PENGEMBANGAN HIDDEN SEMI MARKOV MODEL
DENGAN DISTRIBUSI DURASI STATE EMPIRIS UNTUK
PREDIKSI STRUKTUR SEKUNDER PROTEIN
TOTO HARYANTO
SEKOLAH PASCASARJANA
INSTITUT PERTANIAN BOGOR
PERNYATAAN MENGENAI TESIS DAN SUMBER
INFORMASI
Dengan ini saya menyatakan bahwa tesis : Pengembangan Hidden Semi
Markov Model dengan Distribusi Durasi State Empiris untuk Prediksi Struktur Protein Sekunder adalah karya saya dengan arahan dari komisi pembimbing dan
belum diajukan dalam bentuk apapun kepada perguruan tinggi manapun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir tesis ini.
Bogor, Januari 2011
Toto Haryanto
ABSTRACT
HARYANTO, TOTO. The Development of Hidden Semi Markov Model with Empirical State Duration Distribution for Protein Secondary Structure Predictions. Under direction of AGUS BUONO and ANTO SATRIYO NUGROHO
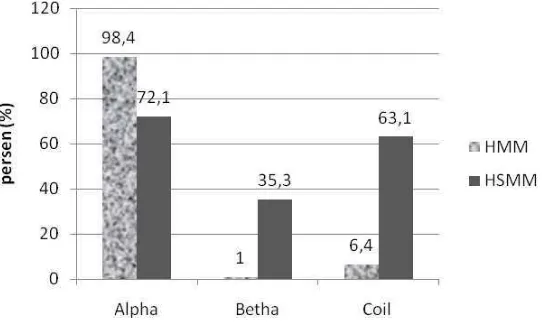
This research aimed to develop Hidden Semi Markov Model (HSMM) with long-duration distribution of state empirically and theoretically to predict protein secondary structure. Data used in this study are subset data taken from database of secondary protein structure in DSSP program with three secondary protein structures of alpha-helix (H), betha-sheet (B), and coil (C). Accuracy of the predicted protein structure with HSMM is compared with Hidden Markov Model (HMM) standard. The results showed that the HSMM generally provides a prediction accuracy 72. 1% , 35.3% and 63,1% for H, B and C respectively. The accuracy of standard HMM is 98.4% for (B), 1% for (B) and 6.4% for (C). The use of empirical state duration distribution gives better accuracy on average compared a theoretical state duration distribution. Use of the empirical state duration distribution provides accuracy 71.2% for (H), 37.1% for (B) and 64.0% for (C). Use of theoretical state duration distribution provides accuracy 75% for (H), 30.2 % for (B) and 60.6% for (C). Less accuracy in betha-sheet prediction with HSMM was caused by high of the dispute between distribution in training data and testing data.
RINGKASAN
HARYANTO, TOTO. Pengembangan Hidden Semi Markov Model dengan Distribusi Durasi State Empiris untuk Prediksi Struktur Sekunder Protein. Dibimbing oleh AGUS BUONO dan ANTO SATRIYO NUGROHO.
Protein merupakan unsur yang sangat esensial dalam makhluk hidup. Secara hierarki, struktur protein dapat dikelompokkan menjadi protein primer, protein sekunder dan protein tersier. Struktur primer berupa untaian asam amino pembentuk protein, sedangkan struktur sekunder adalah segmentasi asam amino berupa alpha-helix (H), betha-sheet (B) dan coil (C). Adapun struktur protein tersier adalah struktur sekunder yang telah mengalami proses pelipatan atau
folding. Fungsi protein akan terlihat, apabila telah membentuk struktur tersiernya. Pembentukan struktur protein dari primer sampai tersier dapat dilakukan dengan menggunakan X-Ray Crystallography dan Nuclear Magnetic Resonance (NMR) spectroscopy. Meskipun kedua teknik tersebut mampu mendapatkan struktur baru, akan tetapi membutuhkan biaya yang relatif mahal. Oleh karena itu, berbagai teknik komputasi banyak digunakan untuk menentukan struktur suatu protein terutama struktur sekunder.
Di sisi lain, karakteristik dari sekuens asam amino sebagai struktur protein primer sangat cocok dengan tipe data yang digunakan pada pembuatan model dengan menggunakan Hidden Markov (HMM) untuk memprediksi struktur sekunder suatu protein. Pada prediksi struktur sekunder protein dengan Hidden Markov Model (HMM) informasi state hanya diperoleh dari satu observasi yang dalam hal ini adalah asam amino. Padahal, pada kenyataannya bisa saja suatu
state dapat menyimpan informasi lebih dari satu observasi atau bahkan suatu
sekuens observasi sebagai informasi. Oleh karena itu, dalam prediksi struktur sekunder protein terdapat pendekatan yang dikenal dengan Hidden Semi Markov Model (HSMM). Pada HSMM, satu state dapat membangkitkan suatu sekuens observasi. Ciri dari HSMM yang terpenting adalah adanya durasi state sebagai informasi dalam proses pelatihan dalam membuat model.
Pada penelitian kali ini, prediksi struktur sekunder protein diimplementasikan dengan algoritme yang telah memiliki kompleksitas
O((MD+M2)T) dan menggunakan beberapa jenis distribusi durasi maksimum
yang akan diperoleh secara empiris dari data latih yang diambil maupun dengan menggunakan distribusi teoritis. Hasil akurasi yang didapatkan akan dibandingkan dengan hasil akurasi HMM standar. Di samping itu, penelitian ini akan mengkaji pengaruh selisih distribusi durasi terhadap nilai akurasi yang dihasilkan pada model Hidden Semi Markov Model (HSMM).
Data untuk proses pelatihan terdiri atas 42556 residu asam amino, sedangkan data pengujian sebanyak 14057 residu. Dari data latih tersebut dibuat beberapa model baik dengan model HMM maupun model HSMM dengan delapan distribusi durasi yang digunakan. Enam dari delapan menggunakan distribusi empiris dan dua menggunakan distribusi teoritis dengan fungsi kepekatan peluang. Selanjutnya model yang telah didapatkan tersebut divalidasi dengan melakukan pengujian.
Pengujian HSMM dilakukan dengan 8 skenario pengujian dengan menggunakan 100% , 90%, 75% dan 50% panjang durasi. Pengujian HSMM juga dilakukan dengan menggunakan durasi yang ditetapkan yaitu 21 dan 15. Pada HSMM dengan menggunakan distribusi teoritis yaitu distribusi dengan fungsi kepekatan peluang eksponensial negatif dan fungsi kepekatan peluang dengan distribusi normal.
Hasil penelitian menunjukkan bahwa secara umum prediksi dengan menggunakan model HSMM memberikan akurasi yang lebih tinggi bila dibandingkan dengan akurasi prediksi menggunakan HMM standar. Ini terlihat dari nilai akurasi prediksi untuk HSMM sebesar 56,9% sedangkan pada prediksi struktur sekunder protein menggunakan HMM menghasilkan akurasi sebesar 35,3%. Penggunaan distribusi durasi secara empiris mampu merepresentasikan kondisi data yang sebenarnya sehingga dalam prediksi akurasi struktur sekunder protein menghasilkan akurasi sebesar 57,4% sementara penggunaan distribusi teoritis menghasilkan akurasi sebesar 55,2%. Secara detail akurasi masing-masing struktur protein dengan distribusi empiris adalah 71,2% , 37,1% dan 64,0% untuk alpha-helix (H), betha-sheet (B) dan coil (C). Adapun prediksi dengan distribusi teoritis menghasilkan akurasi 75,0% untuk struktur alpha-helix(H), 30,2% untuk betha-sheet(B) dan 60,6% untuk coil (C). Pada hasil prediksi, struktur betha-sheet memberikan nilai akurasi yang paling rendah. Setelah dilakukan perbandingan selisih distribusi data latih dan data uji diperoleh bahwa struktur betha-sheet memiliki selisih yang paling besar dibandingkan dengan selisih distribusi alpha-helix dan coil. Hal ini juga didukung oleh besarnya perbedaan distribusi empiris dan teoritis yang dimiliki oleh struktur betha-sheet.
Prediksi struktur sekunder protein dengan Hidden Markov Model dan Hidden Semi Markov Model (HSMM) belum dapat mengakomodasi kondisi data yang tidak seimbang. Dengan demikian, jumlah data uji yang paling sedikit akan menyebabkan akurasi yang rendah. Oleh karena itu, penelitan berikutnya sebaiknya dilakukan metode pada HSMM yang dapat menanggulangi ketidakseimbangan data ini.
©
Hak Cipta Milik IPB, tahun 2011 Hak Cipta dilindungi Undang-UndangPENGEMBANGAN HIDDEN SEMI MARKOV MODEL
DENGAN DISTRIBUSI DURASI STATE EMPIRIS UNTUK
PREDIKSI STRUKTUR SEKUNDER PROTEIN
TOTO HARYANTO
Tesis
sebagai salah satu syarat untuk memperoleh gelar Magister Sains pada
Program Studi Ilmu Komputer
SEKOLAH PASCASARJANA
INSTITUT PERTANIAN BOGOR
Judul Penelitian : Pengembangan Hidden Semi Markov Model dengan Distribusi Durasi State Empiris untuk Prediksi Struktur Sekunder Protein
Nama : Toto Haryanto
NIM : G651080091
Disetujui Komisi Pembimbing
Dr. Ir. Agus Buono, M.Si, M.Kom. Dr. Eng Anto Satriyo Nugroho
Ketua Anggota
Diketahui
Ketua Program Studi Dekan Sekolah Pascasarjana
Ilmu Komputer
Dr. Ir. Agus Buono, M.Si, M.Kom Prof. Dr. Ir. Khairil A. Notodiputro, M.S.
PRAKATA
Alhamdulillah segala puji penulis panjatkan ke hadirat Allah Subhanahu wa
ta‘ala atas segala rahmat, kasih sayang dan cinta-Nya sehingga tesis ini berhasil diselesaikan. Shalawat dan salam semoga senantiasa tercurah pada teladan kita Nabi Muhammad Shallallahu alaihi wasallam, kerabat, sahabat dan umatnya. Judul yang dipilih dalam karya ilmiah ini ialah Pengembangan Hidden Semi Markov Model dengan Distribusi Durasi State Empiris untuk Prediksi Struktur Sekunder Protein.
Terima kasih penulis ucapkan kepada Bapak Dr. Ir. Agus Buono, M.Si, M.Kom dan Bapak Dr. Eng Anto Satriyo Nugroho selaku pembimbing atas arahan dan masukannya. Ucapan terima kasih juga penulis sampaikan kepada Bapak Dr. Ir. Iman Rusmana, M.Si selaku penguji pada sidang tesis kali ini. Penulis mengucapkan terima kasih kepada Departemen Ilmu Komputer IPB yang telah memberikan beasiswa selama melaksanakan studi S2. Penulis juga mengucapkan terima kasih kepada dosen dan seluruh staf Departemen Ilmu Komputer IPB atas dukungan dan bantuan yang telah diberikansertarekan-rekan S2 (Pak Aziz, Pak Defiana, Pak Altien, Pak Aristoteles dan Pak Rossy) atas kebersamaannya selama penulis menyelesaikan studi.
Di samping itu, penulis mengucapkan terima kasih kepada keluarga khususnya istri dan anakku tercinta atas kesabaran dan kasih sayang selama penulis melakukan studi S2. Ucapan terima kasih juga penulis ucapkan kepada Ibunda tercinta atas Do’a dan segala perhatiannya. Penulis juga mengucapkan terima kasih kepada semua pihak yang telah membantu proses penelitian ini. Semoga karya ilmiah ini bermanfaat.
Bogor, Januari 2011
RIWAYAT HIDUP
Penulis lahir di Kota Mangga, Indramayu pada tanggal 17 November 1982, anak dari pasangan Caca Goembira (alm) dan Iyus Rusyati. Penulis merupakan anak kedua dari empat bersaudara.
Tahun 2001 penulis lulus dari SMU Negeri 1 Cirebon dan pada tahun yang sama melanjutkan program sarjana ke Institut Pertanian Bogor (IPB), Departemen Ilmu Komputer, Fakultas Matematika dan Ilmu Pengetahuan Alam (FMIPA) melalui jalur Undangan Seleksi Masuk IPB (USMI) dan lulus pada tahun 2006. Pada Tahun 2008, penulis meneruskan studi S2 dan diterima di Program Studi Ilmu Komputer Pascasarjana IPB.
DAFTAR ISI
DAFTAR TABEL ………...iv
DAFTAR GAMBAR ……...v
I PENDAHULUAN ... viii
1.1 Latar Belakang ... 1
1.2 Penelitian Terkait ... 4
1.3 Tujuan Penelitian ... 4
1.4 Ruang Lingkup Penelitian ... 4
II TINJAUAN PUSTAKA ... 5
2.1 Struktur Protein ... 5
2.2 Prediksi Struktur Sekunder Protein ... 9
2.3 Hidden Markov Model (HMM) ... 10
2.4 Algoritme Baum-Welch ... 11
2.5 Algoritme Viterbi ... 13
2.6 Hidden Semi Markov Model ... 14
III METODE PENELITIAN ... 17
3.1 Kerangka Pemikiran ... 17
3.2 Studi Pustaka ... 18
3.3 Perumusan Masalah dan Hipotesa Awal ... 19
3.4 Pendefinisian Metode Penelitian ... 19
3.5 Pengambilan Data Struktur Sekunder Protein ... 19
3.6 Pembuatan Model dengan HMM Standar ... 20
3.7 Pembuatan Model dengan Hidden Semi Markov Model (HSMM) ... 21
3.8 Pengujian ... 21
IV HASIL DAN PEMBAHASAN ... 23
4.1 Praproses dan Pengkodean Data ... 23
4.2 Skenario Pengujian ... 23
4.2.1Pengujian skenario 1 dengan panjang durasi state 100% dari total panjang dengan alpha = 54, betha = 15 dan coil = 35 ... 25
4.2.2Pengujian skenario 2 dengan panjang durasi state 90% dari total panjang dengan alpha = 48, betha = 14 dan coil = 31 ... 25
4.2.3Pengujian skenario 3 dengan panjang durasi state 75 % dari total panjang dengan alpha = 40, betha = 11 dan coil = 26 ... 26
4.2.4Pengujian skenario 4 dengan panjang durasi state 50 persen dari total panjang dengan alpha = 27, betha = 8 dan coil = 18 ... 27
4.2.5Pengujian skenario 5 dengan panjang durasi state maksimum 21 ... 28
4.2.6Pengujian skenario 6 dengan panjang durasi state maksimum 15 ... 28
4.2.7Pengujian skenario 7 dengan panjang durasi state menggunakan distribusi fungsi kepekatan peluang eksponensial negatif ... 29
4.2.8Pengujian skenario 8 dengan panjang durasi state menggunakan distribusi fungsi kepekatan peluang normal dengan rataan geometrik . 30 4.3 Perbandingan akurasi Hidden Semi Markov Model (HSMM) ... 31
4.4 Analisis Error Identifikasi Struktur sekunder protein ... 32
4.4.1Matriks Konfusi Skenario 1 ... 32
4.4.2Matriks Konfusi Skenario 2 ... 33
4.4.3Matriks Konfusi Skenario 3 ... 34
4.4.4Matriks Konfusi Skenario 4 ... 35
4.4.5Matriks Konfusi Skenario 5 ... 36
4.4.6Matriks Konfusi Skenario 6 ... 37
4.4.7Matriks Konfusi Skenario 7 ... 38
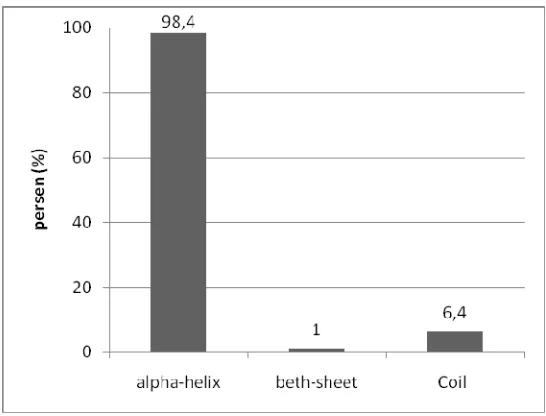
4.6 Matriks Konfusi Hidden Markov Model (HMM) Standar ... 42
4.7 Perbandingan Akurasi Prediksi HSMM dan HMM Standar ... 43
4.8 Pengaruh Distribusi Empiris Durasi State terhadap Akurasi HSMM ... 43
4.9 Perbandingan Distribusi Teoritis dan Empiris pada Data Pelatihan ... 47
V SIMPULAN DAN SARAN ... 51
5.1 Simpulan ... 51
5.2 Saran ... 51
DAFTAR PUSTAKA ... 53
DAFTAR TABEL
Halaman
1. Asam amino, singkatan, simbol dan karakteristik ... 6
2. Codon pembentuk protein ... 7
3. Skenario pengujian struktur sekunder protein ... 24
4. Perbandingan akurasi tertinggi setiap skenario pengujian ... 31
5. Matriks konfusi pengujian skenario 1 ... 32
6. Matriks konfusi pengujian skenario 2 ... 33
7. Matriks konfusi pengujian skenario 3 ... 34
8. Matriks konfusi pengujian skenario 4 ... 35
9. Matriks Konfusi Skenario 5 ... 36
10. Matriks Konfusi Skenario 6 ... 37
11. Matriks Konfusi Skenario 7 ... 38
12. Matriks Konfusi Skenario 8 ... 39
DAFTAR GAMBAR
Halaman
1. Proses pembentukan protein ... 5
2. Contoh urutan asam amino pembentuk suatu protein ... 6
3. Contoh susuan asam amino hormon prolaktin pada manusia ... 8
4. Gambar struktur sekunder protein : (a). alpha-helix (H), (b). betha-sheet (B) dan (c). coil (C) ... 8
5. Struktur tersier protein ... 9
6. Bentuk umum HSMM (Yu 2009) ... 15
7. Diagram alur penelitian prediksi struktur sekunder protein ... 18
8. Contoh Format Data Struktur sekunder protein dari file dengan ekstensi. dssp (Define Secondary Structure of Protein) ... 20
9. Ilustrasi HMM untuk prediksi strukutr protein sekunder (Martin et al. 2005) ... 20
10. Pemodelan prediksi struktur sekunder protein dengan menggunakan Hidden Semi Markov Model (HSMM) ... 21
11. Visualisasi hasil praproses dan pengkodean ... 23
12. Perbandingan akurasi prediksi struktur protein total pada skenario1 model HSMM dan HMM standar ... 25
13. Perbandingan akurasi prediksi struktur sekunder protein total pada skenario 2 model HSMM dan HMM standar ... 26
14. Perbandingan akurasi prediksi struktur sekunder protein total pada skenario 3 model HSMM dan HMM standar ... 27
16. Perbandingan akurasi prediksi struktur sekunder protein total pada skenario 5 model HSMM dan HMM standar ... 28
17. Perbandingan akurasi prediksi struktur sekunder protein total pada skenario 6 model HSMM dan HMM standar ... 29
18. Perbandingan akurasi prediksi struktur sekunder protein total pada skenario 7 model HSMM dan HMM standar ... 30
19. Perbandingan akurasi prediksi struktur sekunder protein total pada skenario 8 model HSMM dan HMM standar ... 30
20. Perbandingan akurasi setiap skenario model HSMM ... 31
21. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HSMM skenario 1 ... 33
22. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HSMM skenario 2 ... 34
23. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HSMM skenario 3 ... 35
24. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HSMM skenario 4 ... 36
25. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HSMM skenario 5 ... 37
26. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HSMM skenario 6 ... 38
27. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HSMM skenario 7 ... 39
28. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HSMM skenario 8 ... 40
29. Perbandingan akurasi prediksi struktur sekunder protein model HSMM dengan durasi empiris dan teoritis ... 41
31. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HMM standar ... 42
32. Perbandingan Akurasi prediksi struktur sekunder protein model HSMM dan HMM standar ... 43
33. Visualiasi distribusi empiris struktur alpha-helix (H) pada data latih dan data uji ... 44
34. Visualiasi distribusi empiris struktur betha-sheet (B) pada data latih dan data uji ... 44
35. Visualiasi distribusi empiris struktur coil (C) pada data latih dan data uji ... 45
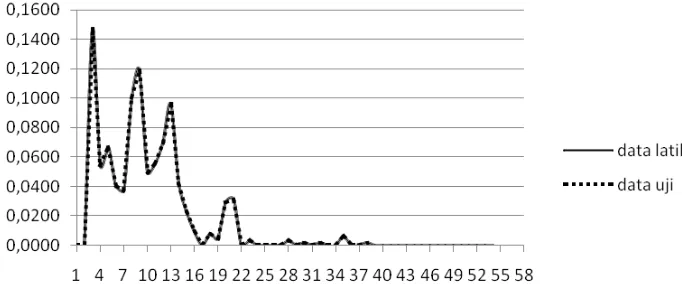
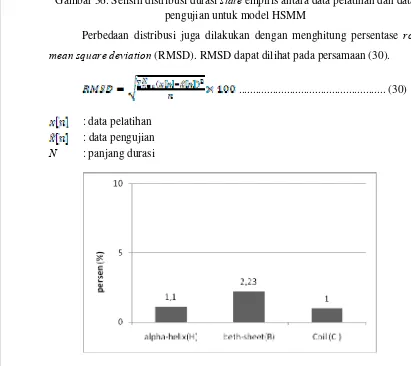
36. Selisih distribusi durasi state empiris antara data pelatihan dan data pengujian untuk model HSMM ... 46
37. Selisih distribusi durasi state empiris antara data pelatihan dan data pengujian untuk model HSMM dengan RMSD ... 46
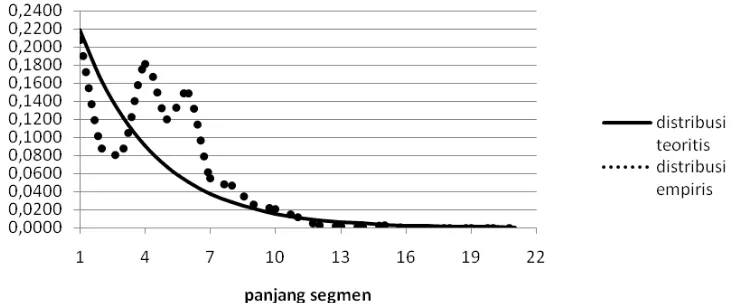
38. Visualiasi perbandingan distribusi teoritis dan empiris struktur alpha-helix (H) pada data latih ... 47
39. Visualiasi perbandingan distribusi teoritis dan empiris struktur betha-sheet (B) pada data latih ... 48
40. Visualiasi perbandingan distribusi teoritis dan empiris struktur coil (C) pada data latih ... 48
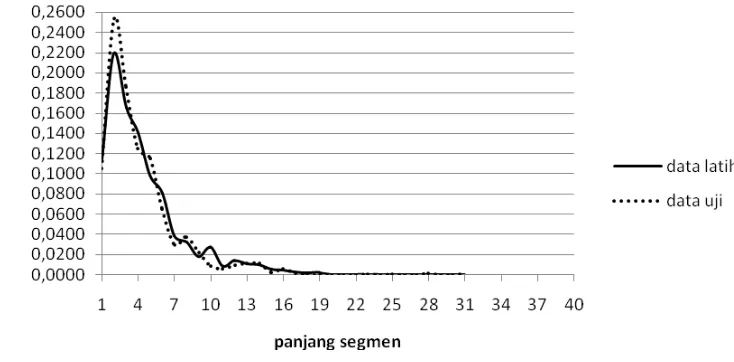
41. Perbandingan selisih distribusi durasi state teoritis dan empiris pada data latih untuk struktur alpha-helix (H), betha-sheet (B) dan coil (C) ... 49
I PENDAHULUAN
1.1 Latar Belakang
Protein, RNA dan berbagai fitur dalam genome dapat diklasifikasikan menjadi suatu keluarga tertentu sesuai dengan sekuensnya. Protein merupakan aktor utama pada makhluk hidup yang memiliki berbagai fungsi yang sangat penting. Protein terbentuk dari sekuens asam amino pembentuknya dengan karakteristik yang berbeda. Struktur protein dapat dilihat secara hierarki sebagai struktur primer, sekunder dan tersier (Polanski dan Kimmel 2007). Para ahli di bidang Biologi mengatakan bahwa protein berasal dari kombinasi tiga huruf (triplet) dari Asam Deoksiribosa (DNA) yang disebut dengan codon (Jones dan Pevzner 2004).
Struktur primer dari protein adalah urutan sekuens asam amino penyusun protein yang dihubungkan melalui ikatan peptida. Struktur sekunder adalah sejumlah rangkaian asam amino yang membentuk struktur tiga dimensi lokal baik struktur alpha-helix (H), betha-sheet (B) maupun coil (C). Adapun struktur tersier adalah gabungan dari berbagai struktur sekunder setelah terjadi proses pelipatan (folding).
Peranan protein sangat terlihat pada saat telah melakukan pelipatan (protein folding) dalam bentuk tiga dimensi (3D) sebagai struktur tersier. Namun, struktur tersier (3D) tersebut ditentukan oleh struktur sebelumnya baik primer maupun struktur sekundernya. Oleh karena itu, penentuan struktur sekunder protein ini menjadi bidang kajian yang banyak dilakukan di bidang Bioinformatika.
komputasi digunakan untuk melakukan prediksi struktur sekunder protein berbasis model komputasi, salah satunya adalah Hidden Markov Models (HMM).
Di sisi lain, karakteristik dari sekuens asam amino sebagai struktur protein primer sangat cocok dengan tipe data yang digunakan pada pembuatan model dengan menggunakan Hidden Markov untuk memprediksi struktur sekunder suatu protein. Menurut Eddy (1998), Hidden Markov Model (HMM) merupakan suatu kelas dari model probabilistik yang secara umum dapat diaplikasikan untuk permasalahan deret waktu atau sekuens yang bersifat linear. Sejalan dengan itu, HMMs merupakan metode yang dianggap memiliki kesuksesan dalam menyelesaikan permasalahan di dalam analisis sekuens meskipun dari sisi kompleksitas masih sulit untuk ditentukan secara manual (Won et al. 2007).
Martin et al. (2005) mengatakan bahwa di dalam memprediksi struktur sekunder protein dapat dilakukan dengan dua metode, yaitu:
Membandingkan model yang telah ada dengan struktur yang akan
diprediksi atau dikenal dengan comparative modelling
Metode de novo, yaitu apabila tidak terdapat model yang tersedia untuk dibandingkan dengan struktur yang akan diklasifikasikan.
Pada penelitian ini yang akan dilakukan adalah membuat model untuk mengklasifikasikan struktur sekunder protein. Untuk membangkitkan model tersebut, akan digunakan HMM akan yang telah secara luas diimplementasikan untuk menyelesaikan permasalahan dalam analisis sekuens. Di antara beberapa permasalahan yang terdapat di dalam prediksi struktur sekunder protein ialah masih terbatasnya model untuk dijadikan acuan dalam memprediksi protein sekunder karena ukuran data sangat besar. Penambahan jumlah data model bisa saja dilakukan, namun besarnya ukuran data akan membutuhkan waktu yang lama dan sulit untuk mencapai konvergen dari model yang bangkitkan pada saat melakukan proses pelatihan atau training.
adalah asam amino. Padahal, pada kenyataannya bisa saja suatu state dapat menyimpan informasi lebih dari satu observasi atau bahkan suatu sekuens observasi sebagai informasi. Oleh karena itu, dalam prediksi struktur sekunder protein terdapat pendekatan yang dikenal dengan Hidden Semi Markov Model (HSMM). Pada HSMM, satu state dapat membangkitkan suatu sekuens observasi. Ciri dari HSMM yang terpenting adalah adanya durasi state sebagai informasi dalam proses pelatihan dalam membuat model.
Hidden Semi Markov Model (HSMM) diperkenalkan pertama kali oleh Ferguson (1980) yang diaplikasikan untuk konversi teks ke suara. Yu dan Kobayashi (2003) melakukan penelitian untuk memperbaiki Algoritme pada HSMM yang dikenalkan oleh Ferguson. Hasilnya adalah kompleksitas algoritme yang diusulkan lebih efisien dibandingkan dengan yang dibuat pertama kali oleh Ferguson. Awalnya, kompleksitas algoritme pada HSMM yang diperkenalkan oleh Ferguson adalah O((MD2+M2)T). Dengan M adalah banyaknya state, D adalah durasi maksimum antar state dan T adalah panjangnya observasi. Yu dan Kobayashi berhasil mengefisienkan algoritme tersebut sehingga kompleksitasnya menjadi O((MD+M2)T) dan mengimplementasikannya untuk menganlisa trafik suatu alamat website. Pada tahun 2006, Yu dan Kobayashi kembali melakukan penelitian dan mengimplementasikan algoritme yang diusulkannya tersebut pada
chipfield-progammable gate-array (FPGA).
Prediksi struktur sekunder protein dengan Hidden Semi Markov Model (HSMM) pertama kali diperkenalkan oleh Schmidler et al. (2000) sebagai metode baru dalam prediksi struktur sekunder protein. Aydin et al. (2006) juga melakukan penelitian untuk memprediksi struktur sekunder protein dengan distribusi durasi maksimum sebanyak 50. Namun, keduanya masih menggunakan algoritme dengan kompleksitas sebagaiamana yang dijelaskan oleh Ferguson.
Pada penelitian kali ini, prediksi struktur sekunder protein diimplementasikan dengan algoritme yang telah memiliki kompleksitas
O((MD+M2)T) dan menggunakan beberapa jenis distribusi durasi maksimum
menggunakan distribusi secara teoritis. Hasil akurasi yang didapatkan akan dibandingan dengan hasil akurasi HMM standar.
1.2 Penelitian Terkait
Prediksi struktur sekunder protein dengan Hidden Semi Markov Model (HSMM) pertama kali diperkenalkan oleh Schmidler et al. (2000). Hasil akurasi prediksi struktur sekunder protein adalah 61,8 % untuk alpha-helix(H) dan 61,3 % untuk betha-sheet (B) dan 65,9 % untuk coil (C). Pada penelitian tersebut panjang durasi yang digunakan adalah 30 dengan kompleksitasnya adalah
O((MD2+M2)T). Aydin et al. (2006) melakukan penelitian untuk melakukan
prediksi struktur sekunder protein pada sekuens tunggal. Hasil akurasi yang diperoleh adalah 65, 9 % untuk alpha-helix(H), 45,4 % untuk betha-sheet(B) dan 81,3 % untuk coil (C).
1.3 Tujuan Penelitian
Penelitian ini bertujuan untuk mengembangkan Hidden Semi Markov Model (HSMM) yang telah memiliki kompleksitas lebih efisien untuk memprediksi struktur sekunder protein kemudian membandingkan hasil akurasi tersebut dengan penggunaan Hidden Markov Models (HMM) standar. Di samping itu, penelitian ini juga bertujuan untuk melihat pengaruh distribusi panjang durasi
state sebagai informasi biologi dalam proses pelatihan model HSMM yang
dilakukan terhadap hasil akurasi yang diperoleh.
1.4 Ruang Lingkup Penelitian
II TINJAUAN PUSTAKA
2.1 Struktur Protein
Protein merupakan bagian yang sangat penting pada setiap makhluk hidup. Proses untuk mendapatkan protein dinamakan dengan translasi. Setiap makhluk hidup memiliki kode genetik yaitu DNA (deoxyribonucleic acid) yang tersusun dari basa nitrogen adenin (A), guanin (G), thymine (T) dan cytosine (C). Melalui proses transkripsi, DNA tersebut ditranskripsikan menjadi RNA (ribonucleic acid). RNA mengalami proses translasi untuk kemudian menghasilkan protein (Jones dan Pevzner 2004). Secara ringkas proses terbentuknya protein dapat dilihat pada Gambar 1.
Gambar 1. Proses pembentukan protein
Protein merupakan elemen dasar dari suatu organisme yang dibentuk dari asam amino. Terdapat 20 asam amino dengan struktur kimia yang berbeda (Polanski dan Kimmel 2007). Susunan asam amino pembentuk protein dapat dilihat pada Tabel 1.
Asam amino tersebut terbentuk dari tiga huruf (triplet) dari kombinasi Asam Deoksirobosa (DNA) yang disebut dengan codon. Codon triplet pembentuk protein dapat dilihat pada Tabel 2. Satu protein protein terdiri atas sejumlah sekuens asam amino. Ilustrasi dari pembentukan satu protein berdasarkan sekuensnya dapat dilihat pada Gambar 2.
translasi transkripsi
DNA : TAC CGC GGC TAT TAC TGC CAG GAA GGA ACT
RNA : AUG GCG CCG AUA AUG ACG GUC CUU CCU UGA
Protein : Met Ala Pro Ile Met Thr Val Leu Pro Stop
Gambar 2. Contoh urutan asam amino pembentuk suatu protein
Gambar 2 merupakan ilustrasi proses pembentukan protein mulai dari urutan sekuens DNA sampai dengan proses translasi yang dapat menghasilkan suatu protein.
Tabel 1. Asam amino, singkatan, simbol dan karakteristik Asam Amino Singkatan Simbol Karakteristik
Alanine Ala A Nonpolar, hydrophobic
Arginine Arg R Polar, hydrophilic
Asparagine Asn N Polar, hydrophilic
Aspartic acid Asp D Polar, hydrophilic
Cystein Cys C Polar, hydrophilic
Glutamine Gln Q Polar, hydrophilic
Glutamic acid Glu E Polar, hydrophilic
Glycine Gly G Polar, hydrophilic
Histidine His H Polar, hydrophilic
Isoleucine Ile I Nonpolar, hydrophobic
Leucine Leu L Nonpolar, hydrophobic
Lysine Lys K Polar, hydrophilic
Methionine Met M Nonpolar, hydrophobic
Phenylalanine Phe F Nonpolar, hydrophobic
Proline Pro P Nonpolar, hydrophobic
Serine Ser S Polar, hydrophilic
Threonine Thr T Polar, hydrophilic
Tryptophan Trp W Nonpolar, hydrophobic
Tyrosine Tyr Y Polar, hydrophilic
Tabel 2. Codon pembentuk protein
U C A G
U
UUU Phe
UUC Phe
UUA Leu
UUG Leu
UCU Ser
UCC Ser
UCA Ser
UCG Ser
UAU Tyr
UAC Tyr
UAA Stop
UAG Stop
UGU Cys
UGC Cys
UGA Stop
UGG Trp
C
CUU Leu
CUC Leu
CUA Leu
CUG Leu
CCU Pro
CCC Pro
CCA Pro
CCG Pro
CAU His
CAC His
CAA Gln
CAG Gln
CGU Arg
CGC Arg
CGA Arg
CGG Arg
A
AUU Ile
AUC Ile
AUA Ile
AUG Met
ACU Thr
ACC Thr
ACA Thr
ACG Thr
AAU Asn
AAC Asn
AAA Lys
AAG Lys
AGU Ser
AGC Ser
AGA Arg
AGG Arg
G
GUU Val
GUC Val
GUA Val
GUG Val
GCU Ala
GCC Ala
GCA Ala
GCG Ala
GAU Asp
GAC Asp
GAA Glu
GAG Glu
GGU Gly
GGC Gly
GGA Gly
GGG Gly
Struktur protein terdiri atas struktur primer, struktur sekunder dan struktur tersier ( Polanski dan Kimmel 2007). Struktur primer dari suatu protein adalah rangkaian asam amino pembentuknya di sekitar rantai polipeptida. Database
(b)
(a) (c)
Gambar 3. Contoh susunan asam amino hormon prolaktin pada manusia
Struktur sekunder adalah struktur protein yang ditentukan oleh bentuk alpha-helix (H), betha-sheet (B) dan coil (C) penyusunnya. Struktur sekunder diperoleh dari sekuens asam amino, yang terikat dengan ikatan péptida. Dari sekuens asam amino tersebut, akan membentuk tiga kemungkinan segmen yaitu alpha-helix (H) yang bentuknya berpilin, betha-sheet (B) yang bentuknya lurus atau coil (C). Contoh bentuk ketiganya dapat dilihat pada Gambar 4. Gambar struktur sekunder protein tersebut didapatkan dengan menggunakan perangkat lunak RasMol Versi 2.7.4.2 sebagai perangkat lunak untuk memodelkan struktur protein yang bersifat free software.
Gambar 4. Gambar struktur sekunder protein : (a). alpha-helix (H), (b). betha-sheet (b) dan (c). coil (c)
tersier protein seperti dilihat pada Gambar 5 dengan menggunakan perangkat lunak RasMol Versi 2.7.4.2 .
Gambar 5. Struktur tersier protein
Data struktur tersier protein dapat diperoleh dari suatu database yang bernama Protein Data Bank (PDB). Dari data ini protein dapat divisualisasikan dalam bentuk tiga dimensi. Fungsi protein akan terlihat apabila sudah melakukan pelipatan atau protein folding. Protein merupakan molekul kompleks dengan struktur tiga dimensi (3D) yang dikenal dengan struktur tersier. Namun, demikian struktur kompleks tersebut dibangun dari struktur primer yang terdiri atas sekuens asam amino pembentuknya ( Polanski dan Kimmel 2007).
2.2 Prediksi Struktur sekunder protein
Struktur sekunder protein merupakan tahap awal dari proses prediksi struktur tiga dimensi (3D) suatu protein. Prediksi struktur sekunder protein bertujuan untuk mendapatkan informasi segmen alpha-helix (H), betha-sheet (B) atau coil (C) dari untaian asam amino primer yang membentuknya.
Crystallography dan Nuclear Magnetic Resonance (NMR) spectroscopy. (Albert
et al. 2007). Cara ini akan menghasilkan struktur protein secara tepat dan sangat memungkinkan menghasilkan struktur baru. Cara kedua adalah dengan pendekatan heuristic yaitu dengan comparative modelling yang hasilnya berupa prediksi. Prediksi struktur protein dengan menggunakan pendekatan heuristik ini, tidak akan menghasilkan penemuan struktur protein baru karena pendekatan ini hanya melakukan perbandingan antara data yang kita miliki dengan model yang sudah ada.
2.3 Hidden Markov Model (HMM)
Hidden Markov Model (HMM) merupakan model probabilistik yang dapat diaplikasikan untuk menganalisis model deret waktu atau sekuens linear (Eddy 1998). Pada sekitar tahun 1990, untuk membandingkan dua buah sekuens data biologi baik DNA atau RNA digunakan perbandingan pasangan antara dua sekuens yang akan disamakan. Namun, terdapat kendala yang ada apabila dua sekuens tersebut tidak sama di samping kesulitan apabila adanya sekuens baru (Baldi dan Brunak 2001). HMMs adalah salah satu pendekatan yang digunakan untuk memodelkan kumpulan sekuens tersebut. HMMs telah banyak dikembangkan pada banyak permasalahan seperti speech recognition (Rabiner 1989).
Menurut Rabiner (1989), aplikasi pada HMMs pada akhirnya akan direduksi untuk menyelesaikan tiga jenis permasalahan, yaitu :
1. Jika diberikan suatu model λ=(A,B,π), bagaimana menghitung
peluang dari sukuens observasi O = O1,O2,...OT yang dinotasikan dengan P(O|λ)
2. Jika diberikan suatu model λ=(A,B,π), bagaimana memilih state
sekuens I = I1,I2,...IT sehingga P(O,I|λ) sebagai peluang bersama
dari sekuens observasi O = O1,O2,...OT dan state sekuens tersebut memiliki nilai maksimum
dengan
λ
adalah model HMMA adalah Matriks peluang transisi,
B adalah Matriks peluang emisi dan
π
adalah Matriks peluang awal / Matriks priority O = O1,O2,...OT adalah variabel observasiλ) |
P(O adalah peluang variabel observasi jika diberikan model
Hidden Markov Model (HMMs) menggambarkan distribusi peluang dari sejumlah sekuens yang tidak terbatas (Eddy 1998). Nama "Hidden Markov Model" berawal dari fakta bahwasannya state dari sekuens merupakan orde pertama dari rantai Markov sebagai variabel yang tidak teramati. Adapun sekuens dari simbol (seperti A,C,G,T/U) merupakan variabel yang secara langsung dapat diobservasi. Pada kasus analisis sekuens dari data biologi, state sekuens akan berasosiasi dengan label biologis yang bermakna (seperti: struktur pada posisi lokus 42) (Eddy 1998).
2.4 Algoritme Baum-Welch
Algoritme Baum-Welch merupakan salah satu algoritme yang digunakan untuk melakukan pelatihan dalam melakukan estimasi parameter model dari HMMs yang dinotasikan sebagai . Algoritme ini mampu melakukan perbaruan nilai Matrikss peluang transisi A, Matriks peluang emisi B dan Matriks prioritas
π . Algoritme ini disebut juga dengan nama forward-backward algorithm.
Berikut adalah prosedur Algoritme Baum-Welch (Dugad dan Desai 1996)
Inisialisasi : set nilai λ=(A,B,π). Algoritme ini akan memperbaiki
nilai λsecara iteratif sampai konvergen.
prosedur forward : definisikan αt(i)= p(O1=O1,O2,...Ot,it=i|,λ)sebagai
) (o b π = (i)
αt i i 1 ………...……….………...……….………..(1)
N= i ij t + t j +
t (j)=b (o ) α(i)a
α
1 1
1 . ………...….…....……(2)
prosedur backward :definisikan t(i)= P(Ot+1,Ot+2,...,OT |it = i,λ)adalah
peluang observasi parsial sekuens dari t + 1 sampai T dengan state i pada saat t dan model λ. Secara efisien dapat dihitung :
1
= (i)
T , 1iN ………...………...…...(3)
N = j + t + t j ijt(i) = a b (o ) (j)
1
1
1 ...(4)
Dengan menggunakan α dan , akan ditentukan dua variabel, yaitu t(i) dan
j) (i,
ξt dengan persamaan sebagai berikut:
N = i t t t t t (i) (i) α (i) (i) α = (i) 1 ...(5)
N = i N = j + t j + t ij t + t j + t ij t t ) (o (j)b (i)a α ) (o (j)b (i)a α = j) (i, ξ 1 1 1 1 1 1 ...(6)Dengan mengasumsikan model saat inisialisasi adalah λ=(A,B,π), maka, update
nilai baru untuk mereestimasi parameter adalah:
(i) =
πi 1 , 1 i N …...(7)
1 1 1 1 T = t t T = t t ij(i)
j)
(i,
ξ
=
T = t t T vk O t t j (i) (i) k b t 1 1 )( 1 j N , 1k M …...(9)
dengan
λ
adalah model HMMA adalah matriks peluang transisi,
B adalah matriks peluang emisi dan
π
adalah matriks peluang awal / matrik priority O = O1,O2,...OT adalah variabel observasiλ) |
P(O adalah peluang variabel observasi jika diberikan model
λ
) (i
t
adalah peluang parsial dari sekuens observasi O1, O2,… Ot sampai denganstate ke-i pada saat ke-t
(i)
T adalah peluang parsial dari sekuen dari t + 1 sampai dengan T (dimana T
= t-1) pada state ke-i pada saat t
2.5 Algoritme Viterbi
Algoritme Viterbi digunakan untuk mendapatkan state yang optimal sehingga peluang suatu observasi adalah yang paling maksimal. Berikut adalah langkah-langkah algoritme Viterbi.
Inisialisasi
……… ………...………..(10)
Proses rekusif
untuk dan
Terminasi
………(12)
Proses menemukan kemungkinan nilai peluang paling besar dari suatu observasi yang
berakhir pada saat t = T
Backtracking
sehingga , …….(13)
Membaca (decoding) path sekuens terbaik dari vektor
2.6 Hidden Semi Markov Model
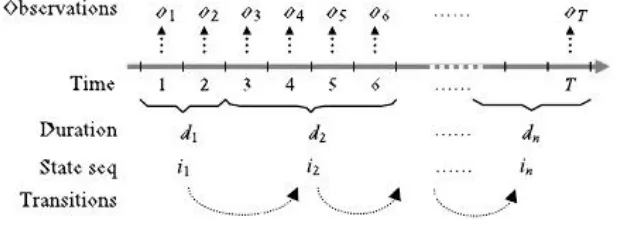
Hidden Semi Markov Model (HSMM) adalah bentuk perluasan dari Hidden Markov Model (HMM) yang mempertimbangkan durasi suatu observas terhadap state yang dimiliki. Pada HSMM, panjang durasi D pada suatu state
Gambar 6. Bentuk umum HSMM (Yu 2009)
Pada HSMM terlihat bahwa suatu state dapat membangitkan lebih dari satu observasi atau bahkan sejumlah sekuens observasi. Sepanjang observasi T, setiap state pada HSMM akan memiliki panjang durasi state dengan nilai tertentu. Pada prediksi struktu protein sekunder observasi ini merupakan sekuens dari asam amino yang membentuk protein sedangkan state adalah struktur protein yang akan diidentifikasi atau diprediksi. Berikut ini adalah tahap algoritme pada HSMM (Yu dan Kobayashi 2006)
Proses Forward
Proses Backward
Penentuan State Optimal
III METODE PENELITIAN
3.1 Kerangka Pemikiran
Protein merupakan aktor utama pada makhluk hidup yang memiliki berbagai fungsi yang sangat penting. Protein terbentuk dari asam amino pembentuknya dengan karakteristik yang berbeda. Struktur protein dapat dilihat secara hierarki sebagai struktur primer, sekunder dan tersier (Polanski dan Kimmel 2007). Prediksi struktur sekunder protein memiliki peranan yang penting sebelum protein tersebut mengalami pelipatan. Di antara model yang digunakan untuk melakukan prediksi struktur protein ini adalah Hidden Markov Model (HMM).
Umumnya proses pelatihan untuk membuat model pada HMMs dapat dilakukan dengan Algoritme Baum-Welch. Proses pelatihan ini tidak mempertimbangkan durasi state atau panjang segmen dari setiap struktur baik alpha-helix (H), betha-sheet (B) atau coil (C). Aydin et al. (2006) melakukan penelitian untuk memprediksi struktur sekunder protein dengan mempertimbangkan panjang durasi state atau panjang segmen tiap struktur protein tersebut yang dikenal dengan Hidden Semi Markov Model (HSMM). Namun demikian, pada penelitian tersebut panjang segmen ditetapkan dengan nilai tertentu dan bagaimana menentukan nilainya tidak disebutkan. Padahal panjang segmen struktur bisa diperoleh secara empiris dari data latih yang digunakan.
Di sisi lain, Hidden Semi Markov Model (HSMM) yang pertama kali diperkenalkan oleh Ferguson memiliki kompleksitas sebesar O((MD2+M2)T)
dengan M adalah Matriks transisi, T adalah sekuens observasi dan D adalah panjang durasi state (Yu dan Kobayashi 2003). Algoritme ini kemudian diefisienkan sehingga kompleksitasnya menjadi O((MD+M2)T dan kebutuhan alokasi memori memiliki kompleksitas O(MT) (Yu dan Kobayashi 2003).
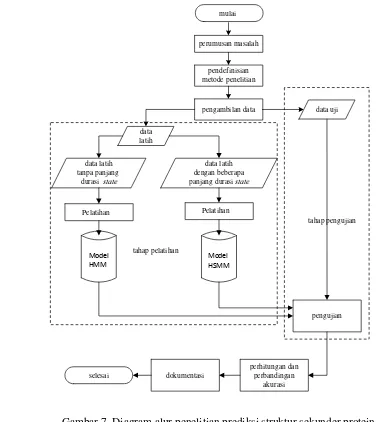
dan dibandingkan dengan akurasi Hidden Markov Model (HMM) standar. Model HSMM pada penelitian ini menggunakan distribusi panjang durasi state baik secara empiris maupun teoritis. Penelitian ini juga akan melihat pengaruh distribusi panjang durasi state terdapat nilai akurasi prediksi struktur sekunder protein. Diagram alur penelitian ini secara umum dapat dilihat pada Gambar 7.
Gambar 7. Diagram alur penelitian prediksi struktur sekunder protein
3.2 Studi Pustaka
Studi pustaka dilakukan untuk mencari riset-riset terbaru yang terkait dengan penelitian yang akan dilakukan. Dari riset yang telah dilakukan tersebut, penggunaan Hidden Markov Model (HMM) memang telah banyak digunakan
perumusan masalah pendefinisian metode penelitian pengambilan data data latih
data uji
data latih tanpa panjang
durasi state
data latih dengan beberapa panjang durasi state
Pelatihan Pelatihan
pengujian perhitungan dan perbandingan akurasi dokumentasi selesai Model HMM Model HSMM tahap pelatihan
untuk komputasi dalam bidang Biologi khususnya melakukan prediksi struktur sekunder protein. Akan tetapi, penggunaan durasi state dan bagaimana pengaruh panjang durasi state atau panjang segmen struktur protein terhadap akurasi proses prediksi masih belum banyak dilakukan.
3.3 Perumusan Masalah dan Hipotesa Awal
Perumusan masalah dilakukan sebagai kerangka untuk melakukan penelitian ini. Berdasar permasalahan tersebut, akan diusulkan suatu pendekatan untuk mencari solusinya. Dalam hal ini, penggunaan panjang durasi state pada Hidden Semi Markov Model (HSMM) dapat meningkatkan akurasi dibandingkan dengan HMM standar dan penggunaan beberapa nilai panjang segmen atau panjang durasi state akan mempengaruhi nilai akurasi.
3.4 Pendefinisian Metode Penelitian
Setelah dilakukan perumusan masalah, maka dipilih metode penelitian yang sesuai dengan permasalahan yang ditemukan. Metode penelitian yang digunakan dalam penelitian ini dapat dilihat pada Gambar 7.
3.5 Pengambilan Data Struktur sekunder protein
Proses prediksi struktur sekunder protein ini diawali dengan pengambilan data sekuens protein sekunder yaitu DSSP dari alamat
webstitehttp://swift.cmbi.ru.nl/gv/dssp/ yang merupakan database assignment
struktur sekunder protein. Pemilihan ini didasarkan bahwa DSSP mencakup semua data protein yang ada di Protein Data Bank (PDB). Data struktur sekunder protein yang diambil terdiri atas 42556 residu yang telah tersegmentasi menjadi 8 segmen struktur sekunder. Namun pada penelitian ini, segmen direduksi menjadi tiga, yaitu alpha-helix (H), betha-sheet (B) dan coil (C) (Aydinet al. 2006). Segmen hasil reduksi adalah {I,H,G} alpha-helix(H), {E,B} betha-sheet (B) , segmen lainnya menjadi coil (C).
sebelum digunakan untuk data pelatihan dan data pengujian terlebih dahulu dilakukan proses parsing dan pengkodean. Kedua proses tersebut dilakukan dengan memudahkan dalam proses komputasi. Hasil dari parsing dan pengkodean adalah pasangan asam amino dan assigment struktur sekunder protein yang telah dikodekan dalam bentuk bilangan integer. Format data dengan ekstensi .dssp merupakan file teks yang terdiri atas beberapa bagian. Gambar 8 merupakan contoh format data .dssp
Gambar 8. Contoh Format Data Struktur sekunder protein dari file dengan ekstensi. dssp (Define Secondary Structure of Protein)
3.6 Pembuatan Model dengan HMM standar
Pada tahap ini dilakukan proses pembentukan model dari data latih baik dengan HMMs standar dan Hidden Semi Markov Model (HSMM). Prediksi struktur sekunder protein ini, model arsitektur HMM dapat diilustrasikan pada Gambar 9.
Gambar 9. Ilustrasi HMM untuk prediksi strukutr protein sekunder (Martin et al. 2005)
Barisan H-B dan C menunjukkan model hidden state yang merepresentasikan alpha-helix (H), betha-sheet (B) dan coil (C), sedangkan barisan di bawah panah adalah barisan sekuens asam amino sebagai sekuens observasi (observable sequence). Hasil dari proses pemodelan ini adalah matriks transisi dan matriks emisi yang telah memiliki nilai tertentu untuk dijadikan
# RESIDUE AA STRUCTURE BP1 … … … X-CA Y-CA Z-CA
… … I H … … … … … …
… … D H … … … … … …
… … E H … … … … … …
… … G C
model dalam proses prediksi. Model tersebut kemudian akan disimpan dalam repositori.
3.7 Pembuatan Model dengan Hidden Semi Markov Model (HSMM)
Pemodelan prediksi struktur sekunder protein dengan HSMM berbeda dengan HMM standar. Pada pemodelan dengan HSMM, durasi state oleh observasi tertentu sangat dipertimbangkan. Pada HSMM tidak terjadi transisi suatu state terhadap dirinya sendiri. Ilustrasi pemodelan prediksi struktur sekunder protein dengan HSMM dapat dilihat pada Gambar 10.
Gambar 10. Pemodelan prediksi struktur sekunder protein dengan menggunakan Hidden Semi Markov Model (HSMM)
3.8 Pengujian
Tahap pengujian dilakukan untuk melihat akurasi dari prediksi yang dilakukan. Pengujian dilakukan baik dengan menggunakan data uji untuk melihat hasil klasifikasi. Hasil pengujian dilakukan dengan menghitung presentase data yang benar dikelaskan dibandingkan dengan semua data uji. Formula untuk melakukan pengujian dapat dilihat pada persamaan (28).
...(28)
3.9 Riset Pendahuluan
Riset pendahuluan dilakukan untuk mengetahui distribusi setiap segmen baik alpha-helix (H), betha-sheet (B) dan coil (C). Distribusi pada awalnya dilakukan dengan pembangkitan menggunakan pendekatan teoritis. Akan tetapi
…….. ……..
OT
d1 d2
1 2 3 4 5 6 ……...
H B
dn
in
observasi
waktu
durasi state sekuens transisi
permasalahannya adalah dengan pembangkitan nilai distribusi ini masih belum merepresentasikan kondisi data yang sebenarnya.
IV HASIL DAN PEMBAHASAN
4.1 Praproses dan Pengkodean Data
Data struktur sekunder protein yang diperoleh dari database masih memiliki format tertentu berekstensi (.dssp) sehingga harus dilakukan praproses dan pengkodean terlebih dahulu. Setiap satu file berekstensi .dssp akan diambil sekuens asam amino dan struktur sekundernya. Praproses dan pengkodean dilakukan agar data tersebut dilakukan untuk memudahkan proses komputasi pada tahap berikutnya. Adapun data tersebut pada memiliki format sebagai berikut:
# RESIDUE AA STRUCTURE BP1 … … … X-CA Y-CA Z-CA.
Header pada kolom ketiga dan keempat yaitu {AA} dan {STRUCTURE}
yang akan diambil sebagai pasangan data asam amino dan struktur sekunder protein . Baik {AA} maupun {STRUCTURE} akan dikodekan ke dalam suatu bilangan integer. Gambar 11 adalah visualisasi praproses dan pengkodean data.
Gambar 11. Visualisasi hasil praproses dan pengkodean
Hasil dari praproses dan pengkodean adalah pasangan sekuens asam amino dan struktur sekunder protein dari setiap residu asam amino. Format data lengkap struktur sekunder protein dapat dilihat pada Lampiran 1.
4.2 Skenario Pengujian
Skenario pengujian dilakukan untuk melakukan kombinasi pengujian sehingga dapat diketahui model Hidden Markov untuk mendapatkan hasil akurasi yang terbaik. Dengan demikian, model tersebut yang nantinya akan diambil untuk melakukan proses prediksi struktur sekunder protein .
# RESIDUE AA STRUCTURE BP1 … … … X-CA Y-CA Z-C … … D H … … … … … … … … E H … … … … … … … … G C … … … … … … … … L C … … … … … …
AA = [ D E G L ... ... ... ... ]
ST = [ H H C C ... ... ... ... ]
AA = [ 4 7 8 11 ... ... ... ... ]
Pada penelitian ini, skenario pengujiannya adalah melakukan pengujian dengan mempertimbangkan persentase beberapa distribusi panjang durasi state
yang diperoleh secara empiris dari data pelatihan dan pengujian dengan distribusi secara teoritis. Panjang durasi state yang didapatkan dari hasil pelatihan atautraining yaitu segmen alpha-helix (H) dengan panjang durasi maksimal adalah 54 residu, betha-sheet (B) dengan panjang durasi maksimum 15 residu dan coil dengan panjang durasi maksimum adalah 35 residu. Dari panjang masing-masing segmen ini kemudian diujikan dengan kombinasi mulai 100% panjang segmen, 90% , 75% dan 50%.
Di samping menggunakan persentase distribusi durasi state, skenario pengujian juga dilakukan dengan pengunaan panjang durasi state tertentu. Dalam hal ini pengujian dilakukan dengan panjang durasi state adalah 21 dengan mengasumsikan frekuensi state yang berjumlah kurang dari 10 diabaikan. Adapun pada Hidden Markov Model standar pengujian dilakukan dengan menggunakan Algoritme Viterbi. Hasil akurasi baik dengan HMM standar dan HSMM kemudian akan dibandingkan.
Data uji yang dilakukan pada pengujian ini sebanyak 43 sekuens yang merupakan 23,5 persen dari data secara keseluruhan. Adapun sebanyak 76,5 persen digunakan sebagai data latih. Skenario pengujian detail dapat dilihat pada Tabel 3. Jumlah residu dalam pengujian struktur alpha-helix (H) sebanyak 6053 residu, betha-sheet (B) sebanyak 2834 dan coil (C) sebanyak 5153 residu.
Tabel 3. Skenario pengujian struktur sekunder protein
Skenario Panjang durasi state Distribusi durasi state
1 100 %
Empiris
2 90 %
3 75 %
4 50 %
5 21
6 15
7 fungsi kepekatan peluang eksponensial negatif
4.2.1 Pengujian skenario 1 dengan panjang durasi state 100% dari total panjang dengan alpha = 54, betha = 15 dan coil = 35
Pengujian pada skenario 1 ini bertujuan untuk membandingkan akurasi prediksi struktur sekunder protein dengan menggunakan panjang durasi maksimum dari setiap segmen baik alpha, betha maupuan coil. Panjang maksimum segmen alpha pada skenario ini adalah 54 residu, segmen betha 15 residu dan segmen coil 35 residu yang diperoleh secara empiris dari data latih. Prediksi pada skenario 1 ini membandingkan hasil akurasi berdasarkan model yang diperoleh pada HMM standar dan model setelah dilakukan perbaikan parameter dengan menggunakan Hidden Semi Markov Model (HSMM). Akurasi pada HSMM ditentukan dengan membandingkan seluruh struktur yang diidentifikasi dengan struktur pada data sebenarnya tanpa melihat akurasi tiap segmen. Akurasi prediksi yang diperoleh dengan HSMM adalah 64,2% sedangkan dengan HMM standar menghasilkan akurasi 53,8%. Perbandingan akurasi tersebut dapat dilihat pada grafik Gambar 12.
Gambar 12. Perbandingan akurasi prediksi struktur protein total pada skenario1 model HSMM dan HMM standar
4.2.2 Pengujian skenario 2 dengan panjang durasi state 90% dari total panjang dengan alpha = 48, betha = 14 dan coil = 31
coil adalah 31. Perbandingan akurasi untuk HMM standar dan Hidden Semi Markov Model (HSMM) skenario 2 dapat dilihat pada grafik Gambar 13.
Gambar 13. Perbandingan akurasi prediksi struktur sekunder protein total pada skenario 2 model HSMM dan HMM standar
Berdasar hasil percobaan, terlihat bahwa model yang diperoleh dengan menggunakan HSMM lebih baik dibandingkan dengan HMM standar. Hal ini terlihat dari rata – rata akurasi proses prediksi struktur sekunder protein yang secara umum menghasilkan nilai lebih tinggi bila dibandingkan dengan rata-rata akurasi HMM standar. Gambar 12, dapat dilihat bahwa akurasi prediksi struktur sekunder protein dengan HSMM memiliki akurasi 64,1%, sedangkan akurasi dengan HMM standar menghasilkan akurasi 53, 8%.
4.2.3 Pengujian skenario 3 dengan panjang durasi state 75 % dari total panjang dengan alpha = 40, betha = 11 dan coil = 26
Pengujian pada skenario 3 dilakukan dengan menggunakan 75% dari panjang state masing-masing segmen baik alpha-helix, betha-sheet dan coil. Masing-masing panjang segmen tersebut adalah 40 residu alpha-helix, 11 residu betha-sheet dan 26 residu coil. Perbandingan hasil pengujian skenario 3 dapat dilihat pada Gambar 14.
Gambar 14. Perbandingan akurasi prediksi struktur sekunder protein total pada skenario 3 model HSMM dan HMM standar
4.2.4 Pengujian skenario 4 dengan panjang durasi state 50 persen dari total panjang dengan alpha = 27, betha = 8 dan coil = 18
Pengujian pada skenario 4 dilakukan dengan menggunakan panjang durasi
state 50 persen dari panjang total dengan durasi alpha-helix adalah 27 residu, betha-sheet 8 residu dan coil 18 residu. Hasil akurasi prediksi struktur sekunder protein model HSMM pada skenario adalah 63,8% sedangkan akurasi dengan model HMM standar sebesar 53,8%. Perbandingan hasil akurasi model HSMM dan HMM standar untuk skenario 4 dapat dilihat pada Gambar 15.
4.2.5 Pengujian skenario 5 dengan panjang durasi state maksimum 21
Berbeda dengan keempat skenario sebelumnya, pada skenario 5 ini, pengujian dilakukan dengan menggunakan panjang durasi state tertentu dengan mengasumsikan bahwa segmen/state yang memiliki frekuensi kurang dari 10 tidak diperhitungkan. Dalam skenario ini, pengujian dilakukan dengan menggunakan durasi state maksimum 21. Artinya untuk semua segmen baik alpha-helix, betha-sheet maupun coil akan diambil distribusi panjang durasi maksimumnya sama dengan 21. Hasil akurasi prediksi struktur sekunder protein dengan model HSMM sebesar 63,4% sedangkan akurasi prediksi dengan HMM standar sebesar 53,8%. Bila dibandingkan dengan empat skenario sebelumnya hasil prediksi dengan HSMM di skenario 5 ini relatif lebih kecil. Hal ini dikarenakan dengan ditetapkannya distribusi panjang durasi state ada kemungkinan hilangnya informasi dari setiap segmen tersebut. Grafik Gambar 16 memperlihatkan hasil perbandingan akurasi skenario 5.
Gambar 16. Perbandingan akurasi prediksi struktur sekunder protein total pada skenario 5 model HSMM dan HMM standar
4.2.6 Pengujian skenario 6 dengan panjang durasi state maksimum 15
panjang durasi 15 sebesar 63,3%. Hasil ini masih sama dengan pengujian dengan skenario 5. Adapun hasil akurasi dengan model HMM standar menghasilkan akurasi sebesar 53,8%.
Gambar 17. Perbandingan akurasi prediksi struktur sekunder protein total pada skenario 6 model HSMM dan HMM standar
Hasil prediksi yang pada Gambar 17 memperlihatkan bahwa akurasi prediksi model HSMM dengan distribusi panjang durasi 15 sebesar 63,3%. Hasil ini masih sama dengan pengujian dengan skenario 5. Adapun hasil akurasi dengan model HMM standar menghasilkan akurasi sebesar 53,8%.
4.2.7 Pengujian skenario 7 dengan panjang durasi state menggunakan distribusi fungsi kepekatan peluang eksponensial negatif
Pengujian pada skenario 7 ini distribusi panjang state dibangkitkan dengan menggunakan distribusi teoritis. Distribusi yang digunakan adalah fungsi kepekatan peluang eksponensial negatif. Hasil pembangkitan ini kemudian digunakan pada proses pelatihan untuk melakukan reestimasi parameter pada HSMM. Akurasi hasil prediksi struktur sekunder protein pada pengujian skenario 7 dapat dilihat pada grafik Gambar 18.
Gambar 18. Perbandingan akurasi prediksi struktur sekunder protein total pada skenario 7 model HSMM dan HMM standar
4.2.8 Pengujian skenario 8 dengan panjang durasi state menggunakan distribusi fungsi kepekatan peluang normal dengan rataan geometrik
Pada skenario 8, nilai peluang dari panjang durasi state menggunakan distribusi peluang normal, akan tetapi rataan panjang segmen yang digunakan dihitung dengan rataan geometrik. Hal ini disebabkan karena panjang segmen yang diperoleh dari data latih memang tidak menyebar secara merata. Hasil prediksi pada pengujian dari model HSMM pada skenario 8 dapat dilihat pada grafik Gambar 19.
Hasil prediksi dengan model HSMM dengan distribusi teoritis dengan fungsi kepekatan peluang normal adalah 61,7%. Hasil ini relatif kecil diantara skenario yang digunakan dalam proses prediksi struktur sekunder protein .
4.3Perbandingan akurasi Hidden Semi Markov Model (HSMM)
Secara umum, dari seluruh skenario pengujian yang dilakukan, terlihat bahwa model yang dihasilkan dengan menggunakan Hidden Semi Markov Model dapat menambah tingkat akurasi bila dibandingkan dengan Hidden Markov Model Standar. Namun demikian, rataan tingkat akurasi yang dihasilkan dari tujuh skenario pengujian tersebut masih relatif rendah. Perbandingan akurasi tertinggi setiap skenario pengujian dapat dilihat pada Tabel 4 berikut:
Tabel 4. Perbandingan akurasi tertinggi setiap skenario pengujian
Skenario 1 2 3 4 5 6 7 8
Akurasi tertinggi 64,4 64,1 63,8 63,8 63,4 63,3 64 61,7
Pada Tabel 3 terlihat bahwa akurasi yang paling tinggi berada pada skenario 1. Pada skenario tersebut panjang durasi state yang digunakan adalah distribusi empiris dengan presentase 100% distribusi panjang segmen alpha, betha dan coil. Hal ini beralasan karena dengan mengambil 100% panjang durasi ini informasi dari setiap segmen terwakili.
4.4 Analisis Error Identifikasi Struktur Sekunder Protein
Analisis error dilakukan untuk mengetahui bagaimana akurasi dari setiap segmen kelas/struktur yang dihasilkan dan mengetahui distribusi kelas yang salah diprediksi. Akurasi setiap segmen kelas/struktur yang dihasilkan pada proses identifikasi struktur sekunder protein dibuat dalam bentuk matriks konfusi. Matriks konfusi memperlihatkan persentasi struktur alpha-helix (H) , betha-sheet (B) dan coil (C) yang dikenali sesuai dengan kelasnya.
4.4.1 Matriks Konfusi Skenario 1
Matriks konfusi hasil pengujian skenario 1 memperlihatkan persentasi akurasi setiap kelas baik (H), (B) maupun (C) dengan Hidden Semi Markov Model (HSMM). Pada skenario ini durasi state yang digunakan adalah distribusi empiris dengan menggunakan 100% panjang maksimum durasi setiap state. Hasil Matriks konfusi dapat dilihat pada Tabel 5.
Tabel 5. Matriks konfusi pengujian skenario 1
kelas hasil prediksi akurasi prediksi (%) H B C
k
el
as as
al H 4401 295 1357 72, 7
B 978 1040 816 36,7
C 1495 417 3241 62,9
Berdasar hasil prediksi dari Matriks konfusi, terlihat bahwa dari 6053 residu asam amino yang memiliki struktur alpha-helix, ternyata 4401 residu yang terprediksi dengan benar, sedangkan sisanya terprediksi di kelas betha-sheet (B) sebanyak 295 residu dan coil sebanyak 1357 residu. Adapun struktur betha-sheet (B) dan coil (C) masing-masing diprediksi sesuai dengan kelasnya sebanyak 1040 residu dan 3241 residu. Akurasi prediksi setiap segmen struktur dapat dilihat pada grafik Gambar 21.
Gambar 21. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HSMM skenario 1
4.4.2 Matriks Konfusi Skenario 2
Prediksi tiap segmen struktur alpha-helix (H), betha-sheet (B) dan coil (C) pada skenario 2 ini menggunakan 90% panjang durasi state. Matriks konfusi pada pengujian skenario 2 dapat dilihat pada Tabel 6 untuk menggambarkan sebaran hasil identifikasi struktur sekunder protein baik alpha-helix (H), betha-sheet (B) maupun coil (C).
Tabel 6. Matriks konfusi pengujian skenario 2
kelas hasil prediksi akurasi prediksi
(%) H B C
kela
s asal
H 4396 295 1362 72,6
B 973 1043 818 36,6
C 1491 420 3242 62,9
Pada pengujian dengan 90% durasi state, terlihat bahwa dari sebanyak 4396 residu asam amino yang memiliki struktur alpha-helix (H), mampu diprediksi sesuai dengan strukturnya. Adapun struktur betha-sheet (B) dan coil masing-masing diprediksi dengan benar sebanyak 1043 residu dan 3242 residu.
betha-sheet (B) dan coil dengan nilai 72, 6%. Adapun akurasi betha-sheet (B) dan coil (C) masing-masing sebesar 36,8% dan 62,9%.
Gambar 22. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HSMM skenario 2
4.4.3 Matriks Konfusi Skenario 3
Matriks konfusi skenario 3 menunjukkan hasil identifikasi struktur protein dengan model HSMM yang menggunakan panjang durasi state 75 % dari panjang total tiap struktur kelasnya. Hasil prediksi berupa Matriks konfusi dapat dilihat pada Tabel 7.
Tabel 7. Matriks konfusi pengujian skenario 3
kelas hasil prediksi akurasi
prediksi (%) H B C
kela
s asal
H 4394 295 1364 72,6
B 972 1019 843 36
C 1498 412 3243 62,9
Gambar 23. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HSMM skenario 3
4.4.4 Matriks Konfusi Skenario 4
Matriks konfusi skenario 4 menunjukkan hasil identifikasi struktur protein dengan model HSMM yang menggunakan panjang durasi state 50 % dari panjang total tiap struktur kelasnya. Apabila dibandingkan dengan hasil identifikasi pada Matriks konfusi sebelumnya ternyata penggunaan 50 % panjang durasi state tidak memberikan kenaikan akurasi yang siginifikan. Matriks konfusi hasil prediksi pengujian skenario 4 dapat dilihat pada Tabel 8.
Tabel 8. Matriks konfusi pengujian skenario 4
kelas hasil prediksi akurasi
prediksi (%) H B C
kela
s a
sal H 4391 254 1408 72,5
B 1003 966 865 34, 1
C 1525 360 3268 63, 4
Gambar 24. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HSMM skenario 4
4.4.5 Matrik Konfusi Skenario 5
Matriks konfusi skenario 5 menunjukkan hasil identifikasi struktur protein dengan model HSMM yang menggunakan panjang durasi tertentu. Dalam hal ini panjang durasi state yang ditentukan adalah 21. Matriks konfusi pada skenario 5 dapat dilihat pada Tabel 9.
Tabel 9. Matriks Konfusi Skenario 5
kelas hasil prediksi akurasi
prediksi (%) H B C
kela
s asal
H 4274 317 1462 70,6
B 950 1062 822 37,4
C 1421 429 3303 64,1
Gambar 25. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HSMM skenario 5
Penggunaan durasi state tertentu ini, memberikan akurasi prediksi yang berbeda bila dibandingkan dengan penggunaan presentasi panjang duras state. Dapat dilihat bahwa akurasi struktur alpha-helix (H) menurun dari sekitar 72% pada penggunaan presentase durasi state menjadi kurang lebih 70%. Sementara akurasi beta-sheet dan coil justru sedikit menaik masing-masing 37,4% dan 64,1%. Menurunnya akurasi alpha diduga disebabkan oleh hilangnya informasi durasi state dengan pemotongan panjang durasi yang hanya 21 ini. Padahal segmen alpha-helix memiliki distribusi dengan panjang maksimum terbesar.
4.4.6 Matriks Konfusi Skenario 6
Matriks konfusi skenario 6 menunjukkan hasil identifikasi struktur protein dengan model HSMM yang menggunakan panjang durasi tertentu, yaitu 15. Hasil identifikasi dalam Matriks konfusi diperlihatkan pada Tabel 10.
Tabel 10. Matriks Konfusi Skenario 6
kelas hasil prediksi akurasi
prediksi (%) H B C
ke
la
s asa
l H 4007 359 1687 66,2
B 792 1177 865 41,5
C 1228 443 3482 67,6
skenario 6. Dibandingkan dengan skenario durasi empiris lainnya, penggunaan panjang durasi state sebesar 15 ini berdampak pada turunnya akurasi untuk prediksi struktur alpha-helix (H). Ini terlihat dari jumlah residu yang hanya 4007 diprediksi dengan benar atau 66,2% saja. Adapun akurasi betha-sheet (B) dan coil (C) justru mengalami kenaikan mencapai 41,5% dan 67,6%. Grafik akurasi prediksi struktur sekunder protein dapat dilihat pada Gambar 26.
Gambar 26. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HSMM skenario 6
4.4.7 Matriks Konfusi Skenario 7
Matriks konfusi skenario 7 memperlihatkan hasil identifikasi struktur protein untuk model HSMM dengan peluang durasi state yang digunakan adalah distribusi dengan fungsi kepekatan peluang eksponensial negatif. Hasil Matriks konfusi dapat dilihat pada Tabel 11.
Tabel 11. Matriks Konfusi Skenario 7
kelas hasil prediksi akurasi
prediksi (%) H B C
k
el
as as
al H 4717 218 1118 77,9
B 1209 834 791 29,4
C 1800 357 2996 58,1
betha-sheet (B) dan coil justru menurun masing-masing 29,4 % dan 58,1%. Secara visual akurasi prediksi skenario 7 dapat dilihat pada grafik Gambar 27.
Gambar 27. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HSMM skenario 7
4.4.8 Matriks Konfusi Skenario 8
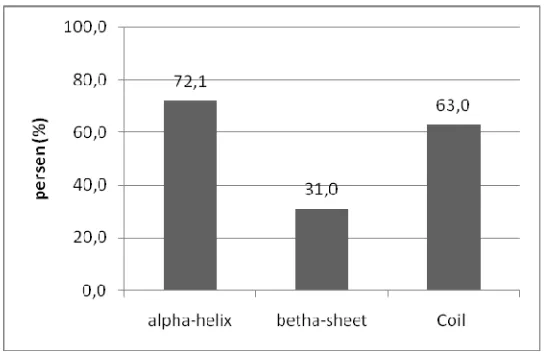
Matriks konfusi skenario 8 memperlihatkan hasil identifikasi struktur protein untuk model HSMM dengan peluang durasi state yang digunakan adalah distribusi peluang normal. Akan tetapi rataan yang digunakan untuk menghitung fungsi kepekatan peluang adalah rataan geometrik. Ini dilakukan karena panjang segmen tiap struktur protein memiliki sebaran yang tidak normal. Hasil Matriks konfusi dapat dilihat pada Tabel 12.
Tabel 12. Matriks Konfusi Skenario 8
kelas hasil prediksi akurasi
prediksi (%) H B C
kela
s asal
H 4362 183 1508 72,1
B 1032 878 924 31,0
C 1577 331 3245 63,0
(B) dan coil (C) menghasilkan akurasi masing-masing sebesar 31% dan 63%. Gambar 28 memperlihatkan grafik akurasi untuk pengujian skenario 8.
Gambar 28. Akurasi prediksi struktur sekunder protein tiap segmen kelas alpha-helix, betha-sheet dan coil model HSMM skenario 8
4.5 Perbandingan Akurasi Prediksi HSMM Distribusi Empiris dan Teoritis
Gambar 29. Perbandingan akurasi prediksi struktur sekunder protein model HSMM dengan durasi empiris dan teoritis
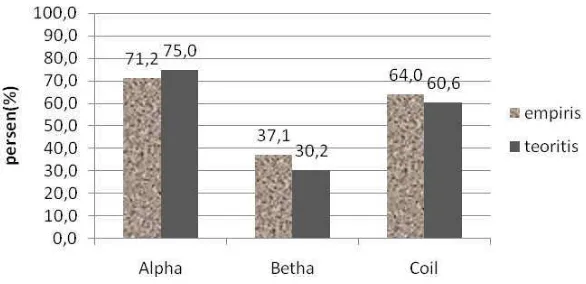
[image:60.612.198.442.444.612.2]Gambar 29 memperlihatkan bahwa hasil akurasi prediksi alpha-helix (H) lebih tinggi pada penggunaan durasi teoritis, namun pada prediksi betha-sheet (B ) dan coil, penggunaan durasi empiris memberikan akurasi yang lebih tinggi. Secara umum perbandingan akurasi rata-rata prediksi alpha-helix(H), betha-sheet (B) dan coil (C) dengan durasi empiris dan teoritis dapat dilihat pada grafik Gambar 30.
4.6 Matriks Konfusi Hidden Markov Model (HMM) Standar
Hasil pengujian juga dilakukan terhadap model dengan HMM standar untuk mengetahui tingkat akurasi yang dihasilkan. Akurasi prediksi setiap struktur protein dapat dilihat pada Matriks konfusi Tabel 13.
Tabel 13. Matriks Konfusi Prediksi dengan HMM Standar
kelas hasil prediksi akurasi prediksi (%)
H B C
kela
s ssa
l H 5957 11 85 98,4
B 2715 28 91 1
C 4815 21 334 6,4
[image:61.612.183.456.406.614.2]Tabel 13 memperlihatkan bahwa proses identifikasi menunjukkan hasil yang tidak seimbang. Hal ini terlihat bahwa untuk struktur betha-sheet (B) dan coil (C) hampir sama sekali tidak dikenali sebagai kelasnya meskipun dalam mengidentifikasi alpha-helix(H) mencapai 98,4%. Dapat dilihat bahwa akurasi betha-sheet (B) hanya 1% dan coil (C) yang hanya 6,4% (Gambar 31).
4.7 Perbandingan Akurasi Prediksi HSMM dan HMM Standar
[image:62.612.188.459.242.405.2]Perbandingan akurasi HSMM dan HMM dilakukan untuk menunjukkan keunggulan HSMM dibandingkan HMM standar. Dapat dilihat (Gambar 32) bahwa meskipun pada prediksi alpha-helix (H) akurasi HMM jauh lebih tinggi, namun tidak diikuti oleh akurasi struktur lainnya. Dengan demikian, secara rata-rata akurasi prediksi struktur sekunder protein dengan HSMM manghasilkan akurasi sebesar 56,9% sedangkan HMM standar menghasilkan akurasi sebesar 35,3%.
Gambar 32. Perbandingan Akurasi prediksi struktur sekunder protein model HSMM dan HMM sSandar
4.8 Pengaruh Distribusi Empiris Durasi State Terhadap Akurasi HSMM Pada proses pelatihan model HSMM dilakukan dengan mempertimbangkan distribusi durasi state yang digunakan baik secara empiris maupun teoritis. Secara keseluruhan rata-rata akurasi struktur alpha-helix (H) dengan distribusi empiris paling tinggi dibandingkan dengan betha-sheet (B) dan coil (C). Rata-rata akurasi struktur (H) dari skenario satu sampai delapan adalah 71,2%, struktur (B) 37,1% dan struktur (C) 64,0%. Terlihat bahwa akurasi struktur betha-sheet (B) paling rendah.
Gambar 33. Visualiasi