
Acknowledgements
The authors would like to thank Indonesia Ministry of Agriculture for funding this research through the KKP3N 2014 program.
References
[1] Liu Y, Schmidt B, Maskell DL. MSA-CUDA. Multiple Sequence Alignment on Graphics Processing Units with CUDA. IEEE International Conference on Application-specific Systems, Architectures and Processors. 2009: 121-128.
[2] Junior SAC. Sequence Alignment Algorithms. Department of Computer Science School of Physical Sciences & Engineering Kingls College London. 2003.
[3] Sujiwo MAP, Kusuma WA. Multiple Sequence Alignment with Star Method in Graphical Processing Unit using CUDA. International Seminar on Science (/SS). Bogar. 2013: 359-363.
[4] Lloyd GS. 2010. Parallel Multiple Sequence Alignment: An Overview.
[5] Edgar RC, Batzoglou S. Multiple sequence alignment. Current opinion in structural biology. 2006; 16(3): 368-373.
[6] Zhou Zm, Chen Z-w. Dynamic Programming for Protein Sequence Alignment. International Journal of Bio-Science and Bio-Technology. 2013; 5(2): 141-150.
[7] Myers EW, Miller W. Optimal alignments in linear space. Oxford Univ Pres. 1988: 1-13.
[8] Sandes EFO, de Melo ACMA. Smith-Waterman Alignment of Huge Sequences with GPU in Linear Space. IEEE International Parallel & Distributed Processing Symposium. 2011: 1199-1211.
[9] Sunarto AA, Kusuma WA, Sukoco H. Paralelisme Of Star Alignment. IEEE International Conference on Instrumentation, Comunications, Information Technologi, and Biomedical Engineering. 2013: 167-171.
[10] Zhou L, Wang H, Wang W. Parallel Implementation of Classification Algorithms Based on Cloud Computing Environment. TELKOMNIKA Indonesian Journal of Electrical Engineering. 2012; 10(5): 1087-1092.
[11] Li H, Gong B, Gao HB, Qian CJ. Parallel Computing Properties of Tail Copolymer Chain.
TELKOMNIKA. 2013; 11 (8): 4344-4350.
[12] "NCBI Home Page,"[Online]. Available: http://www.ncbi.nlm.nih.gov/
[13] Quinn MJ. Parallel Programing in C with MPI and OpenMP. McGraw-Hill Companies, Inc. 2003. [14] Maria A Kartawidjaja. Analisis Kinerja Perkalian Matriks Paralel Menggunakan Metrik lsoefisiensi.