ISOLASI DAN KLONING GEN PENYANDI GLUKANASE
(GLN) DARI
Trichoderma
sp
.
PADA VEKTOR DONOR
DAN VEKTOR EKSPRESI
RISKA AYU PURNAMASARI
DEPARTEMEN BIOKIMIA
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM
INSTITUT PERTANIAN BOGOR
ABSTRAK
RISKA AYU PURNAMASARI. Isolasi dan Kloning Gen Penyandi Glukanase
(GLN) dari
Trichoderma
sp. pada Vektor Donor dan Vektor Ekspresi. Dibimbing
oleh SURYANI dan TETTY CHAIDAMSARI.
Kelapa Sawit (
Elaeis guineensis
Jacq.) merupakan tanaman perkebunan
yang berperan penting dalam perekonomian Indonesia dan dunia. Dalam proses
budidaya tanaman ini terdapat beberapa hambatan yang menurunkan tingkat
produksinya. Salah satu hambatan yakni adanya penyakit busuk pangkal batang
(BPB) yang disebabkan oleh jamur patogen
Ganoderma boninense
Pat. Penyakit
BPB merupakan penyakit yang paling mematikan pada kelapa sawit. Adapun
pengaruh patogenitas jamur
G. boninense
dapat ditanggulagi dengan
memanfaatkan enzim glukanase yang berasal dari jamur
Trichoderma
sp. Isolasi
dan kloning gen penyandi glukanase diharapkan menjadi solusi dalam
menanggulangi penyakit BPB pada kelapa sawit. Penelitian ini bertujuan
mengisolasi dan menyisipkan gen penyandi glukanase (GLN) pada vektor donor
dan vektor ekspresi dengan teknik kloning Gateway untuk tujuan perbanyakan
gen. Setelah proses isolasi dan amplifikasi, gen GLN yang berukuran 1700 bp
disisipkan dalam vektor kemudian ditransformasikan ke dalam
Escherichia coli
.
Hasil penyisipan gen GLN pada vektor donor yang diuji dengan teknik PCR
koloni dan isolasi plasmid menghasilkan pita DNA berukuran 6461 bp.
Sedangkan hasil penyisipan gen GLN pada vektor ekspresi yang diuji dengan
teknik PCR koloni dan isolasi plasmid menghasilkan pita DNA berukuran 4700
bp. Dengan demikian, dapat disimpulkan bahwa gen GLN telah berhasil diisolasi
dan disisipkan ke dalam vektor donor dan vektor ekspresi.
ABSTRACT
RISKA AYU PURNAMASARI. Isolation and Cloning Glucanase Encoding Gene
(GLN) from
Trichoderma
sp. in donor and expression vectors. Under the direction
of SURYANI and TETTY CHAIDAMSARI.
Palm Oil (
Elaeis guineensis
Jacq.) is one of the plantations that have an
important role for national and world economy. In the production process, there
are several obstacles that can decrease production level of palm oil. One of the
obstacles is the presence of basal stem rot (BSR) disease that caused by
Ganoderma boninense.
Basal stem rot disease is the most destructive disease in
palm oil. The effect of pathogen can be solve with utilization of glucanase enzyme
from
Trichoderma
sp. Isolation and cloning of glucanase encoding gene was
expected to be a solution to against BSR disease in palm oil. The objectives of this
research are to isolate and insert the glucanase encoding gene (GLN) inside donor
and expression vectors with Gateway cloning technique. After the isolation and
amplification process, the 1700 bp of GLN gene insert to the vectors
then transformed into
Escherichia coli
. The result of GLN gene insertion into
donor vector was carried out using PCR colony technique and plasmid isolation
which resulted DNA band of 6461 bp. And then, the result of GLN gene insertion
into expression vector was carried out using PCR colony technique and plasmid
isolation which resulted DNA band of 4700 bp. The conclusion of this research
was that the fulllength of GLN gene had been successfully isolated and inserted
into the donor and expression vectors.
`
ISOLASI DAN KLONING GEN PENYANDI GLUKANASE
(GLN) DARI
Trichoderma
sp
.
PADA VEKTOR DONOR
DAN VEKTOR EKSPRESI
RISKA AYU PURNAMASARI
Skripsi
sebagai salah satu syarat memperoleh gelar
Sarjana Sains pada
Departemen Biokimia
DEPARTEMEN BIOKIMIA
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM
INSTITUT PERTANIAN BOGOR
Judul Skripsi : Isolasi dan Kloning Gen Penyandi Glukanase (GLN) dari
Trichoderma
sp. pada Vektor Donor dan Vektor Ekspresi
Nama
: Riska Ayu Purnamasari
NIM
: G84070004
Disetujui
Komisi Pembimbing
Dr. Suryani, M.Sc
Ketua
Dr. Tetty Chaidamsari, M.Si
Anggota
Diketahui,
Ketua Departemen Biokimia
Dr. Ir. I Made Artika, M.App.Sc
NIP : 19630117 198903 1 000
PRAKATA
Syukur Alhamdulillah kehadirat Allah SWT atas segala limpahan
kekuatan dan hidayah-Nya sehingga penulis dapat menyelesaikan penelitian dan
karya ilmiah ini. Shalawat dan salam semoga tercurahkan kepada Rasulullah
SAW yang menjadi kebanggaan dan teladan umat Islam sampai akhir zaman.
Penelitian ini bertujuan untuk mengisolasi dan memperbanyak gen penyandi
glukanase (GLN) pada vektor donor dan vektor ekspresi dengan teknik kloning
Gateway. Penelitian yang dilakukan semenjak bulan Maret 2011 di Balai
Penelitian Bioteknologi Perkebunan Indonesia (BPBPI) ini diharapkan mampu
mengurangi dampak penyakit busuk pangkal batang (BPB) yang menyerang
tanaman kelapa sawit.
Penulis mengucapkan terima kasih kepada Dr. Suryani, M.Sc dan Dr.
Tetty Chaidamsari, M.Si selaku pembimbing yang telah memberikan bimbingan,
dukungan, dan arahan. Ucapan terimakasih juga penulis sampaikan kepada Nina
Yuniar dan Herti Sugiharti selaku teknisi laboratorium yang telah membantu
dalam melakukan penelitian. Tak lupa penulis sampaikan juga kepada ayah, ibu,
dan seluruh keluarga yang senantiasa memberi dukungan serta doa.
Penulis berharap penelitian ini bermanfaat baik bagi penulis maupun bagi
masyarakat pada umumnya serta semua pihak yang membutuhkan.
.
Bogor, Desember 2011
RIWAYAT HIDUP
Penulis dilahirkan di Serang pada tanggal 20 Februari 1989 dari ayah Aan
Supriana, SE dan ibu Yuliasih. Penulis merupakan anak pertama dari dua
bersaudara. Pendidikan penulis dimulai dari TK PGRI Cilegon, SD YPWKS V,
kemudian melanjutkan ke SLTP Negeri 1 Cilegon. Tahun 2007 penulis lulus dari
SMA Negeri 1 Cilegon dan pada tahun yang sama lulus seleksi masuk IPB
melalui jalur Undangan Seleksi Masuk IPB (USMI). Penulis memilih program
studi mayor Biokimia, Fakultas Matematika dan Ilmu Pengetahuan Alam.
Selama mengikuti perkuliahan, penulis aktif dalam beberapa organisasi,
diantaranya menjadi Reporter Koran Kampus IPB pada tahun 2007/2009, menjadi
anggota Himpunan Mahasiswa Biokimia
Community Research and Education of
Biochemistry
(CREBs) pada tahun 2008/2009, dan menjadi wakil ketua unit
kegiatan mahasiswa
Forum for Scientific Studies
IPB (UKM FORCES IPB)
2009/2010. Penulis pernah mendapatkan Beasiswa dan Pelatihan dari Yayasan
Karya Salemba Empat selama dua tahun (2009/2010). Prestasi yang didapat
penulis saat kuliah antara lain, mendapat juara 2 lomba karya tulis ilmiah dengan
tema lingkungan dan air, mendapat juara 2 lomba esai biokimia, dan menjadi
presenter dalam konferensi tentang lingkungan SUSTAIN 2010 di Kyoto, Jepang.
Penulis juga pernah mendapat beasiswa pertukaran pelajar selama dua bulan di
Amerika Serikat dalam program
Indonesian English Language Study Program
(IESLP).
DAFTAR ISI
Halaman
DAFTAR GAMBAR ... viii
DAFTAR LAMPIRAN ... ix
PENDAHULUAN ... 1
TINJAUAN PUSTAKA
Kelapa Sawit ... 2
Penyakit Busuk Pangkal Batang pada Kelapa Sawit ... 2
Pertahanan Tanaman dengan Enzim Glukanase ... 3
Kapang
Trichoderma
sp
.
sebagai Sumber Enzim Glukanase ... 3
Metode
Polymerase Chain Reaction (PCR)
untuk Amplifikasi DNA ... 4
Metode Kloning Gateway ... 5
BAHAN DAN METODE
Bahan dan Alat ... 6
Metode Percobaan ... 7
HASIL DAN PEMBAHASAN
Hasil Desain Primer Gateway ... 9
Kultur Kapang Trichoderma ... 10
Hasil Isolasi DNA Kapang
Trichoderma
sp ... 10
Amplikon Gen Penyandi Glukanase (GLN) ... 12
Rekombinan Hasil Reaksi BP dengan Vektor Donor dan Hasil PCR
Koloni ... 13
Hasil Analisis Urutan Basa Gen GLN pada Vektor Donor ... 14
Rekombinan Hasil Reaksi LR dengan Vektor Ekspresi dan Hasil PCR
Koloni ... 14
SIMPULAN DAN SARAN
Simpulan ... 16
Saran ... 16
DAFTAR PUSTAKA ... 16
DAFTAR GAMBAR
Halaman
1 Perkebunan Kelapa Sawit ... 2
2 Penyakit Busuk Pangkal Batang pada Kelapa Sawit ... 3
3 Konidia pada
Trichoderma
sp
.
... 4
4 Hifa pada
Trichoderma
sp . ... 4
5 Tahapan-tahapan pada PCR ... 5
6 Tahapan-tahapan reaksi BP ... 5
7 Tahapan-tahapan reaksi LR ... 6
8 Hasil Kultur Kapang
Trichoderma
sp ... 10
9 Elektroforegram Isolat DNA
Trichoderma
sp ... 11
10 Elektroforegram amplikon gen GLN dengan primer gateway ... 12
11 Elektroforegram ekstraksi dan purifikasi amplikon gen GLN ... 12
12 Koloni yang tumbuh setelah reaksi BP ... 13
13 Elektroforegram PCR koloni rekombinasi gen GLN pada vektor donor ... 13
14 Elektroforegram hasil isolasi plasmid rekombinan setelah reaksi BP ... 14
15 Koloni yang tumbuh setelah reaksi LR ... 15
16 Elektroforegram PCR koloni rekombinasi gen GLN pada vektor ekspresi .. 15
DAFTAR LAMPIRAN
Halaman
1 Desain primer metode Gateway ... 20
2 Skema pembuatan media dan subkultur kapang. ... 21
3 Skema isolasi DNA kapang ... 22
4 Skema pembuatan
buffer
TBE dan gel agarosa 1% ... 23
5 Perhitungan konsentrasi dan bobot molekul DNA ... 24
6 Marka seleksi vektor donor (pDONR221). ... 25
7 Analisis BLAST sekuen DNA
Trichoderma
sp ... 26
8 Hasil subkultur koloni bakteri ... 27
1
PENDAHULUAN
Kelapa sawit (Elaeis guineensis Jacq.) merupakan salah satu komoditas perkebunan Indonesia yang mempunyai peranan penting dalam perekonomian nasional dan memiliki prospek pengembangan yang cerah. Indonesia memiliki potensi yang sangat besar dalam pengembangan perkebunan dan industri kelapa sawit karena memiliki potensi cadangan lahan yang cukup luas, ketersediaan tenaga kerja, dan kesesuaian agroklimat (Pahang 2010). Luas perkebunan kelapa sawit pada tahun 2007 sekitar 6,8 juta hektar (Heriyadi 2009). Dari luas tersebut sekitar 60% diusahakan oleh perkebunan besar dan sisanya diusahakan oleh perkebunan rakyat (Soetrisno 2008).
Sebanyak 85% lebih pasar dunia kelapa sawit dikuasai oleh Indonesia dan Malaysia (Pahang 2010). Permintaan dunia terhadap kelapa sawit mengalami peningkatan dalam kurun waktu 2000-2005. Kebutuhan minyak dunia yang kian meningkat menempatkan minyak kelapa sawit sebagai sumber energi yang secara perlahan akan menggantikan minyak bumi yang keadaannya tidak dapat diperbaharui. Semenjak tahun 2007 hingga saat ini Indonesia merupakan produsen minyak kelapa sawit terbesar di dunia. Luas kebun kelapa sawit mencapai 7.8 juta hektar, jumlah produksi sebesar 22 juta ton, dan ekspor mencapai 16.5 juta ton pada tahun 2010 (Dahuri 2008).
Proses perkembangan kelapa sawit tak lepas dari beberapa masalah yang menyebabkan penurunan produksi kelapa sawit. Salah satu penyebab penurunan produksi kelapa sawit yakni penyakit busuk pangkal batang (BPB) yang disebabkan oleh jamur patogen, Ganoderma boninense Pat. Penyakit BPB merupakan penyakit paling serius pada tanaman kelapa sawit. Kematian yang diakibatkan mencapai 50% pada tanaman produktif (Turner 1981; Darmono et al. 1993). Jamur ini merupakan jamur tanah hutan hujan tropik yang bersifat saprofitik dan akan berubah menjadi patogenik bila bertemu dengan akar kelapa sawit yang tumbuh di dekatnya. Gejala awal penyakit ini ditunjukkan dengan warna pelepah daun yang berubah seperti kekurangan hara. Selanjutnya, daun mengalami penuaan dini yang dimulai dari daun tua kemudian ke daun yang lebih muda. Umumnya 6-12 bulan setelah gejala terakhir tanaman akan mati. Infeksi terjadi karena adanya kontak akar yang sakit.
Meningkatnya serangan sejalan dengan tingkat umur generasi tanaman (Lubis 1992).
Pengendalian penyakit BPB saat ini dilakukan dengan pengendalian kultur teknis, mekanis, dan kimiawi. Pengendalian tersebut seringkali mengalami kegagalan karena patogen penyebab BPB bersifat tular tanah dan memiliki kemampuan untuk bertahan dalam keadaan lingkungan yang kurang mendukung, serta dapat bertahan lama di dalam tanah tanpa inang (Susanto et al. 2005). Berdasarkan hal itu, maka dalam dasawarsa terakhir ini teknologi pengendalian penyakit BPB berbasis genetik mulai digunakan. Pendekatan yang dilakukan memanfaatkan elemen genetik yang dapat berupa sekuen pengendali yang dimasukkan ke dalam genom tanaman. Selain itu, mekanisme ketahanan tanaman terhadap patogen juga dapat diketahui melalui analisis ekspresi gen yang terdapat dalam tanaman transgenik sehingga pengendalian patogen akan semakin efektif (Gustian 2002).
Enzim glukanase pada tanaman merupakan bagian yang penting dari mekanisme pertahanan tanaman dalam melawan patogen jamur. Enzim glukanase diharapkan mampu mencegah penyakit busuk pangkal batang yang menyerang kelapa sawit. Hal ini karena enzim glukanase mempunyai pertahanan yang mampu menghambat pertumbuhan jamur patogen (De La Cruz et al. 1995; Susanto 2002). Selain terdapat pada tanaman, enzim glukanase juga ditemukan pada kapang, yakni jenis Trichoderma sp. Potensi jamur Trichoderma sp. sebagai agen pengendali hayati sudah banyak diketahui.
Trichoderma sp. mampu menghasilkan metabolit sekunder yang berupa enzim hidrolitik ekstraselular, salah satunya adalah enzim glukanase (Nugroho 2003).
Adapun tujuan dari penelitian ini yakni untuk memperbanyak gen dengan mengisolasi dan menyisipkan gen GLN dengan menggunakan vektor donor dan vektor ekspresi yang dilakukan dalam kloning metode Gateway. Penggunaan kedua vektor ini berfungsi sebagai pembawa gen yang akan diperbanyak dengan pendekatan biologi molekular. Pendekatan biologi molekuler dapat digunakan untuk mempelajari gen-gen penyandi glukanase (GLN) yang terdapat pada kapang Trichoderma sp.
2
Adapun, teknik isolasi yang dilakukan pada penelitian ini adalah isolasi DNA dengan metode Oroczo-Castillo. Gen GLN hasil isolasi yang berukuran 1700 bp (Lo et al.
2001) terlebih dahulu disisipkan dalam vektor donor selanjutnya disisipkan ke dalam vektor ekspresi. Keduanya ditransformasikan ke dalam Escherichia coli.
Hipotesis dari penelitian ini yaitu gen GLN yang terkandung dalam kapang
Trichoderma sp. dapat disisipkan dalam vektor donor dan vektor ekspresi sehingga dapat diperbanyak dengan teknik kloning Gateway. Penelitian ini diharapkan mampu memberikan solusi alternatif penanganan penyakit busuk pangkal batang melalui pemanfaatan teknologi berbasis genetik.
.
TINJAUAN PUSTAKA
Kelapa Sawit
Kelapa sawit (Elaeis guineensis Jacq.) berasal dari Afrika. Tanaman ini dikenal di Indonesia sejak tahun 1915, namun baru ditanam secara komersial pada tahun 1970-an (Lubis 1992; Semangun 2000). Dalam bahasa Yunani, elaeis berasal dari kata elaion yang berarti minyak, sedangkan guineensis berasal dari guinea yaitu nama pantai barat Afrika, dan Jacq adalah nama botanis Amerika yaitu Jacquis.
Kelapa sawit (Elaeis quineensis Jacq.) dari famili Arecaceae merupakan salah satu sumber minyak nabati, dan merupakan primadona bagi komoditi perkebunan. Bagian yang paling utama untuk diolah dari kelapa sawit adalah daging buah. Bagian daging buah menghasilkan minyak kelapa sawit mentah yang diolah menjadi bahan baku minyak goreng. Kelebihan minyak nabati dari sawit adalah harga yang mura dan memiliki kandungan karoten tinggi. Sisa pengolahan buah sawit sangat potensial menjadi bahan campuran makanan ternak dan difermentasikan menjadi kompos (Lubis 1992).
Potensi kelapa sawit di Indonesia cukup besar. Luas perkebunan kelapa sawit mencapai 7.8 juta hektar (Gambar 1), jumlah produksi sebesar 22 juta ton, dan ekspor mencapai 16.5 juta ton pada tahun 2010 (Dahuri 2008). Kelapa sawit sebagai tanaman penghasil minyak sawit merupakan salah satu primadona tanaman perkebunan yang menjadi sumber penghasil devisa non migas bagi Indonesia. Cerahnya prospek komoditi minyak kelapa sawit dalam perdagangan
minyak nabati dunia telah mendorong pemerintah Indonesia untuk memacu pengembangan areal perkebunan kelapa sawit (Pahang 2010).
Gambar 1 Perkebunan Kelapa Sawit (Yanuar 2009).
Salah satu hambatan dalam budidaya kelapa sawit ialah adanya serangan patogen yang menimbulkan penyakit. Beberapa penyakit dan penyebab diantaranya adalah penyakit antraknosa (Botryodiplodia sp., Melaconium sp.), penyakit karat daun (Cephaleurous virescen), penyakit busuk tandan buah (Marasmus palmivorus), dan penyakit busuk pangkal batang (Ganoderma boninense). Dari beberapa penyakit di atas yang paling penting dan sangat merugikan adalah penyakit busuk pangkal batang (BPB) yang disebabkan oleh G. boninense (Turner 1981; Darmono 1993).
Penyakit Busuk Pangkal Batang pada Kelapa Sawit
Penyakit busuk pangkal batang (BPB) kelapa sawit pertama kali ditemukan pada tahun 1915 di Zaire. Pada saat itu penyakit ini dianggap tidak menimbulkan kerugian yang berarti (Turner 1981). Kemudian pada tahun 1920 patogen penyebab BPB di Afrika Barat diidentifikasi berasal dari genus Ganoderma. Spesies yang menyebabkan penyakit BPB dilaporkan berbeda-beda di setiap negara (Arifin et al. 2000). Di Indonesia diketahui penyebab BPB adalah spesies Ganoderma boninense.
3
lanjut atau sudah membentuk tubuh buah, sehingga tindakan pengendalian sulit dilakukan.
Pada tanaman gejala eksternal ditandai dengan menguningnya sebagian besar daun. Perkembangan penyakit menyebabkan tanaman tampak lebih pucat, pertumbuhannya terhambat, dan memiliki daun pedang yang tidak membuka (Arifin et al 2000).
Nekrosis dimulai dari daun tua kemudian menyebar ke daun yang lebih muda hingga ke mahkota tanaman dan akhirnya daun mati (Arifin et al 2000). Pada pangkal batang dekat pelepah terlihat gejala kecokelatan yang semakin lama akan menyebar ke bagian atas. Hal ini menandakan bahwa penampang pangkal batang telah mengalami pembusukan sekitar 50% atau lebih. Busuk kering yang merupakan gejala khas dari penyakit BPB terjadi pada jaringan dalam batang berwarna cokelat terang dengan jalur-jalur tidak teratur yang berwarna lebih gelap. Serangan lebih lanjut dapat mengakibatkan tanaman kelapa sawit tumbang karena jaringan kayu pada bagian pangkal batang mengalami pelapukan (Gambar 2) (Yanti & Susanto 2004).
Perkembangan penyakit selanjutnya ditunjukkan oleh pembentukan tubuh buah atau basidiospora dari kapang G. boninense
pada pangkal batang. Pada tanaman terserang tidak selalu ditemukan basidiospora karena basidiospora akan dibentuk setelah penyakit berkembang cukup lanjut. Ukuran dari basidiospora yang terus bertambah besar menunjukkan perkembangan penyakit semakin lanjut dan akhirnya menyebabkan kematian pada tanaman (Arifin et al. 2000).
Penularan penyakit BPB ini sebagian besar terjadi melalui mekanisme kontak akar dan batang sawit yang sakit. Sisa-sisa tanaman sakit yang kontak dengan akar tanaman sehat dapat meningkatkan penyebaran penyakit meskipun tidak melibatkan basidiopora.
Gambar 2 Penyakit Busuk Pangkal Batang pada Kelapa Sawit (Yanuar 2009).
Pertahanan Tanaman dengan Enzim Glukanase
Glukanase pada tanaman merupakan bagian yang penting dari mekanisme pertahanan tanaman dalam melawan patogen jamur. Penelitian yang dilakukan Donzelli et al. (2001) menyatakan bahwa glukanase mempunyai peranan penting dalam mekanisme pertahanan melawan patogen.
Glukanase berperan sebagai enzim autolitik (De la Cruz et al. 1λλ5). Enzim β -1,3-glukanase ditemukan secara luas di dunia tumbuhan (Buchner et al. 2002). Selain pada tumbuhan tingkat tinggi, enzim ini secara luas ditemukan pada sebagian besar bakteri dan jamur. Terdapat dua jenis glukanase yakni eksoglukanase dan endoglukanase (Cohen et al. 1999).
Glukanase diinduksi dalam responnya terhadap serangan patogen atau cekaman lingkungan. Bersama dengan kitinase, glukanase mempunyai pertahanan yang berhubungan secara fungsi biologis melalui penghambatan pertumbuhan jamur patogenik (Marco & Felix 2007). Enzim β-1,3-glukanase akan menghidrolisis polisakarida yang terdiri atas ikatan glikosidik pada dinding sel jamur patogen (Lahsen et al. 2001). Substrat khusus dari enzim ini adalah 1,3-glukan ditemukan di dinding sel jamur patogen.
Fakta bahwa glukanase dapat digunakan untuk meningkatkan respon pertahanan tanaman ditunjukkan oleh enzim laminarinase
(β-1,3-glukanase) yang berasal dari alga coklat (Laminaria digitata). Enzim ini efektif dalam melakukan pertahanan tanaman dengan menurunkan pertumbuhan Botrytis cinerea
dan Plasmopara viticola pada infeksi tanaman. Keragaman fungsi enzim ini menyediakan berbagai macam keuntungan pada pertahanan tanaman tingkat tinggi melawan serangan mikroorganisme (De la Cruz et al. 1995).
Kapang Trichoderma sp. sebagai Sumber Enzim Glukanase
Kapang Trichoderma sp. memiliki kemampuan sebagai pelindung tanaman. Spesies ini memiliki potensi dalam industri bioteknologi dan kesehatan karena kemampuannya menghasilkan berbagai enzim hidrolitik ekstraselular dan juga antibiotik. Kemampuan berbagai spesies ini untuk menghasilkan enzim hidrolitik dan senyawa-senyawa antifungi, antikhamir dan antibakteri, tak lepas dari kemampuannya untuk melindungi tanaman dari berbagai penyakit (Lahsen et al. 2001).
4
Kemampuan Trichoderma sp. untuk melindungi tanaman melibatkan beberapa mekanisme yang terkait dengan sifat biokimiawi spesies tersebut. Semua galur
Trichoderma sp. merupakan fungi biokontrol efektif yang akan tumbuh efektif di sekitar perakaran tanaman yang sehat, sehingga terjadi simbiosis mutualisme antara fungi biokontrol tersebut dengan tanaman yang dilindunginya. Oleh karena itu, mekanisme perlindungan tanaman oleh Trichoderma sp. tidak hanya melibatkan serangan terhadap patogen pengganggu, tetapi juga melibatkan produksi beberapa metabolit sekunder yang berfungsi meningkatkan pertumbuhan tanaman dan akar, dan memacu mekanisme pertahanan tanaman itu sendiri (Marco & Felix 2007). Mekanisme penyerangan terhadap patogen tanaman antara lain adalah melalui proses mikoparasitisme yang melibatkan produksi berbagai enzim hidrolitik (Kucuk & Kivank 2003). Salah satu golongan enzim hidrolitik yang dianggap cukup penting peranannya pada proses mikoparasitisme dari beberapa fungi patogen adalah enzim glukanase (Brunner et al. 2005).
Kapang Trichoderma sp. termasuk fungi yang mempunyai filamen. Sebagian besar diketahui sebagai pelindung tanaman terhadap penyakit tanaman yang disebabkan oleh jamur patogen (Howell 2003; Kucuk & Kivanc 2003). Koloni dari kapang Trichoderma
sp. berwarna putih, kuning, hijau muda, dan hijau tua (Alexopoulus & Mims 1979).
Susunan sel Trichoderma sp. bersel banyak berderet membentuk benang halus yang disebut dengan hifa. Hifa pada jamur ini berbentuk pipih, bersekat, dan bercabang-cabang membentuk anyaman yang disebut miselium. Miselium tersebut dapat tumbuh dengan cepat dan dapat memproduksi berjuta-juta spora, karena sifatnya inilah Trichoderma
sp. dikatakan memiliki daya kompetitif yang tinggi (Alexopoulus & Mims 1979). Dalam pertumbuhannya, bagian permukaan akan terlihat putih bersih, dan berwarna kusam pada miselium. Setelah dewasa, miselium memiliki warna hijau kekuningan (Howell 2003). Kapang ini memiliki bagian yang khas antara lain miselium bersekat, bercabang banyak dan konidia spora bersekat. Pada bagian ujungnya tumbuh sel yang bentuknya menyerupai botol (fialida) (Gambar 3). Sel ini dapat berbentuk tunggal atau berkelompok. Konidianya bergerombol membentuk seperti bola dan berkas-berkas hifa terlihat menonjol dengan jelas. Kapang Trichoderma sp. berkembangbiak secara aseksual dengan
membentuk cabang dari hifa (fialida)
(Gambar 4) (Frazier & Westhoff 1981; Samuels 2006).
Gambar 3 Konidia pada Trichoderma sp (Harman 2004).
Gambar 4 Hifa pada Trichoderma sp (Harman 2004).
Metode Polymerase Chain Reaction (PCR)
untuk Amplifikasi DNA
5
kultur bakteri di dalam tabung PCR (Bintang 2010). Proses PCR membutuhkan beberapa komponen. Empat komponen utama pada proses PCR adalah (1) DNA template/cetakan, yaitu fragmen DNA yang akan dilipatgandakan, (2) oligonukleotida primer, yaitu sekuen oligonukleotida yang digunakan untuk mengawali sintesis rantai DNA, (3) deoksiribonukleotida trifosfat (dNTP), terdiri atas dATP, dCTP, dGTP, dTTP, dan (4) enzim DNA polimerase yang melakukan katalisis reaksi sintesis rantai DNA. Komponen lain yang juga penting adalah larutan penyangga (buffer) (Bintang 2010).
Reaksi PCR umumnya terdiri atas reaksi denaturasi, penempelan primer (annealing), dan pemanjangan utas (extension) (Gambar 5) (Gilbert 2000; Jorde 2000). Reaksi pelipatgandaan suatu fragmen DNA dimulai dengan melakukan denaturasi DNA cetakan sehingga rantai DNA yang berantai ganda akan terpisah menjadi rantai tunggal. Denaturasi DNA dilakukan pada suhu sebesar 95oC selama 1-2 menit, kemudian suhu diturunkan menjadi 50oC sehingga primer akan menempel pada cetakan yang telah terpisah menjadi rantai tunggal. Primer akan membentuk jembatan hidrogen dengan cetakan pada daerah sekuen yang komplementer dengan sekuen primer. Proses PCR diakhiri dengan pemanjangan pada suhu 72oC. Banyaknya siklus amplifikasi tergantung pada konsentrasi DNA target didalam campuran reaksi (Yuwono 2006).
Gambar 5 Tahapan-tahapan dalam PCR (Jorde 2000).
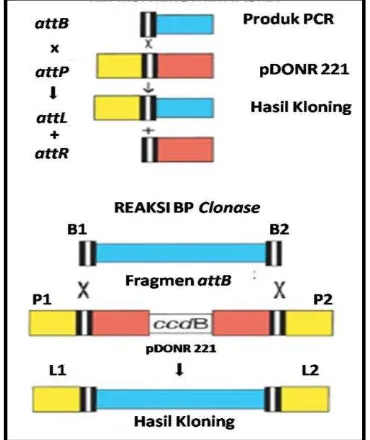
Metode Kloning Gateway
Sistem pengklonan Gatewaypertama kali dikembangkan oleh para peneliti di lembaga penelitian Life Technologies, Inc. Kemudian, teknik itu dikomersialisasikan oleh Invitrogen. Cara kerja sistem ini dengan mengonstruksi gen sisipan dengan rekombinasi situs spesifik pada bakteriofage lambda (Curtis & Grossniklaus 2003; Magnani et al. 2006; Karimi et al. 2007; Dubin et al. 2008). Secara umum sistem kloning gateway terdiri atas reaksi BP dan LR (Gambar 6 dan 7). Reaksi BP dikatalisis oleh enzim BP clonase II. Enzim ini membantu proses rekombinasi antara produk PCR (dilengkapi oleh situs
attB) dengan vektor donor yang mengandung situs attP. Setelah proses rekombinasi yang mempertemukan dua situ attB dan attP, produk PCR yang berupa fragmen DNA akan tersisipi pada vektor donor. Reaksi BP ini menghasilkan vektor plasmid rekombinan yang mengandung situs attL (Entry clones) (Karimi et al. 2007).
Hasil dari reaksi BP yang berupa entry clones merupakan substrat kunci pada reaksi selanjutnya, yakni reaksi LR. Reaksi LR dikatalisis oleh enzim LR clonase. Enzim ini membantu proses rekombinasi antara DNA fragmen yang terdapat dalam entry clones
(mengandung situs attL) dengan vektor ekspresi yang mengandung situs attR. Hasil dari reaksi ini adalah plasmid rekombinan yang mengandung DNA fragmen yang dilengkapi dengan situs attB seperti semula (Karimi et al. 2007).
6
Gambar 7 Tahapan-tahapan reaksi LR (Karimi et al. 2007)..
Komponen penting yang digunakan baik dalam metode kloning Gateway maupun metode kloning pada umumnya adalah vektor. Vektor adalah agen pembawa fragmen DNA agar bisa masuk ke dalam sel makhluk hidup. Vektor berfungsi memperbanyak fragmen DNA. Beberapa vektor kloning yang umum digunakan diantaranya adalah plasmid, vektor lamda, virus, kromosom, dan kosmid (Jorde 2000).
Vektor merupakan molekul DNA yang bisa bereplikasi secara mandiri dan dapat digunakan sebagai pembawa molekul DNA lain yang tidak mampu bereplikasi sendiri dalam sel. Oleh karena itu, dalam rekayasa genetika, vektor pengklonan digunakan sebagai pembawa DNA asing (DNA target) yang akan dimasukkan ke dalam sel inang (Yuwono 2006).
DNA target perlu disisipkan ke dalam vektor agar DNA tersebut dapat direplikasi di dalam sel inang. DNA target tetap dipertahankan keberadaannya di dalam sel inang. Vektor yang telah disisipi gen asing dinamakan DNA rekombinan. Vektor pengklonan dirancang dan dikembangkan secara khusus untuk digunakan pada sel inang tertentu. Sel inang dibedakan menjadi empat, yaitu sel bakteri, khamir, tumbuhan, dan hewan. Namun, sebagian besar percobaan menggunakan Escherichia coli sebagai inang. Secara umum, vektor untuk bakteri (E. coli)
dapat dibedakan antara lain vektor berbasis
plasmid, bakteriofaga , bakteriofaga M13,
dan gabungan antara plasmid dan bakteriofaga (Koolman 2005).
Vektor yang digunakan dalam teknik kloning Gateway adalah vektor donor dan vektor ekspresi dari Invitrogen. Vektor donor yang digunakan dalam kloning gateway adalah pDONR221. Vektor pDONR221 berukuran 4761 bp dan resisten terhadap kanamisin. Sedangkan vektor ekspresi yang digunakan adalah pGWB5 yang berukuran 3000 bp dan resisten terhadap kanamisin.
BAHAN DAN METODE
Bahan dan Alat
Bahan-bahan yang digunakan untuk media pertumbuhan kapang Trichoderma sp. antara lain, isolat kapang Trichoderma sp. dan media MEA (Malt Extract Agar) yang terdiri atas ekstrak malt, ekstrak khamir, dan agar. Bahan-bahan yang digunakan untuk isolasi dan amplifikasi DNA kapang antara lain
Trichoderma sp., larutan cethyl trimethyl ammonium bromide (CTAB) 10%, Tris-HCl 1 M pH 8.0, larutan etilenediamine tetraasetate
(EDTA) 0.5 M pH 8.0, larutan NaCl 5 M, etanol 80%, isopropanol, etanol absolut
dingin, β-merkaptoetanol, campuran
kloroform: isoamilalkohol (24:1), natrium asetat 3 M pH 5.2, nitrogen cair, polyvinyl-pyrolidone (PVP), bufer TE (Tris-HCl dan EDTA), molecular water (MW), RNase, Enzim Taq Polymerase, primer Gateway, primer M13, vektor pDONR221, dan vektor pGWB5. Bahan-bahan untuk elektroforesis antara lain loading buffer (bromfenol blue
2.5%: sukrosa 40%), agarosa, EtBr 1% (w/v), bufer TBE (Tris Boric EDTA) 0.5×, marker 1 kb Plus DNA ladder. Selain itu, Pure Link Quick Gel Extraction Kit dari Invitrogen digunakan untuk isolasi plasmid.
Alat-alat yang digunakan adalah sentrifus 5417R, spektrofotometer UVmini-1240, pH meter Cyberscan, penangas air, pipet mikro, pipet Mohr, bulp, sudip, tabung Eppendorf, tabung sentrifus 30 mL, tip plastik, neraca analitik Scaltec, inkubator, oven, kertas aluminium, mortar, perangkat elektroforesis
Toylab, cetakan gel, power supply, dan alat untuk dokumentasi hasil pengamatan elektroforesis UV (UV Transilluminator 2201
7
Metode Desain Primer
Primer disusun berdasarkan perancangan primer pada metode Gateway. Empat basa nukleotida GGGG diikuti situs attB kemudian ditambahkan 18-25 urutan basa nukleotida spesifik gen GLN. Situs attB ditambahkan pada masing-masing primer, yakni attB1 pada bagian forward dan attB2 pada bagian
reverse. Primer spesifik gen GLN dirancang secara manual dengan terlebih dahulu mencari sekuen DNA gen GLN yang diperoleh dari situs web http://www.ncbi.nlm.nih.gov/. Kualitas hasil rancangan diuji dengan bantuan program Gene Runner.
Pembuatan Media Kultur Kapang
Media MEA (Malt Extract Agar) ditimbang dalam neraca analitik, lalu dilarutkan dengan akuades hingga volumenya 50 mL dalam labu Erlenmeyer 100 mL. Pelarutan bahan dan media dibantu dengan pengaduk otomasis agar media larut seluruhnya. Setelah larut, media dan tabung reaksi serta cawan Petri disterilisasi dalam autoklaf dengan suhu 121oC tekanan 1 atm selama 15 menit.
Subkultur Kapang
Media MEA yang telah disterilisasi dituang ke cawan Petri steril. Penuangan dilakukan di laminar air flowcabinet secara aseptik. Setelah dituang ke dalam cawan Petri, media dibiarkan hingga padat. Setelah media MEA padat, isolat kapang Trichoderma sp. yang telah tersedia ditanam pada media tersebut. Isolat yang tersedia digores dan dipindahkan ke dalam media MEA secara steril. Setelah selesai, cawan ditutup dan diinkubasi selama 5 hari.
Isolasi DNA Kapang
Isolasi DNA dilakukan berdasarkan metode Orozco-Castillo et al. (1994). Sebanyak 0.5 g miselia kapang digerus dengan PVP dan N2 cair dalam mortar sampai
halus. Serbuk kapang halus yang dihasilkan dimasukkan ke dalam 1 mL buffer ekstraksi (CTAB 2%, EDTA 20mM, Tris-HCL 100 mM, dan NaCl 1.26 M) yang telah dipanaskan selama 10 menit dalam suhu 65oC dan diberi 10 L β-merkaptoetanol. Campuran tersebut dikocok, lalu dipanaskan selama 30 menit pada suhu 65oC sambil dikocok perlahan setiap 10 menit. Setelah itu, larutan buffer
ekstraksi diinkubasi selama 5 menit pada suhu kamar. Kemudian ditambahkan 1 mL larutan
kloroform: isoamilalkohol. Larutan disentrifus selama 10 menit dengan kecepatan 11.000 rpm pada suhu 25oC. Hasil sentrifugasi akan membentuk dua lapisan. Supernatan dipipet ke dalam tabung baru dan diekstrak kembali dengan 1 volume larutan kloroform: isoamilalkohol, selanjutnya disentrifus selama 10 menit dengan kecepatan 11.000 rpm pada suhu 25oC. Lapisan bagian atas dipipet ke dalam tabung baru dan ditambahkan isopropanol dingin sebanyak 1 volume. Larutan dalam tabung baru tersebut dikocok perlahan hingga homogen kemudian disimpan dalam kulkas (4oC) selama 30 menit. Setelah itu, disentrifus 10 menit 11.000 rpm pada suhu 25oC. Supernatan dibuang dan pelet DNA dikeringanginkan dengan cara membalikkan tabung. Pelet DNA dilarutkan dalam 500 L bufer TE. Lalu ditambahkan CH3COONa 3 M
pH 5.2 sebanyak 2 volume. Kocok hingga homogen dan simpan dalam suhu -20oC selama 30 menit atau semalam. Campuran tersebut kemudian disentrifus selama 10 menit 12.000 rpm pada suhu 4oC. Supernatan dibuang, pelet DNA dicuci dengan etanol 80% sebanyak 500 L kemudian disentrifus selama 2 menit 12.000 rpm pada suhu 4oC dan hasilnya dikeringkanginan dalam alat vakum berkecepatan tinggi. Pelet DNA yang sudah kering dilarutkan dalam 30 l buffer TE, kemudian ditambahkan RNase 25 g/mL, setelah itu simpan dalam suhu -20oC. Kualitas DNA diuji dengan elektroforesis gel agarose 1%. Sedangkan konsentrasinya diukur dengan spektrofotometer pada panjang gelombang 230, 260, dan 280 nm.
Isolasi
Elektroforesis Gel Agarosa
Pembuatan Gel Agarosa 1%. Sebanyak 0.3 gram agarosa ditambah dengan 30 mL TBE 0.5X, lalu dipanaskan hingga larut. Setelah larut dengan sempurna, larutan tersebut dibiarkan sampai hangat, kemudian
tambahkan 1.5 L EtBr dan kocok perlahan hingga homogen, lalu dituang ke dalam cetakan yang dilengkapi dengan sisir. Campuran tersebut didiamkan selama kira-kira 30 menit sampai gel tersebut benar-benar beku. Gel tersebut kemudian dimasukkan dalam alat elektroforesis dan direndam dengan bufer TBE 0.5X.
Elektroforesis Gel Agarosa. Sebanyak 1 L sampel DNA dicampurkan dengan 1 L
8
dengan bantuan sinar Ultra Violet (UV). Profil DNA yang terlihat kemudian disimpan dalam perangkat dokumentasi gel ( gel-documentation). Perangkat dokumentasi gel adalah alat yang terdiri atas kotak berbahan metal tempat penyinaran sinar UV yang dihubungkan dengan kamera digital. Kamera digital tersebut terpasang pada komputer atau laptop yang sudah terdapat software yang dapat menyimpan fotografi dari hasil elektroforeis. Dokumentasi gel berfungsi untuk mengambil foto gel hasil elektroforesis kemudian menyimpannya dalam bentuk data fotografi yang dapat dicetak.
Amplifikasi Gen Penyandi Glukanase
Proses PCR ini digunakan untuk memilih suhu annealing yang paling optimum untuk penempelan primer Gateway di antara suhu 45oC, 50oC, atau 55oC. Sebanyak 1 L DNA yang telah diisolasi dilarutkan dalam campuran berisi 17.5 L MW (Molecular Water), 2.5 L Bufer 10X, 0.75 L MgCl2, 1
L dNTPs, 1 Lprimer forward, 1 Lprimer reverse dan 0.25 L Taq polymerase. Campuran tersebut kemudian dimasukkan dalam mesin PCR dengan diawali pradenaturasi pada suhu 94oC selama 7 menit. Kemudian dilanjutkan dengan denaturasi pada suhu 94oC selama 45 detik,
annealing pada 45oC, 50oC, 55oC dan 60oC selama 45 detik, dan suhu pemanjangan 72oC selama 2 menit. Program ini dilakukan sebanyak 35 siklus. Proses PCR diakhiri dengan pemanjangan akhir pada suhu 72oC selama 5 menit. Hasil amplifikasi diverifikasi dengan elektroforesis gel agarosa 1% dengan tegangan 75 volt selama ± 45 menit. Pita DNA yang terbentuk dibandingkan dengan marker.
Ekstraksi dan Purifikasi DNA Hasil Amplifikasi
Bagian gel yang menunjukkan adanya fragmen DNA hasil amplifikasi dipotong menggunakan pemotong steril dan dimasukkan dalam tabung mikro. Satu tabung mikro berisi 400 mg gel yang menunjukkan DNA spesifik hasil amplifikasi. Sampel DNA dalam gel yang telah dimasukkan dalam tabung dimurnikan dengan menggunakan
Pure Link Quick Gel Extraction Kit dari Invitrogen. Gel dilarutkan dengan GSI (Gel Solubilization/larutan pemurni gel) sebanyak 1.200 L, selanjutnya dilakukan inkubasi pada suhu 50oC selama 15 menit dengan pengocokan setiap 3 menit hingga gel benar-benar larut.
Purifikasi dilakukan menggunakan prinsip sentrifugasi. Pertama, kolom gel dimasukkan dalam tabung. Gel yang sudah larut dipipet ke tengah kolom kemudian disentrifus 11000 g selama 2 menit pada suhu 25oC lalu dibuang supernatannya. Selanjutnya, ditambahkan 500-700 Lwash buffer kemudian disentrifus 11000 g pada suhu 25oC selama 2 menit lalu dibuang supernatannya. Kolom gel ditempatkan kembali pada tabung dan sentrifus 11000 g pada suhu 25oC selama 2-3 menit dalam keadaan kosong untuk membuang residu. Kolom gel dipindahkan ke tabung baru. Kemudian, sebanyak 30 µ L larutan penyangga untuk elusi dipipet ke tengah kolom lalu diinkubasi 2 menit pada suhu ruang. Selanjutnya dilakukan proses sentrifus dengan kecepatan 11000 g pada suhu 25oC selama 2 menit. Setelah itu, kolom gel dilepaskan dari tabung. Tabung yang berisi hasil elusi disimpan pada suhu -20oC Hasil elusi diverifikasi dengan elektroforesis gel agarosa 1%.
Pengklonan Fragmen DNA pada Vektor Donor
Rekombinasi Fragmen DNA Pada
Vektor Donor. Sebanyak 1 L (150 ng/ L) PCR produk hasil elusi dari reaksi amplifikasi dan 1 L (150 ng/ L) plasmid pDONR221 dilarutkan dengan 6 L buffer TE pH 8 sampai volume total 8 L. Selanjutnya, ditambahkan 2 LBP Clonase lalu diinkubasi pada suhu 25oC selama 2 jam/ Semalam. Setelah itu, 1 L proteinase K ditambahkan ke dalam campuran lalu diinkubasi pada suhu 37oC selama 15 menit. Hasil reaksi BP kemudian disimpan pada suhu -4oC sebelum dilakukan proses selanjutnya.
Transformasi ke Escherichia coli.
Transformasi diawali dengan penambahan hasil reaksi BP sebanyak 5 L ke dalam tabung yang berisi 200 L sel kompeten
E.coli XL-1 blue. Kemudian diinkubasi dalam es selama 30 menit. Selanjutnya dilakukan perlakuan kejut panas (heat shock) pada suhu 42oC selama 50 detik lalu diinkubasi kembali dalam es selama 10 menit. Setelah itu, 800 L campuran LB dan glukosa 20 mM ditambahkan ke dalam tabung kemudian dilakukan inkubasi pada inkubator bergoyang pada suhu 37oC selama 1.5 jam. Sebanyak 100
9
supernatan sisa dicampur kemudian disebar pada media seleksi yang mengandung kanamisin. Selanjutnya, dilakukan proses inkubasi pada suhu 37oC selama 16 jam lalu diamati koloni putih yang terbentuk. Kemudian, dipilih sejumlah koloni tunggal lalu dilakukan duplikasi pada media LA padat dan dikonfirmasi dengan PCR koloni.
Konfirmasi Koloni Transforman yang membawa Fragmen Sisipan. Sedikit koloni berwarna putih dipindahkan ke media LA yang mengandung antibiotik kanamisin dalam cawan Petri (duplikat koloni). Koloni dipindahkan pada tabung mikro. Pemindahan koloni dilakukan secara steril dalam laminar air flow cabinet. Komponen-komponen untuk melakukan PCR terdiri atas, MW, buffer complete, dNTP’s, Primer M13-F, Primer M13-R, dan Taq Polymerase. Kemudian dilakukan PCR untuk melisiskan dinding sel kemudian dilanjutkan dengan program lisis 96oC selama 5 menit, 50oC selama 1 menit 30 detik, 96oC selama 1 menit 30 detik, 45oC selama 1 menit 30 detik, 96oC selama 1 menit, 40oC selama 1 menit. Program dihentikan sejenak untuk penambahan komponen-komponen PCR yang telah dicampur sebanyak 5 L ke dalam masing-masing tabung. Kemudian program PCR dilanjutkan kembali dengan program pada suhu 94oC selama 30 detik, 55oC selama 1 menit, 72oC selama 2 menit. Setelah proses PCR selesai kemudian dielektroforesis dengan gel agarosa 1%.
Pengklonan Fragmen DNA dengan Vektor Ekspresi
Rekombinasi Fragmen DNA Pada
Vektor Ekspresi. Tahapan yang dilakukan hampir sama dengan rekombinasi dan transformasi fragmen DNA pada vektor donor sebelumnya. Perbedaannya, rekombinasi pada vektor ekspresi, sampel yang digunakan ialah amplikon hasil elusi dari reaksi BP.
Konfirmasi Koloni Transforman yang membawa Fragmen Sisipan. Tahapan yang dilakukan hampir sama dengan tahapan konfirmasi pada vektor donor. Perbedaannya, konfirmasi pada vektor ekspresi, primer yang digunakan ialah primer GTW GLN-forward, Primer GTW GLN-reverse.
HASIL DAN PEMBAHASAN
Primer Spesifik Gen GLN
Pemilihan primer oligonukleotida berguna untuk polymerase chain reaction
(PCR), hibridisasi oligo, dan sekuensing DNA. Desain primer yang tepat merupakan salah satu faktor penting dalam keberhasilan isolasi gen dan sekuensing DNA. Oleh karenanya, desain primer penting dilakukan sebelum isolasi dan amplifikasi DNA. Primer spesifik gen GLN dirancang secara manual. Situs attB ditambahkan pada masing-masing primer, yakni attB1 pada bagian forward dan
attB2 pada bagian reverse. Sekuen GGGG pada situs attB adalah bentuk efisiensi saat amplifikasi gen agar primer dapat melekat kuat pada gen target (lampiran 1).
Proses PCR menggunakan sepasang primer (forward dan reverse) yang dirancang untuk menempel secara spesifik pada daerah tertentu dalam DNA cetakan Trichoderma sp.
Kedua primer ini juga berfungsi untuk membatasi daerah DNA yang akan diamplifikasi. Situs attB akan mengarahkan gen saat proses amplifiksi sehingga arah penempelan primer akan benar.
Efektifitas primer dapat dilihat dari nilai TM yakni temperatur pada proses penempelan yang dihitung manual dengan rumus tertentu (lampiran 1). Adapun basa nitrogen yang digunakan dalam perhitungan nilai TM adalah basa nitrogen selain situs attB dan sekuen GGGG. Hal ini karena situs attB dan sekuen GGGG digunakan hanya untuk efektifitas reaksi.
Urutan primer spesifik Gateway yang mengandung situs attB dan urutan basa nukleotida gen GLN dapat dilihat pada Tabel 1 dan pada Lampiran 1. Parameter yang digunakan adalah persentase GC dan suhu Tm. Secara teori, persentase GC yang baik yakni lebih dari 50% dan suhu Tm tidak melebihi 70oC (Yuwono 2006) . Pada primer GLN-F persen GC dan suhu Tm yang didapat sebesar 55% dan 60oC. Sedangkan pada primer GLN-R persen GC dan suhu Tm yang didapat sebesar 45% dan 58oC. Secara umum, dapat dikatakan primer yang sudah didesain dapat digunakan pada proses PCR.
Tabel 1 Urutan primer gen GLN
Sekuen %GC;Tm
GLN R
5’GGGGCCAACTTTGTAC
AAGAAAGCTGGGTCTAA
AGGCATTGCGAGTAGT’3
45%; 58oC
GLN F 5’GGGGCCAACTTTGTAC AAAAAAGCAGGCTATGG
CGCCCTCAGTTACACT’3
55%; 60oC
Kultur Kapang Trichoderma sp.
10
2009). Untuk mendapatkan kondisi steril semua media dan alat yang akan digunakan terlebih dahulu disterilisasi dalam autoklaf dengan suhu 121oC dan tekanan 1 atm selama 15 menit. Selain itu, subkultur kapang dilakukan secara steril dalam laminar air flow cabinet.
Kontaminasi dapat terjadi apabila lingkungan kurang streril (Rachmawan 2001). Kontaminasi diketahui dengan perubahan warna media menjadi kuning keruh. Pertumbuhan koloni mikroba lain dan koloni kapang Trichoderma sp. dapat dibedakan berdasarkan bentuk koloninya. Umumnya, koloni kontaminan berupa koloni putih atau serabut putih kehitaman. Sedangkan, pertumbuhan Trichoderma sp. menghasilkan warna putih dan kuning kehijauan setelah 5 hari inkubasi pada kisaran suhu 25-28oC (Gambar 8). Kapang menghasikan enzim yang diproduksi secara ekstraseluler. Produksi glukanase oleh kapang dipengaruhi oleh beberapa faktor diantaranya, pH, suhu, sumber karbon dan sumber nitrogen (Rani & Panneerselvam 2009).
Pemilihan sumber karbon dan energi menjadi hal yang penting dalam proses produksi enzim oleh filamen jamur. Menurut Rachmawan (2001) Peran utama sumber karbon dan nitrogen adalah sebagai sumber energi dan bahan pembangun sel. Kekurangan sumber-sumber nutrisi ini dapat mempengaruhi pertumbuhan kapang dan dampak yang paling buruk adalah dapat menyebabkan kematian dari kapang yang ditumbuhkan pada media. Dalam hal ini ekstrak pati digunakan sebagai sumber karbon dan ekstrak khamir sebagai sumber nitrogen. Komposisi sumber karbon dan nitrogen yang dipakai dalam penelitian ini cukup memenuhi kebutuhan pertumbuhan kapang sehingga kapang dapat dengan baik tumbuh pada media.
Gambar 8 Hasil Kultur Kapang
Trichoderma sp.
DNA Kapang Trichoderma sp.
Keberhasilan isolasi DNA kapang dilihat dari tingkat kemurnian dan konsentrasi DNA yang diperoleh. Isolasi total DNA merupakan tahapan penting yang menentukan tahapan selanjutnya yakni amplifikasi DNA hasil isolasi. Agar hasil yang didapat pada tahap ini maksimal, perlu dipertimbangkan pemilihan metode isolasi yang sesuai dengan sampel sumber DNA. Isolasi DNA pada dasarnya dapat dilakukan dengan merusak dinding dan membran sel serta membran inti. Kemudian dilanjutkan dengan ekstraksi, pengendapan, dan pemurnian (Chang et al. 1993; Anam 2009). Adapun, metode yang digunakan penelitian ini adalah metode Orozco-Castillo
et al. (1994).
Kapang memiliki sel yang berfilamen sehingga dibutuhkan nitrogen cair yang berfungsi untuk memudahkan penggerusan sampai menjadi serbuk dan menjaga agar DNA tidak mengalami kerusakan (Kotake 1997). Bentuk serbuk akan mudah larut secara homogen dalam pelarut yang akan digunakan dalam proses isolasi. Perusakan dinding dan membran sel dilakukan dengan cara kimia dan fisika. Perusakan secara kimia dilakukan dengan penambahan buffer ekstraksi yang telah ditambahkan merkaptoetanol 1%. Sedangkan proses perusakan secara fisik dilakukan pengocokan dengan vorteks dan pemanasan pada suhu 65oC. Kemudian dilakukan proses sentrifugasi yang bertujuan memisahkan DNA dari dinding dan membran. Prinsip sentrifugasi memisahkan organel berdasarkan ukuran dan densitasnya. Selain itu, pemisahan didasarkan atas pengendapan partikel yang tersuspensi dalam satu wadah (Yuwono 2005).
Selanjutnya, campuran kloroform dan isoamilalkohol digunakan untuk mengendapkan pengotor seperti protein dan lemak. Larutan ini digunakan dalam proses ekstrasi DNA. Penambahan isopropanol dilakukan untuk mencuci DNA dari pengotor yang masih tersisa. Natrium asetat digunakan dalam tahapan presipitasi. Tahapan ini merupakan proses pengendapan DNA dari komponen lainnya. Setelah itu, endapan atau pelet RNA dicuci dengan alkohol. Pengendapan DNA dilakukan juga dengan metode sentrifugasi. Nuklease agak sulit dihilangkan dari endapan DNA dan bahkan kadang masih dapat bertahan setelah proses denaturasi panas dan ekstraksi (Zuo et al.
2005). Untuk mencegah aktivitas nuklease, sampel disentrifugasi pada suhu sebesar 4oC. Penggunaan suhu ini digunakan untuk
16
Budiani A et al. 2009. Kloning gen penyandi -1,6-glukanase kapang secara cepat dengan teknik RT-PCR menggunakan primer spesifik.
Menara Perkebunan 77(1): 47-57.
Chang S, Puryear J, Cairney J. 1993 A Simple and Efficient Method for Isolating RNA from Pine Trees. Plant Mol Biol Reporter 11: 113-116.
Cohen et al. 1999. Molecular characterization
of a novel β-1,3-exoglucanase related to mycoparasitism of Trichoderma harzianum. Gene 226: 147-154.
Curtis MD, Grossniklaus U. 2003. A Gateway cloning vector set for High-Througput functional analysis of genes in plants. Plant Physiology.
133: 462-469.
Dahuri R. 2008. Kedaulatan pangan bangsa. [Terhubung berkala]. http://www.tar-getmdgs.org. [17 juli 2011].
Darmono TW, Suharyanto, Darussamin A, Moekti GR. 1993. Antibodi poliklonal terhadap filtrat pencucian kultur miselium Ganoderma sp. Abstrak Hasil Penelitian Kelapa Sawit. Bogor: Pusat Perpustakaan dan Penyebaran Teknologi Pertanian.
De la Cruz J, Pintor JA, Benitzer T, Llobel A. 1995a. Purification and characterization of an endo-β -1,6-glucanase from Trichoderma harzianum that is related to its mycoparasitism. J Biotech 177:64-71.
Donzelli B, Lorito M, Scala F, Harman G. 2001. Cloning, sequence and structure of a gene encoding an antifungal glucan 1,3-beta-glucosidase from Trichoderma atroviride (T. harzianum). Gene 277: 199-208.
Dubin MJ, Bowler C, Benvenuto G. 2008. A modified Gateway cloning strategy for overexpressing tagged proteins in plants. Plant Methods: 1-3.
Frazier WC, Westhoff AC. 1981. Food Microbiology. New Delhi: McGraw Hill. Published Co. Ltd.
Gilbert HF. 2000. Basic Concepts in Biochemistry. New York: McGraw-Hill Companies, Inc.
Gustian. 2002. Transformasi genetik dengan bantuan Agrobacterium dan regenerasi tanaman transgenik pada Kedelai (Glycine max L. Mer). Disertasi. Program Pascasarjana Institut Pertanian Bogor.
Harman GE, Howell CR, Viterbo A, Chet I, Lorito M. 2004. Trichoderma species: opportunistic, avirulent plant symbionts. [Terhubung Berkala] http://www.ncbi.nlm.nih.gov/entrez/q uery/author/LoritoM. [2 Oktober 2011]
Hart H, Craine LE, Hart DJ. 2003. Organic Chemistry: A Short Course. Houghton Mifflin College Div.
Harvey D. 2000. Modern Analitycal Chemistry. New York. McGraw-Hill.
Heriyadi. 2009. Dampak Ekologi Pengembangan Kelapa Sawit untuk Bioenergi.[Terhubung Berkala]. http:/energi.infogue.com/dampak_ek ologi_pengembangan_kelapa_sawit _untuk_bioenergi.
Howell CR. 2003. Mechanisms Employed by
Trichoderma Species in the Biological Control of Plant Diseases: The History and Evolution of Current Concepts. Plant Disease 87 (1): 4-10.
Jorde LB. 2000. Biochemistry Notes. New York. Kaplan, Inc.
Karimi et al. 2007. Recombinational cloning with plant Gateway vectors. Plant Physiology. 145: 1144-1154.
Koolman J, Roehm KH. 2005. Color Atlas of Biochemistry. Stuttgart: Thieme.
Kotake T, Nakagawa N, Takeda K, Sakurai M. 1997. Purification and characterization of a wall-bound exo-1,3- β-glucanase from barley seedlings. Plants Cell Physiol
38:194-200.
17
of Their Antifungal, Biochemical and Physiological Features. Turk J Biol. 27: 247-253
Lahsen HA et al. 2001. An Antifungal Exo-_-1,3-Glucanase (AGN13.1) from the Biocontrol Fungus Trichoderma harzianum. App and Env Microbiol. 67 (12): 5833-5899.
Lo N et al. 2000. Evidence from multiple gene sequences indicates that termites evolved from wood-feeding cockroaches. Current Biology. 10 (13): 801-805.
Lubis AU. 1992. Kelapa Sawit (Elaeis gunensis Jacq.) di Indonesia
Pematangsiantar: PPPM.
Marco JL, Felix CR. 2007. Purification and Characterization of a b-Glucanase Produced by Trichoderma harzianum
Showing Biocontrol Potential.
Journal of Biology and Technology. 50: 21-29.
Magnani E, Bartling L, Hake S. 2006. From Gateway to Multisite Gateway in one recombination event. Molecular Biology: 1-7.
Metzler DE. 2000. Biochemistry, The Chemical ractions of Living Cell.
Elsevier Academic Press.
Mikkelsen SR, Corton E. 2004. Bioanalytical Chemistry. New York: Wiley-Liss, Inc., Publication
Nugroho TT et al. 2003. Isolasi dan karakterisasi sebagian kitinase Trichoderma viridae TNJ63. Jurnal Natur Indonesia. 5(2): 101-106.
Orozco-Castillo C et al. 1994. Detection of genetic diversity and selective gene introgression in coffee using RAPD markers. Theor. Appl. Genet. 87: 934-940.
Pahang I. 2010. Panduan Lengkap Kelapa Sawit; Manajemen Agribsnis dari Hulu hingga Hilir. Jakarta: Penebar Swadaya.
Rachmawan O. 2001. Sumber Kontaminasi dan Teknik Sanitasi. Jakarta: Departemen Penididikan Nasional. Rani, C. and A. Panneerselvam. 2009.
Influence of Environmental and Nutritional Parameters on Lipase Production. ARPN Journal of Agricultural and Biological Science. 4(5). 39-43.
Sambrook J, Fristsch EF, Maniatis T. 1989.
Molecular Cloning: A Laboratory Manual. New York: Cold Spring Harbour Laboratory Press.
Samuels GJ. 2006. Trichoderma: Systematics, the Sexual State, and Ecology.
Phytopathology. 96:195-206
Semangun H. 2000. Penyakit-Penyakit Tanaman Perkebunan di Indonesia. Yogyakarta: Gajah Mada University Press.
Smith AD et. al. 2000. Oxford Dictionary of Biochemistry and Biology Molecular.
New York: Oxford University Press, Inc.
Soetrisno N. 2008. Peranan Industri Sawit dalam Pengembangan Ekonomi Regional : Menuju Pertumbuhan Partisipatif Berkelanjutan. [Makalah] Universitas Sumatera Utara 6 Desember 2008.
Susanto A. 2002. Kajian Pengendalian Hayati
Ganoderma boninese Pat. Penyakit Busuk Pangkal Batang Kelapa Sawit. Disertasi. Program Pascasarjana Institut Pertanian Bogor.
Susanto A, Sudharto PS, Purba RY. 2005. Enchancing biological control of basal stem root disease (Ganoderma boninense) in oil palm plantations.
Mycopathologia 159 (1): 153-157.
Turner PD. 1981. Oil Palm Disease and Disorders. Oxford University Press.
Yanti F, Susanto A. 2004. Cara praktis isolasi tubuh buah Ganorderma boninense
pada medium Potato Dextrose Agar
18
Yanuar. 2009. Pengembangan Kelapa Sawit Nasional, Mewujudkan Visi Indonesia 2020. [Terhubung Berkala]. http://ditjenbun.deptan.go.-id/index.php?option=com_content&v iew=article&id=85:pengembangan- kelapa-sawit-nasional-mewujudkan-visi-indonesia-2020&catid=36:news. [15 Juli 2011].
Yuwono T. 2005. Biologi Molekular. Jakarta: Erlangga.
Yuwono T. 2006. Teori dan Aplikasi Polymerase Chain Reaction.
Yogyakarta: Penerbit Andi.
Zuo Y et al. 2007. Crystal structure of Rnase T, and exoribonuclease involved in tRNA maturation and end turnover.
19
20
Lampiran 1 Desain primer metode Gateway
Forward primer
:
attB1
Gateway GLN-
F 5’
-GGGG
CCA ACT TTG TAC AAA AAA GCA GGC T
ATGGCGCCCTCAGTTACACT- 3
’
18-25 basa nukleotida gen GLN yang digunakan untuk menghitung Tm
Reverse primer
:
attB2
Gateway GLN-
F 5’ –
GGGG
CC AAC TTT GTA CAA GAA AGC TGG GT
CTAAAGGCATTGCGAGTAGT- 3
’
18-25 basa nukleotida gen GLN yang digunakan untuk menghitung Tm
Perhitungan Tm:
Forward primer
: 4(G+C) + 2(A+T) = 4(4+7) + 2(3+5) = 60
Maka, Tm untuk
Forward primer
60
oC
21
Lampiran 2 Skema pembuatan media dan subkultur kapang
Media
Malt Extract Agar
(
Malt Extract
5 g, Ekstrak
Khamir 1.25 g, Agar 5 g)
Pembuatan media dan
sterilisasi dengan autoklaf
Penuangan media pada cawan
Petri
Subkultur kapang dengan
teknik sebaran pada
media
Inkubasi kapang selama 5
hari pada suhu kamar
22
Lampiran 3 Tahapan isolasi DNA Kapang
1 g sampel kapang
Trichoderma
sp.
Pembuatan serbuk sampel dengan 0.1 g PVPP
dan N
2Ekstraksi sampel dengan 5 mL buffer
ekstraksi
Ditambahkan isopropanol (1:1)
Supernatan diambil dan ditambahkan 5 ml larutan
Kloroform: Isoamilalkohol
Ditambahkan 5 mL Kloroform: Isoamilalkohol
Resuspensi pelet dengan 1 mL buffer TE
Pelet dicuci dengan etanol 70 %
dan 2,5 mL etanol absolut
Resuspensi pelet 100 l buffer TE atau
ddH
2O, ditambahkan RNase 25 g/ml
Inkubasi 65oC, 30 menit
Sentrifugasi 11.000 rpm, 10 menit
Sentrifugasi 11.000 rpm, 10 menit
Disimpan suhu
4
oC, 30 menit
Sentrifugasi 11.000 rpm, 10 menitDitambahkan CH
3COONa 3 M pH 5.2, 2,5
mL etanol absolut
Disimpan suhu
-20
oC, 30 menit
Sentrifugasi4
oC
, 12.000 rpm, 10 menit23
Lampiran 4 Skema pembuatan
buffer
TBE dan gel agarosa 1% (Sambrook
et al.
1989)
-
Pembuatan Bufer TBE
-
Pembuatan Gel Agarosa 1%
108 gram Tris Base, 55 gram Asam
Borat, dan 40 ml EDTA 0.5 M
disiapkan
Dilarutkan dalam aquades
Ditera sampai 1 liter
Sebanyak 0.3 gram agarosa ditimbang dan
dilarutkan dalam 30 mL bufer TBE 0.5x.
Dilakukan pemanasan kemudian
diamkan sampai hangat
Ditambahkan 1.5 µL EtBr
Larutan dituang dalam cetakan yang
telah disusun bersama sisirnya.
24
Lampiran 5 Perhitungan konsentrasi dan bobot molekul DNA
Perhitungan konsentrasi DNA Total
DNA 1
[ ] RNA total= 0,005 x 50 x 100
25
26
27
Lampiran 8 Hasil subkultur koloni bakteri
Duplikat dari koloni hasil reaksi BP dengan sisipan gen penyandi glukanase
(GLN) pada vektor donor