PEMODELAN PEMROGRAMAN DINAMIS PADA
MULTIPLE
SEQUENCE ALIGNMENT
UNTUK PERANCANGAN PRIMER
SELULASE
M.
BAHRUL ULUM
SEKOLAH PASCASARJANA INSTITUT PERTANIAN BOGOR
PERNYATAAN MENGENAI TESIS DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA
*Dengan ini saya menyatakan bahwa tesis berjudul pemodelan pemrograman dinamis pada multiple sequence alignment untuk perancangan primer selulase adalah benar karya saya dengan arahan dari komisi pembimbing dan belum diajukan dalam bentuk apa pun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir tesis ini.
Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
Bogor, Agustus 2013
M.Bahrul Ulum NIM G651110111
*
RINGKASAN
M. BAHRUL ULUM. Pemodelan Pemrograman Dinamis Pada Multiple Sequence Alignment Untuk Perancangan Primer Selulase. Dibimbing oleh WISNU ANANTA KUSUMA dan JONI PRASETYO.
Selulase mempunyai peranan utama dalam pemanfaatan limbah biomassa yang mengandung lignin, hemicellulose dan cellulose (lignocellulose). Limbah biomassa ini sangat banyak terdapat dilingkungan dan sampai saat ini masih belum dimanfaatkan secara maksimal. Selulase berperan menguraikan cellulose atau hemicellulose menjadi gula. Beberapa mikroorganisme mempunyai kemampuan untuk memproduksi selulase, terutama extracellular enzim selulase. Tetapi kebanyakan mikroorganisme dari alam memproduksi enzim selulase dengan jumlah terbatas (aktifitasnya rendah). Dalam rangka meningkatkan produktivitas mikroorganisme dalam menghasilkan selulase, salah satu metode yang bisa diterapkan adalah memasukan sekuens gen penghasil selulase ke dalam mikroorganisme tersebut. Data untuk menyusun sekuens gen tersebut bisa dilakukan dengan merangkum sekuens gen dari mikroorganisme penghasil selulase. Langkah pertama untuk memprediksi sekuens gen penyandi selulase tersebut adalah merancang primer sekuens gen penyandi selulase yang dirangkum dari beberapa mikroorganisme penghasil selulase.
Tujuan penelitian ini adalah merancang potensial primer selulase dari sekuen DNA penyandi selulase yang akan dilakukan penyejajaran dengan teknik multiple sequence alignment menggunakan metode progresif. Metode penelitian ini terdiri atas beberapa tahapan proses yaitu pengambilan data, multiple sequence alignment dengan metode progresif dan perancangan primer. Data yang digunakan berupa gen penyandi selulase yang diambil dari GenBank NCBI (National Center for Biotechnology Information)
Penelitian ini, kami melakukan pendekatan untuk mencari potensial primer yang berpotensi untuk meningkatkan produktivitas enzim selulase. Dalam perancangannya, digunakan metode multiple sequence alignment (MSA) yang menggunakan algoritme progressive alignment. Pada tahap penyejajaran, dari 5 sekuens penyandi selulase didapatkan adanya 3 daerah konservatif (conserved regions). Sedangkan template yang digunakan untuk merancang potensial primer yaitu sekuens Solanum lycopersicum NM-001247953 dari daerah yang memiliki similaritas yang tinggi (conserved region). Diperoleh 46 pasang primer dari 3 conserved region. Masing-masing dari conserved region ke-1 sebanyak 28 pasang primer, conserved region ke-2 sebanyak 13 pasang primer dan conserved region ke-3 sebanyak 5 pasang primer.
SUMMARY
M. BAHRUL ULUM. Modeling of the Dynamic Programming Multiple Sequence Alignment for Design Primer Cellulase. Supervised by WISNU ANANTA KUSUMA and JONI PRASETYO.
The role of cellulase is absolutely very important to degrade cellulose which is abundant in environment like biomass waste. Many microorganisms have capability to produce cellulase. In order to improve the cellulase production by the microorganism, one of methods can be done by inserting the sequence gene of cellulase. The information of the sequence gene can be extracted from microorganisms of cellulase producer. The first step to predict the sequence gene of cellulase, we should design the primer of the sequence gene by summarizing the information from many microorganism which are cellulase producer.
The purpose of this study is the potential cellulase primers design from DNA sequence encoding cellulase alignment to be performed by multiple sequence alignment techniques using progressive methods. The procedure of this research in general divided into 3 parts, which are data preparation, multiple sequence alignment with progressive methods and primer design. The data used in the form of genes encoding cellulase retrieved from NCBI GenBank (National Center for Biotechnology Information).
This study, our approach to search for potential primer to increase the productivity of cellulase enzymes. In its design, the method used multiple sequence alignment (MSA), which uses progressive alignment algorithm. In the alignment phase, from 5 obtained a cellulase coding sequences conserved region 3 (conserved regions). While the template that is used to design the primer sequences potentially Solanum lycopersicum NM-001247953 from areas that have a high similarity (conserved region). Retrieved 46 primer pairs from conserved region 3. Each of the conserved region-1 to as many as 28 pairs of primers, conserved region-2 to as many as 13 pairs of primers and the conserved region of the 3 by 5 primer pairs.
Keywords: bioinformatics, cellulase, multiple sequence alignment, primer design
© Hak Cipta Milik IPB, Tahun 2013
Hak Cipta Dilindungi Undang-Undang
Dilarang mengutip sebagian atau seluruh karya tulis ini tanpa mencantumkan atau menyebutkan sumbernya. Pengutipan hanya untuk kepentingan pendidikan, penelitian, penulisan karya ilmiah, penyusunan laporan, penulisan kritik, atau tinjauan suatu masalah; dan pengutipan tersebut tidak merugikan kepentingan IPB
Tesis
sebagai salah satu syarat untuk memperoleh gelar Magister Komputer
pada
Program Studi Ilmu Komputer
PEMODELAN PEMROGRAMAN DINAMIS PADA
MULTIPLE
SEQUENCE ALIGNMENT
UNTUK PERANCANGAN PRIMER
SELULASE
M.BAHRUL ULUM
SEKOLAH PASCASARJANA INSTITUT PERTANIAN BOGOR
Judul Tesis : Pemodelan Pemrograman Dinamis pada Multiple Sequence Alignment untuk Perancangan Primer Selulase
Nama : M.Bahrul Ulum NIM : G651110111
Disetujui oleh Komisi Pembimbing
Dr Eng Wisnu Ananta Kusuma, ST, MT Dr Eng Joni Prasetyo, ST, MT
Ketua Anggota
Diketahui oleh
Ketua Program Studi Dekan Sekolah Pascasarjana Ilmu Komputer
Dr Yani Nurhadryani, SSi, MT Dr Ir Dahrul Syah, MSc. Agr
PRAKATA
Puji dan syukur penulis panjatkan kepada Allah subhanahu wa ta’ala atas segala karunia-Nya sehingga karya ilmiah ini berhasil diselesaikan. Tema yang dipilih dalam penelitian yang dilaksanakan sejak bulan September 2012 ini ialah bioinformatika, dengan judul Pemodelan Pemrograman Dinamis pada Multiple Sequence Alignment untuk Perancangan Primer Selulase.
Terima kasih penulis ucapkan kepada Bapak Dr Eng. Wisnu Ananta Kusuma dan Bapak Dr Eng. Joni Prasetyo selaku pembimbing yang telah banyak memberikan saran dan dukungan. Kepada Bapak Dr Ir Agus Buono, MSi, MKom selaku penguji. Selain itu, penghargaan penulis sampaikan kepada semua dosen dan staf Departemen Ilmu Komputer IPB, dosen dan staf ISTA yang telah membantu selama proses penelitian. Kepada ayah, ibu, bungsu, serta seluruh keluarga atas segala doa dan kasih sayangnya. Ungkapan terima kasih juga disampaikan kepada guru-guru PP.Darut Tafsir atas segala doa dan dukungannya serta teman-teman seperjuangan Ilkom 13 khususnya Nona, Mba Dian, Kang Asril (Tim Bioinformatik) yang telah membantu selama proses penelitian.
Semoga karya ilmiah ini bermanfaat.
Bogor, Agustus 2013
DAFTAR ISI
DAFTAR TABEL iv
DAFTAR GAMBAR iv
DAFTAR LAMPIRAN iv
1 PENDAHULUAN 1
Latar Belakang 1
Rumusan Masalah 2
Tujuan Penelitian 2
Manfaat Penelitian 2
Ruang Lingkup Penelitian 2
2 TINJAUAN PUSTAKA 3
Deoxyribonucleic Acid (DNA) 3
Sequence Alignment 3
Multiple Sequence Alignment 4
Selulase 5
GenBank 5
3 METODE PENELITIAN 7
4 HASIL DAN PEMBAHASAN 12
5 SIMPULAN 22
DAFTAR PUSTAKA 23
LAMPIRAN 25
DAFTAR TABEL
1 Matriks jarak 9
2 Sekuens gen penyandi selulase 12
3 Similarity scores 13
4 Sekuens Conserved Region 18
5 Primer dari region ke-1 21
6 Primer dari region ke-2 21
7 Primer dari region ke-3 19
DAFTAR GAMBAR
1 Deoxyribonucleic Acid (DNA) 3
2 Sequence alignment 4
3 (a) Global alignment dan (b) Local alignment 4 4 Multiple sequence alignment 5
5 Website NCBI 6
6 Prosedur penelitian 7
7 Diagram alir algoritme progressive alignment 8 8 Guide tree / Phylogenetic tree 9 9 Hasil penyejajaran dengan metode progresif 9
10 Proses algoritme progressive alignment 10
11 (a) forward primer (b) reverse primer 10 12 Tiga tahap progressive sequence alignment 12 13 Pseudo-code algoritme Needleman-Wunsch 13 14 Pseudo-code metode Neighbor-Joining 14 15 Proses pembangunan sebuah pohon filogenetik
(phylogenetic tree) menggunakan metode Neighbor-Joining 15 16 Phylogenetic tree dari 5 gen penyandi selulase 15 17 Pseudo-codeprogressive alignment 16 18 Hasil multiple sequence alignment dari 5 gen penyandi selulase 17 19 Waktu yang dibutuhkan oleh algoritme progressive alignment 17 20 Hasil perancangan potensial primer dari
sekuens conserved region ke-1 19
21 Hasil perancangan potensial primer dari
sekuens conserved region ke-2 19
22 Hasil perancangan potensial primer dari
sekuens conserved region ke-3 20
DAFTAR LAMPIRAN
1 Gen penyandi selulase 25
1
PENDAHULUAN
Latar Belakang
Pemanfaatan selulase di Indonesia semakin meningkat, karena saat ini selulase digunakan untuk biokonversi bahan lignoselulosa menjadi sumber energi dari bahan baku yang terbarukan (Anindyawati 2009; Joshi et al. 2011). Selain itu selulase juga sangat berperan dalam berbagai industri, di antaranya industri dengan penggunaan selulase yang cukup besar seperti industri tekstil, pulp dan kertas, deterjen, farmasi, pertanian dan makanan (Kuhad et al. 2011; Mojsov 2012). Berbagai aplikasi dari selulase menjadikannya sangat potensial untuk diproduksi terutama di Indonesia. Umumnya selulase yang digunakan saat ini berasal impor. Selulase dapat diproduksi oleh kelompok bakteri, kapang maupun khamir. Mikroba yang umum digunakan adalah Trichoderma reesei (Sukumaran et al. 2005; Lee dan Koo 2001; Sim dan Oh 1993). Beberapa mikroorganisme mempunyai kemampuan untuk memproduksi selulase, terutama extracellular selulase. Tetapi kebanyakan mikroorganisme dari alam memproduksi enzim selulase dengan jumlah terbatas (aktifitasnya rendah).
Dalam rangka meningkatkan produktivitas mikroorganisme dalam menghasilkan selulase, salah satu metode yang bisa diterapkan adalah memasukan sekuen gen penghasil selulase ke dalam mikroorganisme tersebut. Data untuk menyusun sekuen gen tersebut bisa dilakukan dengan merangkum sekuen gen dari mikroorganisme penghasil selulase. Langkah pertama untuk memprediksi sekuen gen penyandi selulase tersebut adalah merancang primer sekuen gen penyandi selulase yang dirangkum dari beberapa mikroorganisme penghasil selulase. Primer merupakan untai asam nukleat yang berfungsi sebagai titik awal untuk mensintesis DNA, yang diperlukan untuk mereplikasi DNA karena enzim-enzim yang mengkatalisis proses ini, yaitu DNA polimerase, hanya dapat menambahkan nukleotida yang baru ke rantai DNA yang ada. Perancangan primer dapat dilakukan berdasarkan sekuen DNA yang telah diketahui ataupun dari sekuen protein yang dituju. Apabila sekuen DNA maupun sekuen protein yang dituju belum diketahui maka perancangan primer dapat didasarkan pada hasil analisis homologi dari sekuen DNA atau protein yang telah diketahui mempunyai hubungan kekerabatan yang terdekat, salah satunya yaitu dengan menggunakan teknik multiple sequence alignment (Lakshmi et al. 2008). Penyejajaran sekuen nukleotida atau protein adalah proses dasar dalam biologi molekuler. biomolekul, seperti deoxyribonucleic acid (DNA), asam ribonukleat (RNA), dan sekuen protein. Multiple sequence alignment merupakan salah satu masalah yang paling mendasar pada bidang bioinformatika karena merupakan langkah awal untuk menganalisis sekuen biologis, pohon filogenetik, merancang aplikasi dalam pemodelan struktural, dan sekuen pencarian database (Kampke et al. 2001; Lakshmi et al. 2008 ).
penyejajaran beberapa sekuen (multiple sequence alignment). Algoritme ini secara umum diklasifikasikan ke dalam tiga kategori sesuai dengan properti mereka exact algorithms (Stoye 1997), progressive algorithms (Loytynoja dan Goldman 2005; Mount 2009) dan iterative algorithms (Lupyan et al. 2005).
Salah satu dari ketiga algoritma penjajaran tersebut yang memiliki keunggulan besar dari segi kesederhanaan serta cukup sensitif adalah Progressive algorithms (Lakshmi et al. 2008). Progressive alignment algorithms merupakan algoritma pendekatan untuk menemukan sekuen secara global pada penjajaran beberapa sekuen (multiple sequence alignment).
Pada penelitian ini, dilakukan penyejajaran sekuen DNA penyandi selulase dengan teknik multiple sequence alignment menggunakan metode progresif (progressive alignment algorithms) Selanjutnya, hasil dari penyejajaran tersebut akan dirancang untuk mendapatkan potensial primernya.
Perumusan Masalah
Bagaimana melakukan penyejajaran sekuen DNA penyandi selulase dengan teknik multiple sequence alignment menggunakan metode progresif untuk merancang potensial primer selulase.
Tujuan Penelitian
Tujuan penelitian ini adalah merancang potensial primer selulase dari sekuen DNA penyandi selulase yang akan dilakukan penyejajaran dengan teknik multiple sequence alignment menggunakan metode progresif.
Manfaat Penelitian
Adapun manfaat dari penelitian yang dilakukan, yaitu untuk mendapatkan potensial primer selulase yang dapat meningkatkan produktifitas enzim selulase.
Ruang Lingkup Penelitian
2
TINJAUAN PUSTAKA
Deoxyribonucleic Acid (DNA)
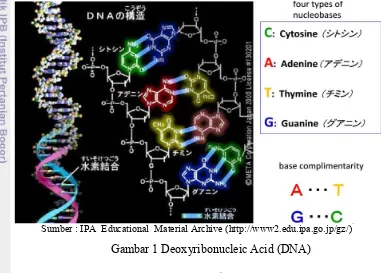
Asam deoksiribonukleat (deoxyribonucleic acid) atau biasa disebut DNA, adalah biomolekul yang berupa asam nukleat yang terdapat dalam inti sel atau nucleus, berfungsi untuk menyimpan informasi genetik pada suatu organisme (Jones dan Pevzner 2004). DNA berbentuk untai ganda (double helix) yang disatukan oleh ikatan hidrogen antara basa-basa di dalam kedua untai tersebut. Basa-basa tersebut adalah adenin (A), sitosin (C), guanin (G), dan timin (T). Adenin berikatan dengan timin, dan sitosin berikatan dengan guanin. DNA dapat melakukan replikasi DNA, di mana ketika replikasi DNA ini dilakukan, terbentuklah DNA baru yang memiliki informasi genetik yang serupa dengan induknya.
Sumber : IPA Educational Material Archive (http://www2.edu.ipa.go.jp/gz/)
Gambar 1 Deoxyribonucleic Acid (DNA)
Sequence Alignment
Ada beberapa tipe al dan global alignement. S pairwise alignment dan mu bahwa pada sekuen baik p sama, dengan relatif sedik daerah lokal yang kecil dar Pairwise alignment adalah alignment adalah memband 2001).
Gambar 3
M
Multiple sequence al yang menjajarkan beberapa yang optimal. Ada khusus
Gambar 2 Sequence alignment
ligment, berdasarkan pada posisi, yaitu local edangkan menurut banyaknya sekuen, dib ultiple alignment. Global alignment dapat di pada awal dan akhir sekuen terletak pada tem kit perbedaan yang nyata. Local alignment m
ri sekuen spesifik dengan tingkat kesamaan y h membandingkan antara dua sekuen, dan dingkan lebih dari dua sekuen (Baxevanis da
(a)
(b)
(a) Global alignment dan (b) Local alignmen
Multiple Sequence Alignment
lignment merupakan perluasan dari pairwise a sekuen terkait untuk mendapatkan pencocok keuntungan dari Multiple sequence alignm
alignment bagi dalam
iasumsikan mpat yang merupakan yang tinggi. n multiple an Ouellete
t
dapat menunjukkan informasi biologis lebih lanjut dari sepasang bijaksana keselarasan (pairwise alignment). Misalnya, memungkinkan identifikasi pola sekuen yang dijajarkan dan motif di seluruh keluarga sekuen. Multiple sequence alignment juga merupakan prasyarat untuk melaksanakan analisis filogenetik sekuen keluarga untuk mengetahui asal hubungan evolusioner yang ada, dapat diketahui juga adanya kesamaan homologi antar sekuen, dan mutasi yang dapat dilihat sebagai adanya perbedaan huruf pada satu kolom, dan adanya insertion atau deletion yang terlihat sebagai gap pada satu atau lebih sekuen pada penjajaran dan memprediksi protein sekunder dan struktur tersier. Multiple sequence alignment juga telah diaplikasikan dalam merancang degenerasi polymerase chain reaction (PCR) primer berdasarkan pada beberapa sekuen terkait (Lakshmi et al. 2008 ).
Gambar 4 Multiple sequence alignment
Selulase
Selulase adalah enzim terinduksi yang disintesis oleh mikroorganisme selama ditumbuhkan dalam medium selulosa (Lee dan Koo 2001). Selulase juga dapat diartikan sebagai nama bagi semua enzim yang memutuskan ikatan glikosidik beta-1,4 di dalam selulosa, sedodekstrin, selobiosa, dan turunan selulosa lainnya. Enzim selulase terdiri dari tiga komponen yaitu endo-1,4-β -D-glukanase, ekso-1,4-β-D- glukanase dan 1,4-β-D-glukosidase yang dapat dihasilkan oleh berbagai macam mikroorganisme. Selulase tidak dimiliki oleh manusia, karena itu manusia tidak dapat menguraikan selulosa. Tetapi hal ini dapat dilakukan oleh beberapa hewan seperti kambing, sapi, dan insekta seperti rayap karena dalam sistem pencernaannya mengandung bakteri dan protozoa yang menghasilkan enzim selulase yang akan menghidrolisis (mengurai) ikatan glikosidik beta-1,4. Oleh karena reaksi yang ditimbulkan oleh selulase saat mengurai selulosa adalah hidrolisis, maka selulase diklasifikasikan ke dalam jenis enzim hidrolase.
Ikatan glikosidik adalah ikatan kovalen yang terbentuk antara dua monosakarida melalui reaksi dehidrasi atau penghilangan gugus air. Ikatan antar glukosa ini dinamakan glikosidik beta-1,4 karena konfigurasi glukosa dalam selulosa semuanya berbentuk beta, yakni ketika glukosa membentuk cincin, gugus hidroksil yang terikat dengan karbon nomor 1 akan terkunci di bagian atas sumbu cincin, sedangkan angka “-1,4” diperoleh dari atom karbon yang menghubungkan antar unit glukosa monosakarida terletak pada atom karbon nomor 1 dan nomor 4.
GenBank
penemuan yang dengan sukarela memberikan hasil temuannya baik belum atau telah dipublikasikan (Baxevanis dan Ouellete 2001).
Terdapat tiga kelompok besar GenBank di dunia yang menjadi acuan dalam pencarian gen, yaitu National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/) di Amerika serikat. DNA Data Bank of Japan (DDBJ) (http://www.ddbj.nig.ac.jp) di Jepang dan European Bioinformatics Institut (EBI) (http://www.ebi.ac.uk) di Inggris.
3
METODE PENELITIAN
Metode penelitian terdiri dari beberapa tahapan, yaitu pengambilan data penelitian, multiple sequence alignment dengan metode progresif dan perancangan primer. Alur penelitian dapat dilihat pada gambar 6.
Gambar 6 Prosedur penelitian
PENGAMBILAN DATA
Data yang digunakan untuk penelitian ini adalah diambil dari GenBank NCBI (National Center for Biotechnology Information).
MULTIPLE SEQUENCE ALIGNMENT
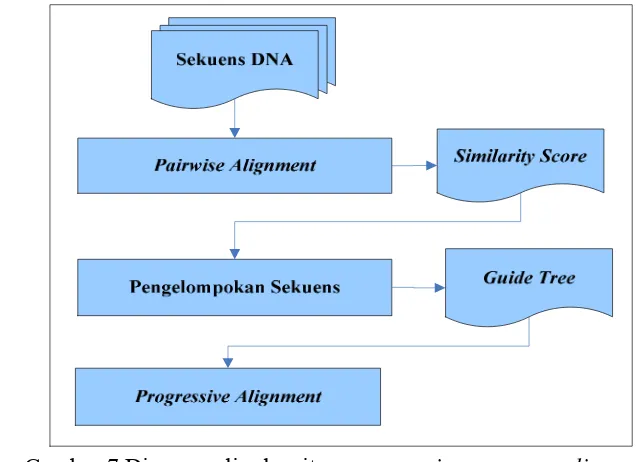
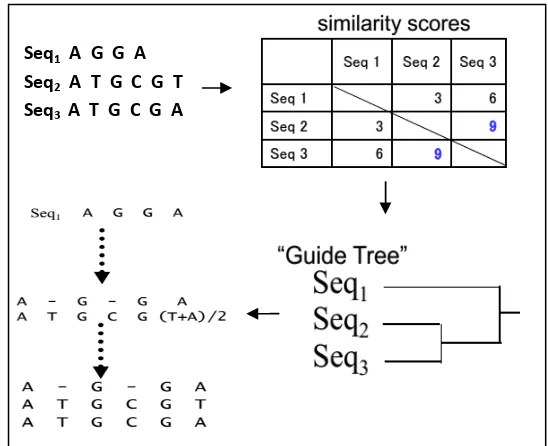
Pada tahap ini dilakukan penyejajaran data DNA dengan metode progresif, tujuannya untuk mendapatkan organisme dan daerah yang memiliki tingkat similaritas yang tinggi (conserved region). Penyejajaran dengan metode progresif dilakukan dalam tiga tahap, yaitu pairwise alignment, pengelompokan sekuen dan progressive alignment. Metode ini merupakan algoritma pendekatan untuk menemukan sekuen secara global pada penyejajaran beberapa sekuen (Gusfield 1997; Sung 2009).
Conserved Region
mulai
Pengambilan data
Multiple sequence alignment dengan metode progresif
Perancangan Primer
Gambar 7 Diagram alir algoritme progressive sequence alignment
1. Pairwise alignment
Proses yang pertama pada penyejajaran progressive sequence alignment adalah membandingkan antara dua sekuen (Pairwise alignment) menggunakan algoritma Needleman-Wunsch. Algoritma needleman & wunsch merupakan metode untuk pencarian sekuen optimal secara global dari 2 sekuen. Algoritma ini terdiri dari memanipulasi matriks, bernama
, . Matriks ini pertama kali dibangun dengan cara yang biasa, kolom
dan baris, ini dipenuhi sebagai berikut (Gautham, 2006) :
, , , (1)
, , ,
Dengan m dan n adalah panjang dari dua sekuen masing-masing dengan kompleksitas waktu komputasi sebesar O (mn). Berikutnya mengatasi sel-sel lainnya dimulai dari pojok kiri atas dari matriks (yaitu sel-sel ( ,
)) dan bergerak ke arah sudut kanan bawah (sel ( , ), mengisi kotak rekursif menurut aturan berikut:
F i,j =Max
F i-1,j-1 + S(Xi ,Yj)
, ,
(2)
dengan , adalah skor match / mismatch, w adalah gap penalty konstan ( ), dan , adalah akumulasi nilai sampai , . Dalam contoh menghitung nilai Matrix (1,1).
F (1,1) = Max{F (0,0) -1, F(0,1) -2, F(1,0) -2} = Max{-1,-4,-4}
Demikian pula, untuk menghitung nilai seluruh matriks.
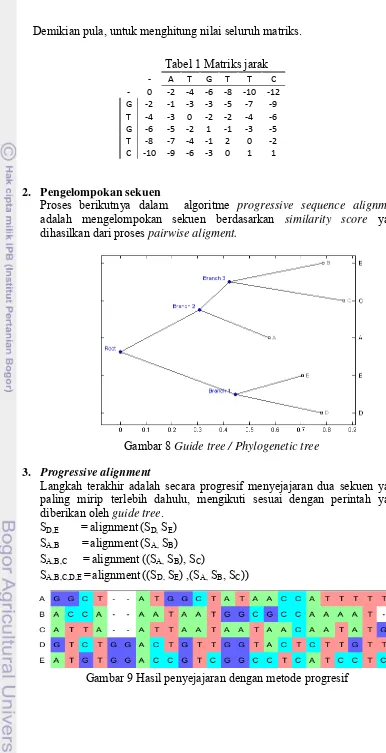
Tabel 1 Matriks jarak
‐ A T G T T C
‐ 0 ‐2 ‐4 ‐6 ‐8 ‐10 ‐12 G ‐2 ‐1 ‐3 ‐3 ‐5 ‐7 ‐9 T ‐4 ‐3 0 ‐2 ‐2 ‐4 ‐6 G ‐6 ‐5 ‐2 1 ‐1 ‐3 ‐5 T ‐8 ‐7 ‐4 ‐1 2 0 ‐2 C ‐10 ‐9 ‐6 ‐3 0 1 1
2. Pengelompokan sekuen
Proses berikutnya dalam algoritme progressive sequence alignment adalah mengelompokan sekuen berdasarkan similarity score yang dihasilkan dari proses pairwise aligment.
Gambar 8 Guide tree / Phylogenetic tree
3. Progressive alignment
Langkah terakhir adalah secara progresif menyejajaran dua sekuen yang paling mirip terlebih dahulu, mengikuti sesuai dengan perintah yang diberikan oleh guide tree.
SD,E =alignment(SD, SE)
SA,B =alignment(SA, SB)
SA,B,C =alignment((SA, SB), SC)
SA,B,C,D,E =alignment((SD, SE) ,(SA, SB, SC))
Gambar 10 Prose
Perancangan primer melakukan amplifikasi ge berdasarkan hasil alignme conserved region yang tin nukleat yang berfungsi seb diartikan sebagai 'oligonu digunakan untuk menginis yang digunakan dalam amp dan reverse. Forward prim sementara reverse primer (Abd-Elsalam 2003).
Gambar 11
Dalam melakukan pe sebagai berikut:
A.Panjang primer
Umumnya panjang prim kurang dari 18 basa ak untuk panjang primer le primer secara bermakna
A C G T C G A
es algoritme progressive sequence alignment
Perancangan Primer
r merupakan faktor penting dalam ke en dengan memperhitungkan pemilihan y ent dari kedekatan sekuen yang diinginka nggi (Abd-Elsalam 2003). Primer adalah u bagai titik awal untuk sintesis DNA. Primer j cleotide' atau rangkaian pendek DNA atau R siasi (memulai) duplikasi DNA (Djamil 200
plifikasi umumnya terdiri dari dua jenis, yak mer bergerak dengan arah 5′ –> 3′ untai DNA
bergerak dengan arah 3′ –> 5′ untai DNA
(a)
(b)
1 (a) Forward primer dan (b) Reverse primer
erancangan primer harus dipenuhi parameter
mer berkisar antara 18 – 30 basa. Primer denga kan menjadikan spesifisitas primer rendah. S ebih dari 30 basa tidak akan meningkatkan s
.
t
eberhasilan yang layak
an dengan untai asam juga dapat RNA yang 05). Primer kni forward
A template, A template
r
-parameter
B.Suhu leleh (Melting temperature / Tm)
Suhu leleh (melting temperatur / Tm) adalah temperatur di mana 50 % untai ganda DNA terpisah. Suhu leleh yang optimal untuk primer di kisaran 52-58oC, umumnya menghasilkan hasil yang lebih baik dibandingkan primer dengan suhu leleh yang lebih rendah (Dieffenbach et al. 1995). Secara teoritis Tm primer dapat dihitung dengan menggunakan rumus [2(A+T) + 4(C+G)] (Wallace et al. 1979).
C.Kandungan GC
Kandungan GC(%) merupakan karakteristik penting dari DNA dan memberikan informasi tentang kekuatan anil. Primer harus memiliki GC konten antara 45 dan 60% (Dieffenbach et al. 1995).
D.Sekuen Akhir 3’ (3' GC Clamp)
Sekuen nukleotida pada akhir 3’ sebaiknya G atau C. Nukleotida A atau T lebih toleran terhadap mismatch dari pada G atau C, dengan demikian akan dapat menurunkan spesifisitas primer (Dieffenbach et al. 1995).
E. Hairpins
Primer sebaiknya menghindari terbentuknya loop (Dieffenbach et al. 1995). F. Cross - Dimer
Primer dapat berikatan dengan primer pasangannya (forward dan reverse) (Dieffenbach et al. 1995).
G.Pengulangan Nukleotida
Primer sebaiknya tidak memiliki urutan pengulangan dari 2 basa dan maksimum pengulangan 2 basa sebanyak 4 kali masih dapat ditoleransi, misalnya GCGCGCGC (Dieffenbach et al. 1995).
Peralatan Penelitian
Alat yang digunakan dalam penelitian ini dibagi dalam perangkat keras dan perangkat lunak, sebagai berikut :
a. Perangkat keras :
• Processor : Pentium(R) Dual-Core CPU T4500 @ 2.30 GHz 2.30 GHz • Memory : RAM 2 GB
• Harddisk : 320 GB b. Perangkat lunak :
• Sistem operasi Windows 7 Starter 32-bit operating system • Notepad
Digunakan untuk melakukan proses pembuatan fasta file (.Fasta) • Matlab 7.11.0 (R2010b)
4
HASIL DAN PEMBAHASAN
Data yang digunakan untuk penelitian ini adalah berupa DNA penyandi selulase yang diambil dari GenBank NCBI (National Center for Biotechnology Information).
Tabel 2 Sekuen gen penyandi selulase
MULTIPLE SEQUENCE ALIGNMENT
Progressive Sequence Alignment
Data yang digunakan diunduh pada basis data NCBI dan disimpan dalam format (.fasta) menggunakan Notepad. Kemudian data tersebut dijajarkan secara Progressive Sequence Alignment menggunakan Matlab, dengan tujuan untuk mengetahui tingkat similaritas dan homology antara sekuen DNA penyandi selulase. Progressive Sequence Alignment terdiri atas tiga tahap (Liu et al. 2009).
Gambar 12 Tiga tahap progressive sequence alignment : (1) pairwise alignment, (2) pengelompokan sekuen dan (3) progressive alignment (Liu et al. 2009)
a. Pairwise Alignment
Tahap pertama pada Progressive Sequence Alignment adalah membandingkan antara dua sekuen (pairwise alignment) gen penyandi selulase menggunakan algoritme Needleman-Wunsch. Algoritme Needleman-Wunsch merupakan metode untuk pencarian sekuen optimal secara global dari 2 sekuen. Pseudo-code dari algoritme Needleman-Wunsch ditampilkan pada gambar 13.
TIPE GEN ORGANISME NOMORAKSESI PANJANG GEN
Cellulase
Solanum Lycopersicum NM_001247953 1895 bp
Triticum Aestivum AY091512 2196 bp Arabidopsis Thaliana NM_124350 2360 bp
Solanum Lycopersicum NM_001247933 1717 bp Solanum Lycopersicum NM_001247938 1780 bp
1. Input: Dua sekuen X dan Y
2. Output: Penyejajaran yang optimal (best alignment)dan skor
α
3. Initialization:
4. Set F(i,0) := −i·w for all i = 0,1,2, . . . ,n
Gambar 13 Pseudo-code algoritme Needleman-Wunsch
Langkah pertama dalam algoritme ini (pseudo-code no 4 dan 5) adalah membuat matriks yang disebut similarity matrix F (i, j). Matriks dibangun dengan cara yang biasa, dengan kolom dan baris , m dan n adalah panjang dari dua sekuen masing-masing dengan kompleksitas waktu komputasi sebesar O (mn). Langkah selanjutnya adalah mengatasi sel-sel lainnya dimulai dari pojok kiri atas dari matriks (yaitu sel ( , )) dan bergerak ke arah sudut kanan bawah (sel ( , ), mengisi kotak rekursif menurut aturan (pseudo-code no 6, 7, 8). , adalah skor match / mismatch, w adalah gap penalty konstan ( ), dan , adalah akumulasi nilai sampai , . Langkah terakhir dari algoritme ini (pseudo-code no 9 s/d 16 ) adalah membangun jejak kembali (traceback), dimulai dari titik di bagian kiri atas dari matriks, dimana sel yang memberikan nilai yang sesuai dengan jalur terbaik yang mungkin akan hadir, yaitu sel dengan skor tertinggi. Prosedur traceback berfungsi untuk mengidentifikasi penyejajaran yang optimal (best alignment).
Dari hasil penyejajaran dua sekuen (pairwise alignment) gen penyandi selulase menggunakan algoritme Needleman-Wunsch diperoleh similarity score (Tabel 3). Similarity score tersebut digunakan untuk tahap selanjutnya yaitu mengelompokan sekuen.
Tabel 3 Similarity scores
1. Variabel:
2. T => node daun 3. d => jarak matriks
4. D => normalisasi matrik jarak 5. r => Array divergensi
6. L => node tambahan
7. Inisialisasi:
8. L = S.
9. Iterasi:
10. Pilih sepasang node i , j yang jarak normalisasi Di j adalah minimum, dengan Di j = di j – (ri + rj), dan ri
adalah perbedaan node i, dengan ∑
11. Tentukan node k baru dan menetapkan dks = ½(dis + djs – dij), untuk semua s di L.
12. Tambahkan k ke S dengan jarak sisi dik = ½ (dij + ri - rj), djk = dij – dik, menghubungkan k ke i dan j, masing-masing. 13. Hapus i dan j dari L dan menambahkan k.
14. Terminasi:
15. Ketika L dibentuk oleh dua daun i dan j, tambahkan sisi yang tertunda antara i dan j dengan jarak dij
S1 = Solanum Lycopersicum NM_001247953
S2 = Triticum Aestivum AY091512
S3 = Arabidopsis Thaliana NM_124350
S4 = Solanum Lycopersicum NM_001247933
S5 = Solanum Lycopersicum NM_001247938
b. Pengelompokan Sekuen
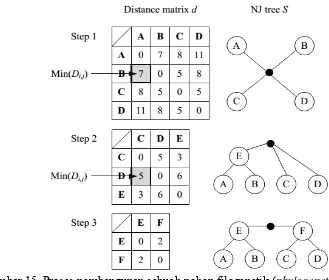
Tahap kedua pada Progressive Sequence Alignment adalah mengelompokan sekuen gen penyandi selulase menggunakan metode Neighbor-Joining. Metode Neighbor-Joining (NJ) didasarkan pada jarak matriks dan secara luas digunakan oleh ahli bioteknologi dan biologi molekuler karena efisien. Metode Neighbor-Joining dimulai dengan pohon berbentuk bintang. Dalam setiap langkah, pasangan node yang paling dekat satu sama lain dipilih dan terhubung melalui simpul baru. Kemudian, jarak dari ini node baru ke seluruh node di pohon dihitung. Algoritma berakhir ketika hanya ada dua node yang tidak terhubung. Berikut adalah pseudo-code dari metode Neighbor-Joining.
Gambar 14 Pseudo-code metode Neighbor-Joining
Gambar 15 Proses pembangunan sebuah pohon filogenetik (phylogenetic tree)
menggunakan metode Neighbor-Joining
Dari hasil pengelompokan sekuen gen penyandi selulase menggunakan metode Neighbor-Joining diperoleh phylogenetic tree (Gambar 16). Phylogenetic tree memainkan peran penting dalam beberapa aplikasi yang relevan dalam bioinformatika, seperti multiple sequence alignment.
c. Progressive Alignment
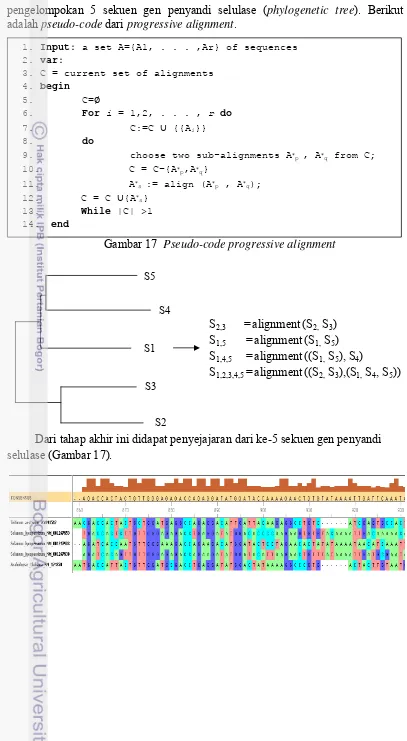
Tahap terahir pada Progressive Sequence Alignment yaitu melakukan penyejajaran dua sekuen yang paling mirip terlebih dahulu yang didapat dari hasil
1. Input: a set A={A1, . . . ,Ar} of sequences 2. var:
3. C = current set of alignments
4. begin
5. C=
6. For i = 1,2, . . . , r do
7. C:=C {{Ai}}
8. do
9. choose two sub-alignments Ap , Aq from C;
10. C = C−{Ap,Aq}
11. As := align (Ap , Aq);
12. C = C {As}
13. While |C| >1
14. end
pengelompokan 5 sekuen gen penyandi selulase (phylogenetic tree). Berikut adalah pseudo-code dari progressive alignment.
Gambar 17 Pseudo-code progressive alignment
Dari tahap akhir ini didapat penyejajaran dari ke-5 sekuen gen penyandi selulase (Gambar 17).
S5
S2,3 =alignment(S2, S3)
S1,5 =alignment(S1, S5)
S1,4,5 =alignment((S1, S5), S4)
S1,2,3,4,5 =alignment((S2, S3),(S1, S4, S5)) S3
S1 S4
Gambar 18 Hasil penyejajaran dari 5 gen penyandi selulase
Dari hasil analisa penyejajaran sekuen gen penyandi selulase, algoritme Progressive Sequence Alignment dapat menghasilkan penyejajaran yang optimal, karena dijajarkan berdasarkan kemiripan dari dua sekuen tetapi dibutuhkan kompleksitas waktu komputasi yang tinggi, terutama pada tahap pairwise alignment (Gambar 19). Dapat diketahui bahwa spesies yang memiliki nilai atau kesamaan tertinggi adalah Solanum lycopersicum NM-001247953, dibandingkan spesies yang lain. Penyejajaran secara multiple sequence alignment menghasilkan daerah yang memiliki similaritas yang tinggi (conserved region), hal itu terlihat pada histogram (Gambar 18). Semakin tinggi histogram berarti semakin tinggi juga tingkat similaritasnya. Conserved region pada basa nukleotida yang mempunyai similaritas dan homologi tinggi, maka daerah tersebut dijadikan template untuk digunakan sebagai primer spesifik. Dalam penelitian ini, hasilnya didapatkan 3 conserved region ditampilkan pada Tabel 4.
Gambar 19 Waktu yang dibutuhkan oleh progressive sequence alignment
Tabel 4 Sekuen conserved region
PERANCANGAN PRIMER SELULASE
Dalam merancang potensial primer selulase digunakan sekuen Solanum lycopersicum NM-001247953 dari daerah yang memiliki similaritas yang tinggi (conserved region) sebagai template. Parameter untuk merancang potensial primer selulase yang digunakan dalam penelitian ini adalah sebagai berikut :
1. Panjang primer sebesar 20 bp (base pair)
2. Melting temperatur (Tm) sebesar min=50oC dan max=60oC 3. Kandungan GC sebesar min=45% dan max=55%
4. Tidak terbentuk hairpin 5. Tidak terbentuk self-dimer
6. Tidak terjadi pengulangan nukleotida 7. 3’ GC clamp
Berdasarkan hasil perancangannya diperoleh sebanyak 46 pasang potensial primer selulase dari ketiga conserved region, ditampilkan pada Gambar 20, 21 dan 22.
REGION POSISI SEKUEN
1 862-1175
GACCACTCTTGTTGGGAGAGACCTGAGGATATGGACACCCCAA GAAGTGTGTACAAAATTGACAAAAACACTCCTGGGACTGAAGT TGCTGCTGAAACTGCTGCTGCTCTCGCTGCTGCTTCCTTAGTCTT TAGGAAATGCAACCCATCTTACTCCAAGATACTAATCAAAAGG GCCATCAGGGTGTTTGCCTTTGCTGATAAGTATAGAGGTTCATA
CAGCAATGGTCTGAGAAAAGTAGTGTGCCCATACTACTGCTCA GTTTCGGGATATGAGGATGAGCTGTTGTGGGGTGCTGCTTGGTT
ACATAGAGC
2 1567-1765
TGTGGTGGAGTTGTTATTACACCAAAGAGGCTTCGAAATGTAG CCAAAAAACAGGTGGACTATTTGTTAGGAGACAATCCACTAA AAATGTCATACATGGTGGGATATGGAGCAAGGTATCCACAGA GGATTCATCACAGGGGATCCTCATTACCCTCAGTCGCGAACCA
TCCAGCAAAGATACAATGCAGGGATGGTT
3 1790-1915
Berdasarkan Gambar 20 diketahui bahwa lokasi potensial primer yang bagus dari conserved region ke-1 setelah diseleksi adalah berada pada posisi antara 150 sampai 210 dari panjang sekuen sebesar 313 bp. Rentang amplifikasi pemilihan primer adalah 5′ –> 3′ = 0 – 169 dan 3′ –> 5′ = 170 – 313. Hasil perancangan dari conserved region 1 didapat sebanyak 28 pasang primer ditampilkan pada Tabel 5.
Gambar 20 Hasil perancangan potensial primer dari
sekuen conserved region ke-1
Gambar 21 Hasil perancangan potensial primer
Berdasarkan Gambar 21 diketahui bahwa lokasi potensial primer yang bagus dari conserved region ke-2 setelah diseleksi adalah berada pada posisi antara 50 sampai 130 dari panjang sekuen sebesar 198 bp. Rentang amplifikasi pemilihan primer adalah 5′ –> 3′ = 0 – 150 dan 3′ –> 5′ = 160 – 198. Hasil perancangan dari conserved region 2 didapat sebanyak 13 pasang primer ditampilkan pada Tabel 6.
Berdasarkan Gambar 22 diketahui bahwa lokasi potensial primer yang bagus dari conserved region ke-3 setelah diseleksi adalah berada pada posisi antara 60 sampai 90 dari panjang sekuen sebesar 125 bp. Rentang amplifikasi pemilihan primer adalah 5′ –> 3′ = 0 – 120 dan 3′ –> 5′ = 80 – 125. Hasil perancangan dari conserved region 3 didapat sebanyak 5 pasang primer ditampilkan pada Tabel 7.
Tabel 7 Primer dari region ke-3
Gambar 22 Hasil perancangan potensial primer
dari sekuen conserved region ke-3
5
SIMPULAN
1. Algoritme progressive alignment dapat menghasilkan penyejajaran yang optimal, karena dijajarkan berdasarkan kemiripan dari dua sekuen tetapi membutuhkan kompleksitas waktu komputasi yang tinggi dan memori yang besar.
2. Kompleksitas waktu komputasi yang dibutuhkan algoritme Needleman-Wunsch dalam melakukan pairwise alignment adalah O(mn).
3. Multiple sequence alignment (MSA) menggunakan algoritme progressive alignment dapat menghasilkan conserved region dari gen penyandi selulase yang digunakan pada perancangan primer.
DAFTAR PUSTAKA
Abd-Elsalam KA. 2003. Bioinformatics tools and guideline for PCR primer design. Afrikan journal of biotechnology. 2 (5): 91-95.
Anindyawati T. 2009. Prospek enzim dan limbah lignoselulosa untuk produksi bioetanol. BS. 44(1): 49-56.
Baxevanis AD, Ouellette BFF. 2001. Bioinformatics A Practical Guide to the Analyses of gene and Proteins. A John Wiley & Sons, Inc., Publication. Dieffenbach CW, Lowe TMJ, Dveksler GS. 1995. General Concepts for PCR
Primer Design. In: PCR Primer, A Laboratory Manual, Dieffenbach CW, Dveksler GS Ed. Cold Spring Harbor Laboratory Press. New York. 133-155.
Djamil. 2005. Studi in silico produksi Hyaluronidase lintah (Hirudo medicinalis) secara rekayasa genetika[Tesis]. Depok (ID): Universitas Indonesia.
Edgar RC, Batzoglou S. 2006. Multiple sequence alignment. Science Direct. 16. Fredslund J, Schauser L, Madsen LH, Sandal N, Stougaard J. 2005. PriFi: using a
multiple alignment of related sequences to find primers for amplification of homologs. Nucleic acids research. 33. doi:10.1093/nar/gki425.
Gautham N. 2006. Bioinformatics Databases and Algorithms. Alpha science Internasional Ltd. Oxford, UK.
Gusfield D. 1997. Algorithms on String, Trees, and Sequences. Computer Science and Computational Biology. New York: Cambridge University Press.
Jones NC, Pevzner PA. 2004. An Introduction to Bioinformatics Algorithm. Series on Computational Molecular Biology. Massachusetts (US) : MIT Press. Joshi B, Bhatt RM, Sharma D, Joshi J, Malla R, Lakshmaiah, Sreerama. 2011.
Lignocellulosic ethanol production: Current practices and recent developments. Biotechnology and Molecular Biology Review. 6(8):172-182 Kampke T, Kieninger M, Mecklenburg M. 2001. Efficient primer design
algorithms. Bioinformatics. 17(3).
Kuhad RC, Gupta R, Singh A. 2011. Microbial Cellulases and Their Industrial Applications. Enzyme Research. doi:10.4061/2011/280696.
Lakshmi PV, Rao AA, Sridhar GR. 2008. An Efficient Progressive Alignment Algorithm for Multiple Sequence Alignment. International Journal of Computer Science and Network Security. 8(10).
Lee SM, Koo YM. 2001. Pilot-scale production of cellulose using Trichoderma reesei Rut C-30 in fed-batch mode. Journal of Microbiology and Biotechnology.11: 229-233.
Liu Y, Schmidt B, Maskell DL. 2009. Parallel Reconstruction of Neighbor-Joining Trees for Large Multiple Sequence Alignments using CUDA. IEEE Loytynoja A, Goldman N. 2005. An algorithm for progressive multiple alignment
of sequences with insertions. Proceeding of the National Academy of Sciences. 102(30):10557–10562. doi:10.1073/pnas.0409137102.
Lupyan D, Macias AL, Ortiz AR. 2005. A new progressive-iterative algorithm for multiple structure alignment. Bioinformatics. 21(15).
Mojsov K. 2012. Microbial cellulases and their applications in textile processing. International Journal of Marketing and Technology. 2(11):12-29.
Sim TS, Oh JCS. 1993. Application of Trichoderma reesei Cellulases for Degradation of Lignocellulosic Compounds. Proceeding of Mie Bioforum. Genetic, Biochemistry and Ecology of Lignocellulose Degradation. Uni Publishers Co. Ltd. 477- 481.
Stoye J, Moulton V, Dress AW. 1997. DCA: An efficient iplementation of the divide and conquer approach to simultaneous Multiple sequence Alignment. Computer Applications in the Biosciences. 13 (6).
Sung WK. 2009. Algorithms in Bioinformatics : A Practical Introduction. Mathematical and Computational Biology Series. US: CRC Press.
Sukumaran RK, Singhania RR, Pandey A. 2005. Microbial cellulases – Production, applications and challenges. Journal of Scientific & Industrial Research. 64:832-844.
29
Lampiran 2 Hasil Multiple Sequence Alignment
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
RIWAYAT HIDUP
M.Bahrul Ulum dilahirkan di Tangerang, 6 April 1988. Penulis merupakan anak keenam dari tujuh bersaudara, dari pasangan H. Syukur Dani dan Hj. Suamah. Tahun 2009, penulis lulus sarjana pada program studi Teknik Informatika Institut Sains dan Teknologi Al-Kamal Jakarta. Penulis melanjutkan jenjang magister pada tahun 2011 di program studi Ilmu Komputer, Fakultas Matematika dan Ilmu Pengetahuan Alam Institut Pertanian Bogor (IPB). Penulis bekerja sebagai staf pengajar di Institut Sains dan Teknologi Al-Kamal Jakarta dari tahun 2011 sampai sekarang.