KLASIFIKASI FRAGMEN
METAGENOME
MENGGUNAKAN
METODE
SUPPORT VECTOR MACHINE
(SVM)
ARINY
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM INSTITUT PERTANIAN BOGOR
PERNYATAAN MENGENAI SKRIPSI DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA
Dengan ini saya menyatakan bahwa skripsi berjudul Klasifikasi Fragmen Metagenome Menggunakan Metode Support Vector Machine (SVM) adalah benar karya saya dengan arahan dari komisi pembimbing dan belum diajukan dalam bentuk apa pun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir skripsi ini.
Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
ABSTRAK
ARINY. Klasifikasi Fragmen Metagenome Menggunakan Metode Support Vector Machine (SVM). Dibimbing oleh WISNU ANANTA KUSUMA dan MUSHTHOFA.
Analisis metagenome merupakan salah satu bidang kajian bioinformatika yang penting. Bidang ini terkait dengan analisis sequences genom yang diperoleh langsung dari lingkungan. Tujuan penelitian ini adalah melakukan klasifikasi fragmen metagenome ke dalam beberapa taksonomi dengan menggunakan metode support vector machine (SVM). Proses ekstraksi fitur dilakukan dengan menggunakan spaced k-mers. Proses klasifikasi diawali dengan membuat model menggunakan data latih dari 381 organisme. Berdasarkan hasil penelitian ini dapat diketahui bahwa nilai akurasi untuk fragmen berukuran pendek (400 bp) ialah 65.3% pada takson genus dan 82.1% pada takson filum. Sementara itu, nilai akurasi meningkat secara signifikan menjadi 95.4% pada takson genus dan 97.6% pada takson filum, ketika menggunakan fragmen yang berukuran panjang (10 Kbp). Dari hasil tersebut dapat disimpulkan bahwa nilai akurasi akan semakin tinggi seiring dengan semakin panjangnya ukuran fragmen dan semakin tingginya tingkat taksonomi. Selain itu, dari hasil penelitian juga dapat disimpulkan bahwa metode ekstraksi fitur yang digunakan sudah sangat baik dan menghasilkan data dengan kondisi linearly separable.
Kata kunci: binning, metagenome, spaced k-mers, SVM
ABSTRACT
ARINY. Metagenome Fragment Binning Using Support Vector Machine (SVM) Method. Supervised by WISNU ANANTA KUSUMA and MUSHTHOFA.
Metagenome analysis is one of the most important bioinformatics field. This field is related to genome which is taken directly from the environment. The purpose of this research is to classify metagenome fragment into some taxonomic levels using support vector machine (SVM) method. Feature extraction is performed using spaced k-mers. Classification process is conducted by creating model using the training data from 381 organisms. The evaluation results show that the accuracies for short fragments (400 bp) are 65.3% and 82.1% at genus level and phylum level, respectively. Meanwhile, the accuracies increase significantly for long fragments (10 kbp), with a value of 95.4% at genus level and 97.6% at phylum level. It can be stated that the accuracy will be increased with the increasing of fragments length and higher taxonomic levels. In addition, the results of the study also conclude that the feature extraction methods used was very good and produce data with linearly separable conditions.
Skripsi
sebagai salah satu syarat untuk memperoleh gelar Sarjana Komputer
pada
Departemen Ilmu Komputer
KLASIFIKASI FRAGMEN
METAGENOME
MENGGUNAKAN
METODE
SUPPORT VECTOR MACHINE
(SVM)
ARINY
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM INSTITUT PERTANIAN BOGOR
Judul Skripsi : Klasifikasi Fragmen Metagenome Menggunakan Metode Support Vector Machine (SVM)
Nama : Ariny NIM : G64090055
Disetujui oleh
Dr Wisnu Ananta Kusuma, ST MT Pembimbing I
Mushthofa, SKom MSc Pembimbing II
Diketahui oleh
Dr Ir Agus Buono, MSi MKom Ketua Departemen
PRAKATA
Puji dan syukur penulis panjatkan kepada Allah subhanahu wa ta’ala atas segala karunia-Nya sehingga karya ilmiah ini berhasil diselesaikan. Shalawat dan salam penulis sampaikan kepada Nabi Muhammad shallallahu ‘alaihi wasallam, keluarganya, sahabatnya, serta umatnya hingga akhir zaman. Tema yang dipilih dalam penelitian yang dilaksanakan sejak bulan September 2012 ini ialah klasifikasi fragmen metagenome, dengan judul Klasifikasi Fragmen Metagenome Menggunakan Metode Support Vector Machine (SVM).
Terima kasih penulis ucapkan kepada seluruh pihak yang telah berperan dalam penelitian ini, yaitu:
1 Ayahanda Arnedy Syamsu, Ibunda Dona Elfira, Kakak Ohayyo Randy Akbar, serta Aditya Ramadhan atas kasih sayang, doa, semangat, dan dorongan kepada penulis sehingga dapat menyelesaikan penelitian ini.
2 Bapak Dr Wisnu Ananta Kusuma, ST MT dan Bapak Mushthofa, SKom MSc selaku pembimbing, yang telah memberikan banyak ide, masukan, dan dukungan kepada penulis.
3 Bapak Prof. Antonius Suwanto yang telah bersedia menjadi penguji, dan memberikan saran yang berharga sehingga tulisan ini menjadi lebih baik dari sebelumnya.
4 Rekan-rekan terdekat Anisaul Muawwanah, Sabarina Hidayat, Husnul Khotimah, Dewi Humaira, Dian Lestari Auliani, Lizza Amini Gumilar, dan Viani Rahmawati yang telah memberi dukungan dan bantuan.
5 Aries Fitriawan, Muhammad Luthfi Fajar, Erwin Musa, dan Aditya Erlangga yang telah membantu mengatasi kesulitan pemrograman yang penulis hadapi. 6 Rekan-rekan Ilmu Komputer angkatan 46 yang saling menyemangati selama
pengerjaan penelitian di tahun yang sama.
7 Seluruh rekan satu bimbingan Bapak Wisnu yang tidak dapat disebutkan satu persatu dan pihak-pihak lainnya.
Semoga penelitian dan tulisan ini dapat memberikan manfaat.
DAFTAR ISI
DAFTAR TABEL vii
DAFTAR GAMBAR vii
DAFTAR LAMPIRAN vii
PENDAHULUAN 1
Latar Belakang 1
Perumusan Masalah 2
Tujuan Penelitian 2
Manfaat Penelitian 3
Ruang Lingkup Penelitian 3
METODE 3
Pengumpulan Data 4
Pembagian Data 4
Praproses Data 4
Ekstraksi Fitur 4
Support Vector Machine (SVM) 5
Grid Search 7
Pelatihan SVM 7
Pengujian SVM 7
Analisis 7
Implementasi 8
HASIL DAN PEMBAHASAN 8
Pembagian Data 8
Praproses Data 9
Ekstraksi Fitur 9
Grid Search 10
Klasifikasi SVM 10
Analisis 11
Implementasi 18
SIMPULAN DAN SARAN 18
Simpulan 18
Saran 19
DAFTAR TABEL
1 Hasil akurasi berdasarkan tingkat taksonomi dan panjang fragmen 11 2 Perbandingan waktu komputasi pembuatan model pada setiap kernel 15 3 Daftar organisme yang memiliki similarity dari hasil alignment
Burkholderia sp JV3 pada BLAST 17
DAFTAR GAMBAR
1 Tahapan penelitian 3
2 Pola spaced k-mers dengan parameter w = 3 dan d = 0, 1, 2 (Kusuma
2012) 5
3 Kondisi linearly separable dengan hyperlane yang memiliki margin
terbesar 5
4 Contoh hasil praproses data dengan jumlah fragmen 9600 dan panjang
fragmen 400 bp 9
5 Hasil grid search mengeluarkan nilai parameter c dan γ terbaik serta
akurasi 5-cross validation 10
6 Hasil akurasi berdasarkan tingkat taksonomi dan panjang fragmen 12
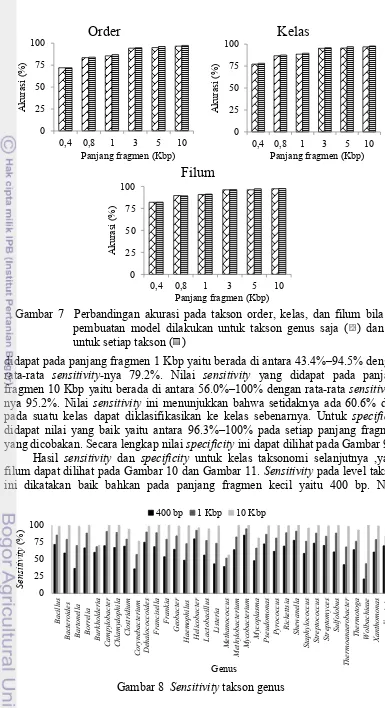
7 Perbandingan akurasi pada takson order, kelas, dan filum bila
pembuatan model dilakukan untuk takson genus saja ( ) dan untuk setiap
takson ( ) 13
8 Sensitivity takson genus 13
9 Specificity takson genus 14
10 Sensitivity takson filum 14
11 Specificity takson filum 14
12 Akurasi menggunakan 4 fungsi kernel berbeda untuk panjang fragmen 10
Kbp dan takson genus 15
13 Hasil klasifikasi data uji Burkholderia sp JV3 (genus Stenotrophomonas)
pada panjang fragmen 400 bp 16
14 Hasil klasifikasi data uji Burkholderia sp JV3 (genus Stenotrophomonas)
pada panjang fragmen 1 Kbp 16
15 Hasil klasifikasi data uji Burkholderia sp JV3 (genus Stenotrophomonas)
pada panjang fragmen 10 Kbp 16
DAFTAR LAMPIRAN
1 Daftar nama organisme data latih 21
2 Daftar nama organisme data uji 30
3 Daftar tingkat taksonomi yang digunakan mulai dari genus, order, kelas
dan filum 35
4 Daftar hasil praproses data yang menyatakan jumlah sequence di setiap tingkat takson dan panjang fragmennya untuk data latih 37 5 Daftar hasil praproses data yang menyatakan jumlah sequence di setiap
6 Nilai parameter c dan γ terbaik yang didapat pada tahap grid search 43 7 Perbandingan akurasi yang dihasilkan dari pembuatan model hanya pada
takson genus dengan pembuatan model disetiap tingkat takson 44
PENDAHULUAN
Latar Belakang
Analisis metagenome merupakan salah satu bidang kajian bioinformatika yang penting dan akan terus berkembang. Studi yang mempelajari metagenome ini disebut metagenomics. Berbeda dengan studi yang mempelajari genom (genomics), metagenomics tidak memerlukan pure clonal cultures dari sequencing individu tertentu. DNA yang berasal dari berbagai organisme dalam suatu komunitas mikrob dapat diperoleh melalui proses sequencing secara langsung (McHardy dan Rigoutsos 2007).
Proses DNA sequencing komunitas mikrob secara langsung ini menghasilkan fragmen-fragmen dari berbagai organisme yang bercampur. Kondisi ini memungkinkan fragmen dari suatu organisme memiliki overlap dengan fragmen dari organisme lain. Kondisi ini dapat menyebabkan kesalahan perakitan fragmen-fragmen yang terkandung di dalam komunitas tersebut dan menghasilkan cymeric contigs (Wooley et al. 2010). Untuk meminimalkan cymeric contigs, salah satu solusinya adalah dengan melakukan sequence assembly dan binning secara berulang. Proses binning dalam persepktif ilmu komputer dapat dilakukan dengan metode supervised atau unsupervised learning. Pada metode supervised learning, fragmen-fragmen diklasifikasikan berdasarkan level taksonomi tertentu, misalnya yang paling rendah ialah level genus, mengingat sulitnya mengklasifikasikan fragmen pada level spesies.
Beberapa peneliti telah melakukan penelitian yang terkait dengan pengklasifikasian fragmen metagenome ini. McHardy et al. (2007) melakukan penelitian untuk mengklasifikasikan fragmen metagenome dengan menggunakan data latih 340 organisme. Metode yang digunakan ialah multiclass support vector machine (SVM) dengan frekuensi k-mers sebagai fiturnya. Aplikasi yang dibangun dinamai PhyloPythia. Hasil akurasi yang diperoleh terbilang cukup tinggi khususnya untuk panjang fragmen ≥ 5 Kbp yaitu antara 60% sampai ˃ 90% di setiap tingkat takson. Tetapi akurasi ini terus menurun dengan signifikan jika menggunakan fragmen dengan panjang ≤ 3 Kbp. Pada fragmen dengan panjang 3 Kbp hanya diperoleh akurasi sebesar 40% sedangkan untuk panjang fragmen 1 Kbp akurasi yang diperoleh < 10%. Selain itu, PhyloPythia menggunakan 5-mers, yang berarti matriks fitur yang dihasilkan memiliki dimensi 45 = 1024. Proses ekstraksi fitur yang melibatkan dimensi yang besar ini memerlukan waktu komputasi yang tinggi.
2
sama. Penelitian ini hanya mengklasifikasikan organisme ke dalam tingkat takson genus. Organisme-organisme tersebut merupakan anggota dari 3 jenis genus berbeda. Hasil akurasi yang diperoleh dari penelitian ini cukup tinggi yaitu 78% untuk panjang fragmen 500 bp sampai dengan 87% untuk panjang fragmen 10 Kbp. Namun, ketika metode ini diterapkan pada dataset berukuran besar (374 organisme), akurasi yang diperoleh menurun secara signifikan, yaitu sebesar 30% untuk panjang fragmen 1 Kbp pada level genus.
Oleh karena itu, untuk mengatasi masalah komputasi yang disebabkan oleh dimensi fitur yang besar dan menurunnya akurasi jika menggunakan dataset dari komunitas organisme yang besar, pada penelitian ini diusulkan metode multiclass SVM dengan frekuensi spaced k-mers sebagai fiturnya. Fitur hasil ekstraksi dengan menggunakan spaced k-mers hanya terdiri atas 192 dimensi. Adapun dataset yang digunakan untuk data latih terdiri atas 381 organisme dan untuk data uji terdiri atas 200 organisme. Fragmen DNA dari organisme tersebut akan diklasifikasikan ke dalam tingkat takson genus, order, kelas, dan filum. Selain itu juga digunakan 4 fungsi kernel berbeda pada pelatihan SVM untuk mengetahui kernel yang dapat menghasilkan model terbaik untuk pengklasifikasian fragmen metagenome.
Perumusan Masalah
Adapun permasalahan yang akan menjadi bahan analisis pada penelitian ini ialah:
1 Berapa akurasi yang dapat diperoleh jika digunakan metode SVM dengan 4 fungsi kernel yang akan diterapkan pada penelitian ini?
2 Bagaimana pengaruh panjang fragmen yang digunakan terhadap hasil akurasi? 3 Bagaimana kinerja metode klasifikasi yang diusulkan ini ketika melakukan
pengklasifikasian fragmen metagenome yang berasal dari organisme-organisme baru?
4 Apakah pembuatan model untuk setiap tingkat takson bisa menghasilkan akurasi yang lebih baik bila dibandingkan dengan pembuatan model pada takson genus saja?
Tujuan Penelitian
Tujuan penelitian ini ialah:
1 Mengklasifikasikan fragmen metagenome ke dalam tingkat taksonominya dengan metode SVM menggunakan 4 fungsi kernel. Keempat kernel yang digunakan yaitu Gaussian radial basis function (RBF), linear (polinomial berderajat 1), kuadratik (polinomial berderajat 2), dan polinomial berderajat 3. 2 Mengetahui pengaruh panjang fragmen yang digunakan terhadap hasil akurasi. 3 Mengetahui kinerja metode pengklasifikasian terhadap fragmen yang berasal
dari organisme baru.
3
Manfaat Penelitian
Manfaat dari penelitian ini diharapkan dapat memberikan kontribusi untuk mendukung proses analisis metagenome sequence.
Ruang Lingkup Penelitian
Ruang lingkup penelitian ini meliputi:
1 Data latih terbatas hanya 381 organisme yang termasuk dalam 48 genus, 31 order, 20 kelas, dan 13 filum.
2 Data uji terbatas hanya 200 organisme yang termasuk dalam taksonomi yang sama dengan data latih, dengan tambahan 1 genus yang tidak ada pada modelnya pada data latih untuk mengetahui kinerja pengklasifikasian SVM. 3 Fragmen yang digunakan dihasilkan dari perangkat lunak MetaSim yang
mensimulasikan Illumina sequencer. Fragmen yang dihasilkan memiliki panjang yang tetap dan tidak mengandung sequencing error.
4 Level taksonomi yang digunakan yaitu genus, order, kelas, dan filum.
METODE
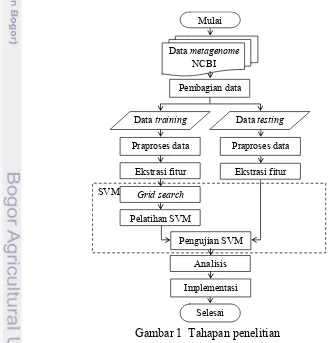
Penelitian ini dilaksanakan dalam beberapa tahapan yang diilustrasikan pada Gambar 1.
Gambar 1 Tahapan penelitian
Mulai
Data metagenome NCBI
Pembagian data
Praproses data Praproses data
Data training Data testing
Ekstrasi fitur Ekstrasi fitur
SVM
Pelatihan SVM
Pengujian SVM Grid search
Selesai Analisis
4
Pengumpulan Data
Data latih dan data uji yang digunakan pada penelitian ini ialah data metagenome yang diunduh dari situs National Centre for Biotechnology Information (NCBI). NCBI merupakan suatu institusi yang fokus di bidang biologi molekuler dan menjadi sumber informasi untuk perkembangan bidang tersebut. Data metagenome ini merupakan sequence DNA organisme dengan format FastA. Alamat untuk mengunduh data ini yaitu ftp://ftp.ncbi.nih.gov/ genomes/Bacteria/.
Pembagian Data
Pada penelitian ini organisme yang digunakan terbatas pada 381 organisme untuk data latih, dan 200 organisme untuk data uji. Pemilihan data uji dilakukan dengan mengambil organisme selain data latih yang juga termasuk ke dalam genus yang sama, serta 1 genus yang tidak termasuk dalam data latih. Pengambilan data uji yang tidak ada modelnya pada data latih ini untuk melihat kinerja hasil pengklasifikasiannya.
Praproses Data
Pada tahap praproses data, sequence DNA metagenome yang sudah dipilih lalu diuraikan fragmennya menggunakan perangkat lunak MetaSim (Richter et al. 2008). MetaSim adalah perangkat lunak untuk mensimulasikan sequencer. Data yang diproses dibaca berulang kali disesuaikan dengan kebutuhan penelitian. Pada penelitian ini data yang disiapkan untuk data latih berjumlah 9600 dan 320 ribu fragmen, sedangkan untuk data uji berjumlah 100 ribu fragmen. Panjang fragmen yang ditetapkan untuk setiap kali pengolahan yaitu 400 bp, 800 bp, 1 Kbp, 3 Kbp, 5 Kbp, dan 10 Kbp. Maka akan dilakukan 12 kali pengolahan untuk data latih dan 6 kali pengolahan untuk data uji, sehingga dihasilkan 18 fail FastA yang berisi fragmen sesuai dengan kebutuhan penelitian. Data latih dengan jumlah fragmen 9600 disiapkan sebagai data pendekatan pencarian parameter terbaik untuk kernel, sedangkan data latih dengan jumlah fragmen 320 ribu menjadi data masukan untuk pembuatan model. Penggunaan data latih kecil sebagai pendekatan pencarian paramater terbaik ini didasarkan pada percobaan yang dilakukan oleh McHardy et al. (2007).
Ekstraksi Fitur
Proses selanjutnya ialah ekstraksi fitur, tahapan ini dilakukan untuk data latih dan data uji. Metode ekstraksi fitur yang digunakan ialah spaced k-mers. Ada 2 buah variabel yang berpengaruh pada metode ekstraksi fitur ini, yaitu w (weight of pattern) adalah banyaknya posisi yang cocok, dan d adalah jumlah posisi don’t care. Mengacu pada penelitian Kusuma (2012), pola terbaik spaced k-mers dengan nilai w = 3 dan d = 0, 1, 2 dapat dilihat pada Gambar 2.
5
maupun G. Kemudian untuk simbol ** berarti diperbolehkan pasangan basa apapun mengisi 2 bit tersebut, sehingga kondisi ini dapat diisi oleh 24 pasang basa mulai dari AA, AC, AT, AG, dan seterusnya hingga GG.
Support Vector Machine (SVM)
SVM merupakan metode pengklasifikasian biner yang dikembangkan oleh Vladimir Vapnik tahun 1995. Konsep dasar pembelajaran SVM ini ialah menemukan hyperplane (bidang pemisah) terbaik yang dapat memisahkan d-dimensi data dengan sempurna ke dalam 2 kelas (kelas +1 dan kelas -1). Secara intuitif, hyperplane yang dicari ialah yang dapat memaksimumkan jarak geometri hyperplane ke support vector-nya. Jarak tersebut diistilahkan dengan margin (Boswell 2002).
Menurut Osuna et al. (2007) linearly separable data merupakan suatu kondisi data yang dapat dipisahkan secara linear. Pada Gambar 3 diilustrasikan kondisi lineraly separable dengan hyperlane yang memiliki margin terbesar. Himpunan n adalah dataset dan i - adalah label kelas dari data i. Kondisi linearly separable terpenuhi jika dapat dicari pasangan (w, b) sedemikian sehingga:
Gambar 3 Kondisi linearly separable dengan hyperlane yang memiliki margin terbesar
6
w i b ≥ i ke as
w i b ≤ i ke as
dengan w adalah bidang normal dan b adalah posisi bidang relatif terhadap pusat koordinat. Kemudian, ruang hipotesis untuk data tersebut ialah set fungsi yang diberikan oleh:
w b sign w b 3
Setelah dilakukan penyelesaian dengan formula Lagrangian menggunakan Lagrange multipier dan normalisasi parameter w, maka fungsi keputusan untuk menentukan kelas dari data uji x adalah:
sign ∑ i i i
l
i
b
dengan = koefisien Lagrange multipier.
Salah satu kendala dalam pengklasifikasian ialah ketersediaan data yang besar dan beragam yang dapat mengakibatkan data tersebut tidak dapat dipisahkan secara linear. Untuk kasus ini SVM memperkena kan “kernel” yang dapat merepresentasikan atau mentransformasikan data ke dimensi lebih tinggi (lebih besar dari 2) dengan fungsi transformasi . Sehingga, data yang sudah berada di dimensi lebih tinggi tersebut dapat dengan mudah dipisahkan dengan hyperplane secara linear (Boswell 2002). Jika terdapat sebuah fungsi kernel K
sehingga i i , maka fungsi transformasi tidak perlu diketahui
secara tepat. Dengan demikian fungsi yang dihasilkan dari pelatihan adalah:
sign ∑ i i i
l
i
b . 5
Terdapat 3 kernel yang biasa digunakan dalam SVM, yaitu sebagai berikut (Osuna et al. 1997) :
1 Gaussian radial basis function (RBF):
e p -‖ - ‖ (6)
2 Polinomial dengan derajat d:
d, (7)
3 Multi layer perceptron (untuk beberapa nilai θ :
tanh - θ . (8)
7 Grid Search
Setelah diperoleh fitur, tahap selanjutnya ialah grid search menggunakan data latih dengan jumlah fragmen 9600. Tahapan ini dilakukan dengan fungsi grid search. Fungsi grid search mengeluarkan nilai parameter terbaik yang dibutuhkan saat pembentukan model (tahap pelatihan) menggunakan kernel RBF dan polinomial. Parameter untuk kernel RBF adalah cost (c) dan gamma (γ), sedangkan untuk kernel polinomial adalah cost (c), gamma (γ), degree (d), dan koeff 0 (r). Akan tetapi, parameter r pada polinomial yang dipakai hanya nilai default-nya saja yaitu 0. Selain mengeluarkan nilai parameter terbaik, fungsi ini juga mengeluarkan akurasi 5-cross validation dari data latih.
Cross-validation merupakan metode statistika untuk mengevaluasi dan membandingkan algoritme pembelajaran dengan membagi data menjadi dua bagian. Satu bagian untuk melatih model dan bagian lainnya untuk memvalidasi model tersebut. Salah satu bentuk cross-validation adalah k-fold cross-validation. K-fold cross-validation akan membagi data menjadi k bagian berukuran sama. Secara bertahap akan dilakukan pelatihan dan validasi sebanyak k ulangan. Sehingga dalam setiap perulangan k-1 bagian akan menjadi data latih, dan 1 bagian sisanya akan digunakan untuk validasi (Refaeilzadeh et al. 2009).
Pelatihan SVM
Proses pelatihan SVM dilakukan untuk data latih hasil ekstraksi fitur dengan jumlah fragmen 320 ribu. Dalam pelatihan ini, akan diterapkan pelatihan menggunakan 4 fungsi kernel, yaitu kernel RBF, linear, kuadratik, dan polinomial berderajat 3.
Pengujian SVM
Hasil dari pelatihan SVM sebelumnya ialah sebuah model yang akan diuji menggunakan hasil ekstraksi fitur dari data uji. Pengujian akan mengklasifikasikan data uji sebanyak 200 organisme ke dalam kelas taksonominya. Semua organisme yang telah dikelaskan menghasilkan persentase hasil pengklasifikasiannya.
Analisis
Dari hasil pelatihan dan pengujian SVM dengan 4 fungsi kernel yang berbeda, akan didapatkan hasil untuk kinerja algoritme SVM ini. Kemudian akurasi untuk hasil klasifikasi dapat dicari dengan menggunakan rumus:
akurasi ∑data uji benar∑ data uji 00% 9
8
kecil, sedang, dan besar. Rumus yang digunakan untuk menghitung nilai sensitivity dan specificity, yaitu:
sensiti it ∑ true ositi es
∑ true ositi es ∑ alse ne ati es 00% 0
s eci icit ∑ true ne ati es∑ true ne ati es
∑ alse ositi es 00% dengan true positive adalah data uji kelas x yang diklasifikasikan ke kelas x, true negative adalah data uji kelas x yang diklasifikasikan ke kelas selain x, false positive adalah data uji kelas selain x yang diklasifikasikan ke kelas x, dan false negative adalah data uji kelas selain x yang diklasifikasikan ke kelas selain x, dengan kelas x adalah kelas yang akan dihitung nilai sensitivity dan specificity-nya.
Setelah seluruh perhitungan nilai akurasi, sensitivity, dan specificity dari hasil kinerja pengklasifikasian fragmen metagenome menggunakan SVM ini didapat, beberapa hal yang akan menjadi bahan analisis ialah:
1 pengaruh panjang fragmen yang digunakan terhadap hasil akurasi, 2 hasil sensitivity dan specificity yang didapat,
3 penggunaan 4 fungsi kernel berbeda pada pelatihan SVM, dan
4 hasil pengklasifikasian data uji yang tidak ada modelnya pada data latih.
Implementasi
Implementasi sistem dilakukan dalam lingkungan pengembangan sebagai berikut:
1 bahasa pemrograman : PHP,
2 library komputasi : LibSVM 3.12, dan 3 database management system (DBMS) : MySQL.
Sistem yang dikembangkan memiliki fungsi utama yaitu melakukan prediksi tingkat taksonomi suatu sequence DNA. Data masukkan untuk sistem ini ialah sebuah sequence DNA, dan keluarannya ialah tingkat taksonominya. Tingkat taksonomi yang akan ditampilkan sebagai hasil prediksi yaitu genus, order, kelas, dan filum. Sistem ini se anjutnya dinamai “Metagenome Binning”.
HASIL DAN PEMBAHASAN
Pembagian Data
9 suatu kelas yang tidak ada modelnya. Untuk daftar taksonomi yang digunakan dapat dilihat pada Lampiran 3.
Praproses Data
Pada tahap praproses data, sequence DNA metagenome yang sudah dibagi menjadi data latih dan data uji akan diuraikan fragmennya menggunakan perangkat lunak MetaSim. Pada penelitian ini data yang dipersiapkan untuk data latih dibaca sebanyak 9600 dan 320 ribu kali. Sehingga didapat 9600 dan 320 ribu fragmen data latih yang diurai dari 381 organisme. Sedangkan untuk data uji dibaca sebanyak 100 ribu kali. Sehingga didapat 100 ribu fragmen data uji yang diurai dari 200 organisme. Hasil praproses data yang menyatakan jumlah sequence di setiap tingkat takson dan setiap panjang fragmen untuk data latih dan data uji dapat dilihat pada Lampiran 4 dan Lampiran 5.
Pada setiap praproses data yang dilakukan, ditentukan 6 panjang fragmen yang akan digunakan yaitu 400 bp, 800 bp, 1 Kbp, 3 Kbp, 5 Kbp, dan 10 Kbp. Keluaran dari pengolahan MetaSim ini ialah fail FastA yang berisi sequence DNA yang sudah terfragmen sesuai dengan nilai parameter yang dimasukkan. Berikut contoh hasil praproses data untuk data latih dengan jumlah fragmen 9600 dan panjang fragmen 400 bp pada Gambar 4.
Ekstraksi Fitur
Metode ekstraksi fitur yang digunakan ialah spaced k-mers dengan nilai w = 3 dan d = 0, 1, 2 yang merupakan pola terbaik yang akan menghasilkan akurasi terbesar dari klasifikasi menurut Kusuma (2012). Hasil dari proses ekstraksi fitur ialah frekuensi tri-nukleotida dari fragmen DNA, sehingga akan terdapat 192 kombinasi tri-nukleotida mulai dari AAA sampai GGG, A*AA sampai G*GG, dan A**AA sampai G**GG.
...
10
Berikut contoh hasil ekstraksi fitur untuk data latih takson genus dengan jumlah fragmen 9600 dan panjang fragmen 400 bp:
1 1:12 2:4 3:5 4:10 5:4 6:3 7:1 8:9 9:4 10:8 11:9 12:3 13:11 ... 190:13 191:6 192:13 1 1:23 2:7 3:11 4:8 5:5 6:5 7:1 8:9 9:8 10:9 11:10 12:11 13:5 ... 190:8 191:4 192:4 1 1:7 2:2 3:11 4:5 5:1 6:5 7:3 8:5 9:7 10:14 11:5 12:5 13:6 ... 190:3 191:3 192:3 ... ... 48 1:18 2:6 3:7 4:8 5:6 6:11 7:7 8:2 9:7 10:5 11:9 12:8 13:2 ... 190:7 191:5 192:3 48 1:14 2:11 3:12 4:5 5:3 6:10 7:6 8:5 9:12 10:5 11:10 12:6 13:2 ... 190:5 191:3 192:5
Grid Search
Fungsi grid search pada LibSVM akan mengeluarkan nilai parameter yang dibutuhkan oleh kernel RBF dan polinomial. Nilai parameter tersebut akan didapat dengan melakukan proses cross validation dengan k = 5. Parameter yang
dibutuhkan untuk RBF ia ah gamma γ sedangkan untuk polinomial ialah
gamma γ degree (d), dan koeff 0 (r). Salah satu hasil grid search yang didapat
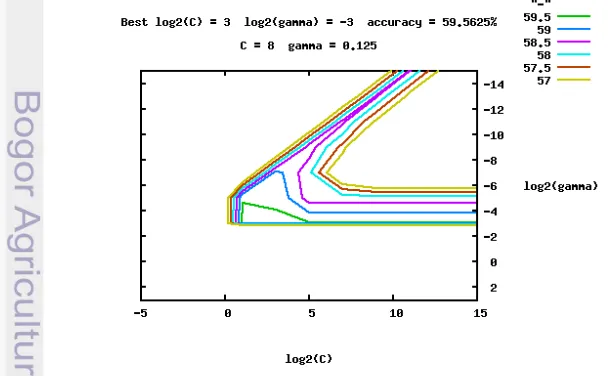
untuk data latih takson genus dengan jumlah fragmen 9600 dan panjang fragmen 400 bp dapat dilihat pada Gambar 5. Dari gambar tersebut ditunjukkan bahwa nilai terbaik untuk c 8 dan γ 0. 5 dengan akurasi 5-cross validation = 59.6%. Hasil grid search lainnya dari setiap data yang digunakan dapat dilihat pada Lampiran 6.
Klasifikasi SVM
Setelah didapatkan fitur untuk data latih dengan banyak fragmen 320 ribu, data uji dengan jumlah fragmen 100 ribu, serta parameter kernel yang dibutuhkan, proses dilanjutkan dengan klasifikasi SVM. Proses klasifikasi SVM diawali dengan menskalakan data latih dan data uji terlebih dahulu sebelum dilakukan pelatihan maupun pengujian. Proses penskalaan ini sangat penting sebelum diterapkan pengklasifikasian dengan SVM. Keuntungan utama dari penskalaan
11 yaitu untuk menghindari atribut atau fitur bernilai besar yang bisa mendominasi fitur lain yang bernilai kecil. Selain itu penskalaan juga dapat mengurangi tingkat kesulitan perhitungan selama proses pengklasifikasian.
Setelah proses penskalaan selesai, proses selanjutnya adalah melakukan pelatihan SVM. Data latih dilatih satu per satu dengan 4 fungsi kernel mulai dari RBF, linear, kuadratik, dan polinomial derajat 3 dengan nilai parameter kernel terkait. Sebanyak 24 pelatihan dilakukan menggunakan fungsi kernel RBF, sedangkan untuk kernel lainnya hanya dilakukan pelatihan 1 kali yaitu pada panjang fragmen 10 Kbp pada tingkat takson genus.
Model yang sudah dihasilkan dari pelatihan sebelumnya digunakan untuk mengklasifikasikan data uji yang merepresentasikan fragmen metagenome dari organisme-organisme baru. Dari pengujian ini diperoleh akurasi dari hasil klasifikasi menggunakan Persamaan 9, sensitivity dan specificity dari setiap kelas yang ada pada takson genus menggunakan Persamaan 10 dan Persamaan 11.
Analisis
Analisis dilakukan atas hasil akurasi yang dihasilkan dengan memvariasikan panjang fragmen, tingkat taksonomi, dan kernel yang digunakan.
Tingkat taksonomi dan panjang fragmen
Analisis pengaruh panjang fragmen terhadap nilai akurasi yang diperoleh merupakan analisis yang penting. Data metagenome yang diambil dari lingkungan terdiri atas banyak organisme di dalamnya, sehingga mengandung jumlah nukleotida yang sangat besar, bahkan bisa mencapai megabases. Sementara itu, teknik untuk melakukan DNA sequencing saat ini hanya berhasil men-sequence
fragmen ≤ 700 bp untuk pembacaan individua atau ≤ 00 bp bi a menggunakan
pyrosequencing (metode sequencing DNA berdasarkan prinsip “sequencing by synthesis” McHardy et al. 2007). Berdasarkan kondisi tersebut, maka diharapkan suatu penelitian terkait dengan metagenome dapat menghasilkan akurasi yang baik bahkan pada panjang fragmen yang pendek.
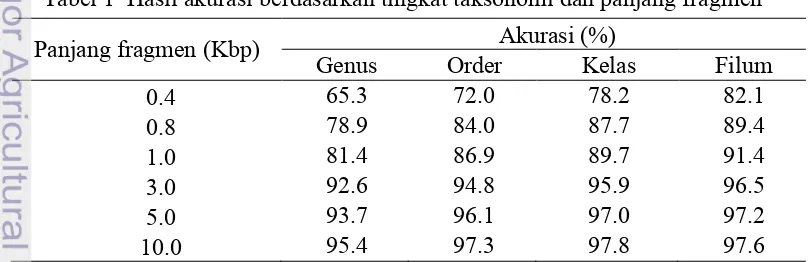
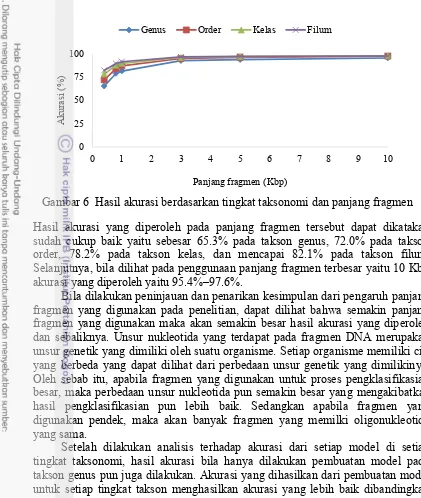
Untuk hasil akurasi berdasarkan tingkat taksonomi, nilai akurasi yang analisis merupakan nilai akurasi dari setiap panjang fragmen yang dicobakan yaitu 400 bp, 800 bp, 1 Kbp, 3 Kbp, 5 Kbp, dan 10 Kbp. Sehingga akan didapatkan 6 akurasi untuk setiap tingkat taksonomi mulai dari genus, order, kelas, dan filum. Hasil akurasi ini ditunjukkan pada Tabel 1, dan divisualisasikan pada Gambar 6.
Pada penelitian ini, panjang fragmen terkecil yang dicobakan adalah 400 bp. Tabel 1 Hasil akurasi berdasarkan tingkat taksonomi dan panjang fragmen
Panjang fragmen (Kbp) Akurasi (%)
Genus Order Kelas Filum
0.4 65.3 72.0 78.2 82.1
0.8 78.9 84.0 87.7 89.4
1.0 81.4 86.9 89.7 91.4
3.0 92.6 94.8 95.9 96.5
5.0 93.7 96.1 97.0 97.2
12
Hasil akurasi yang diperoleh pada panjang fragmen tersebut dapat dikatakan sudah cukup baik yaitu sebesar 65.3% pada takson genus, 72.0% pada takson order, 78.2% pada takson kelas, dan mencapai 82.1% pada takson filum. Selanjutnya, bila dilihat pada penggunaan panjang fragmen terbesar yaitu 10 Kbp akurasi yang diperoleh yaitu 95.4%–97.6%.
Bila dilakukan peninjauan dan penarikan kesimpulan dari pengaruh panjang fragmen yang digunakan pada penelitian, dapat dilihat bahwa semakin panjang fragmen yang digunakan maka akan semakin besar hasil akurasi yang diperoleh dan sebaliknya. Unsur nukleotida yang terdapat pada fragmen DNA merupakan unsur genetik yang dimiliki oleh suatu organisme. Setiap organisme memiliki ciri yang berbeda yang dapat dilihat dari perbedaan unsur genetik yang dimilikinya. Oleh sebab itu, apabila fragmen yang digunakan untuk proses pengklasifikasian besar, maka perbedaan unsur nukleotida pun semakin besar yang mengakibatkan hasil pengklasifikasian pun lebih baik. Sedangkan apabila fragmen yang digunakan pendek, maka akan banyak fragmen yang memilki oligonukleotida yang sama.
Setelah dilakukan analisis terhadap akurasi dari setiap model di setiap tingkat taksonomi, hasil akurasi bila hanya dilakukan pembuatan model pada takson genus pun juga dilakukan. Akurasi yang dihasilkan dari pembuatan model untuk setiap tingkat takson menghasilkan akurasi yang lebih baik dibandingkan pembuatan model hanya untuk takson genus. Perbandingan akurasi ini dapat dilihat pada Gambar 7 dan Lampiran 7. Sehingga disimpulkan bahwa pembuatan model untuk setiap tingkat takson memang lebih baik apabila diinginkan hasil penelitian yang baik.
Sensitivity dan specificity
Perhitungan sensitivity dan specificity pada penelitian ini dibatasi pada takson genus dan filum saja, serta hanya pada panjang fragmen 400 bp, 1 Kbp, dan 10 Kbp. Penelitian dengan ekstasi fitur menggunakan spaced k-mers dan pengklasifikasian menggunakan metode SVM ini dapat menghasilkan sensitivity yang baik pada level takson genus, yang dapat dilihat pada Gambar 8. Nilai sensitivity yang didapat pada panjang fragmen 400 bp yaitu berada di antara 21.1%–85.2% dengan rata-rata sensitivity-nya 60.6%. Nilai sensitivity yang
Gambar 6 Hasil akurasi berdasarkan tingkat taksonomi dan panjang fragmen
0 25 50 75 100
0 1 2 3 4 5 6 7 8 9 10
A
k
u
rasi
(%)
Panjang fragmen (Kbp)
13
didapat pada panjang fragmen 1 Kbp yaitu berada di antara 43.4%–94.5% dengan rata-rata sensitivity-nya 79.2%. Nilai sensitivity yang didapat pada panjang fragmen 10 Kbp yaitu berada di antara 56.0%–100% dengan rata-rata sensitivity-nya 95.2%. Nilai sensitivity ini menunjukkan bahwa setidaknya ada 60.6% data pada suatu kelas dapat diklasifikasikan ke kelas sebenarnya. Untuk specificity didapat nilai yang baik yaitu antara 96.3%–100% pada setiap panjang fragmen yang dicobakan. Secara lengkap nilai specificity ini dapat dilihat pada Gambar 9.
Hasil sensitivity dan specificity untuk kelas taksonomi selanjutnya ,yaitu filum dapat dilihat pada Gambar 10 dan Gambar 11. Sensitivity pada level takson ini dikatakan baik bahkan pada panjang fragmen kecil yaitu 400 bp. Nilai
Gambar 8 Sensitivity takson genus
14
sensitivity yang didapat yaitu 40.8%–88.4% untuk panjang fragmen 400 bp, 60.7%–94.6% untuk panjang fragmen 1 Kbp, dan 66.2%–99.6% untuk panjang fragmen 10 Kbp. Kemudian untuk nilai specificity juga baik yaitu berkisar antara 88.1%–100.0% untuk semua panjang fragmen yang dicobakan.
Bila ditinjau keterkaitan antara hasil sensitivity dengan jumlah data yang ada pada kelas taksonnya, disimpulkan bahwa semakin banyak jumlah data untuk kelas tersebut maka menghasilkan sensitivity yang besar, dan sebaliknya. Dapat dilihat data uji untuk filum Actinobacteria, Firmicutes, dan Sphirochaetes yang memiliki anggota filum tersebar dihasilkan sensitivity yang besar pula. Sensitivity
Gambar 9 Specificity takson genus
Gambar 10 Sensitivity takson filum
15 untuk ketiga filum tersebut yaitu 82.2%–99.6% pada panjang fragmen kecil (400 bp) dan panjang fragmen besar (10 Kbp). Namun untuk filum lainnya yang memiliki anggota filum jauh lebih kecil, hanya menghasilkan sensitivity < 50% pada panjang fragmen kecil (400 bp). Pengaruh semakin besarnya data yang membuat nilai sensitivity juga besar dipengaruhi oleh semakin banyaknya pembelajaran yang dilakukan. Jumlah data uji untuk setiap kelas taksonnya dapat dilihat pada Lampiran 5.
Jenis kernel
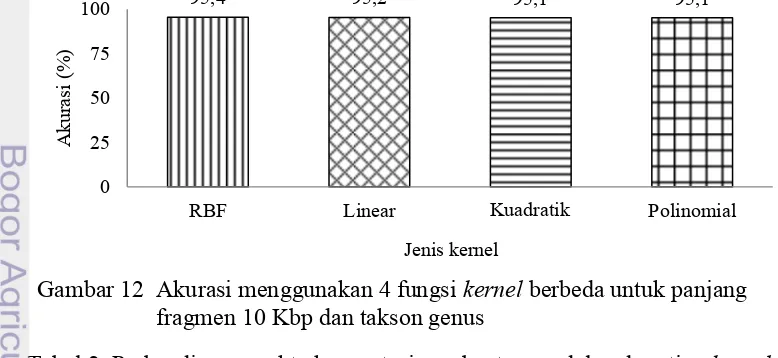
Pengujian pengaruh penggunaan kernel terhadap hasil akurasi dilakukan untuk mengetahui kernel yang dapat menghasilkan model terbaik pada kasus pengklasifikasian fragmen metagenome. Gambar 12 memvisualisasikan hasil akurasi berdasarkan fungsi kernel yang digunakan dari pengklasifikasian data uji dengan panjang fragmen 10 Kbp pada tingkat takson genus. Dapat dilihat bahwa akurasi yang didapatkan dengan menerapkan 4 jenis kernel berbeda menghasilkan persentase akurasi yang tidak jauh berbeda. Akurasi yang dihasilkan terbilang sudah sangat baik yaitu mencapai > 95%.
Dari hasil akurasi ini dapat dikatakan bahwa penggunaan kernel ternyata tidak berpengaruh. Penggunaan kernel linear yang sesungguhnya tidak diterapkan kernel apapun menghasilkan akurasi yang tidak berbeda dengan penggunaan kernel lainnya. Maka pada kondisi ini dapat dikatakan bahwa metode ekstraksi fitur yang digunakan yaitu spaced k-mers sudah baik, sehingga data sudah terpisah secara linear tanpa perlu diterapkan fungsi kernel apapun pada pembutan modelnya.
Setelah itu dilakukan pula pencatatan waktu komputasi pembuatan model dari setiap kernel yang dapat dilihat pada Tabel 2. Dari hasil ini dapat dikatakan bahwa kernel RBF memiliki kinerja terbaik dalam melakukan pelatihan SVM untuk data fragmen metagenome, tetapi membutuhkan waktu komputasi yang sedikit lebih lama.
Gambar 12 Akurasi menggunakan 4 fungsi kernel berbeda untuk panjang fragmen 10 Kbp dan takson genus
Tabel 2 Perbandingan waktu komputasi pembuatan model pada setiap kernel Panjang fragmen Waktu komputasi (menit)
RBF Linear Kuadratik Polinomial (d=3)
10 Kbp 13 10 8 8
95,4 95,2 95,1 95,1
0 25 50 75 100
RBF Linear Quadratic Polinomial
A
k
u
rasi
(%)
Jenis kernel
16
Hasil klasifikasi genus yang tidak ada pada data latih
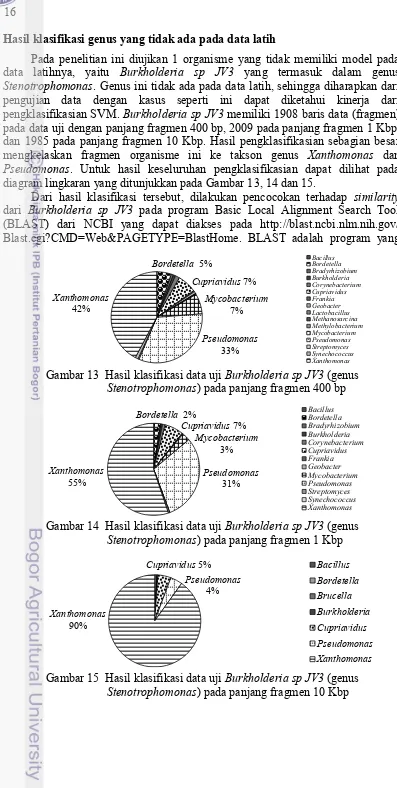
Pada penelitian ini diujikan 1 organisme yang tidak memiliki model pada data latihnya, yaitu Burkholderia sp JV3 yang termasuk dalam genus Stenotrophomonas. Genus ini tidak ada pada data latih, sehingga diharapkan dari pengujian data dengan kasus seperti ini dapat diketahui kinerja dari pengklasifikasian SVM. Burkholderia sp JV3 memiliki 1908 baris data (fragmen) pada data uji dengan panjang fragmen 400 bp, 2009 pada panjang fragmen 1 Kbp, dan 1985 pada panjang fragmen 10 Kbp. Hasil pengklasifikasian sebagian besar mengkelaskan fragmen organisme ini ke takson genus Xanthomonas dan Pseudomonas. Untuk hasil keseluruhan pengklasifikasian dapat dilihat pada diagram lingkaran yang ditunjukkan pada Gambar 13, 14 dan 15.
Dari hasil klasifikasi tersebut, dilakukan pencocokan terhadap similarity dari Burkholderia sp JV3 pada program Basic Local Alignment Search Tool (BLAST) dari NCBI yang dapat diakses pada http://blast.ncbi.nlm.nih.gov/ Blast.cgi?CMD=Web&PAGETYPE=BlastHome. BLAST adalah program yang
Gambar 13 Hasil klasifikasi data uji Burkholderia sp JV3 (genus Stenotrophomonas) pada panjang fragmen 400 bp
Gambar 14 Hasil klasifikasi data uji Burkholderia sp JV3 (genus Stenotrophomonas) pada panjang fragmen 1 Kbp
Gambar 15 Hasil klasifikasi data uji Burkholderia sp JV3 (genus Stenotrophomonas) pada panjang fragmen 10 Kbp
17
dapat menemukan “region of local similarity” antar sequences. Program ini dapat
membandingkan urutan nukleotida atau protein suatu sequence dengan sequence lainnya dan menghitung secara statistik unsur yang signifikan sama. BLAST dapat digunakan untuk menyimpulkan hubungan fungsional dan evolusioner antar sequences serta membantu mengidentifikasi anggota dari gen. Hasil BLAST dari organisme Burkholderia sp JV3 mengeluarkan daftar organisme yang memiliki similarity dengan organisme tersebut. Tabel 3 adalah daftar organisme-organisme yang dihasilkan dari BLAST yang juga merupakan organisme yang digunakan pada data latih.
Dari hasil pengklasifikasian pada penelitian ini dan hasil BLAST, dapat dilihat bahwa benar Xanthomonas dan Pseudomonas memiliki tingkat similarity yang cukup besar dengan Burkholderia sp JV3. Maka hasil klasifikasi SVM pada Tabel 3 Daftar organisme yang memiliki similarity dari hasil alignment
Burkholderia sp JV3 pada BLAST
No Deskripsi Max
score Total score
Query 1 Xanthomonas campestris
pv. campestris complete genome, strain B100
11297 1.484 × 106 39% 0.0 100%
2 Xanthomonas campestris pv. campestris str. ATCC 33913, complete genome
11291 1.473 × 106 39% 0.0 100%
3 Xanthomonas campestris pv. campestris str. 8004, complete genome
11285 1.474 × 106 39% 0.0 100%
4 Xanthomonas campestris pv. vesicatoria complete genom
11068 1.421 × 106 36% 0.0 100%
5 Xanthomonas oryzae pv. oryzae KACC10331, complete genome
8408 1.175 × 106 31% 0.0 100%
6 Xanthomonas oryzae pv. oryzae MAFF 311018 DNA, complete genome
8403 1.178 × 106 31% 0.0 100%
7 Xanthomonas oryzae pv. oryzae PXO99A,
9 Pseudomonas putida KT2440 complete genome
3166 2.618 × 106 5% 0.0 100%
10 Pseudomonas putida GB-1, complete genome
3160 2.624 × 106 5% 0.0 100% 11 Pseudomonas putida
W619, complete genome
18
penelitian ini bergantung pada unsur nukleotida yang dimiliki oleh setiap data, baik data latih yang akan menjadi model maupun data uji yang akan diprediksi kelasnya.
Implementasi
Tahap akhir yaitu implementasi yang menghasilkan sistem bernama Metagenome Binning. Sistem ini dapat melakukan prediksi tingkat taksonomi dari sequence DNA yang menjadi masukkan sistem sebelumnya. Tingkat taksonomi yang akan ditampilkan sebagai keluaran sistem yaitu genus, order, kelas, dan filum. Tahapan dan tampilan dalam menggunakan sistem ini dapat dilihat pada Lampiran 8.
SIMPULAN DAN SARAN
Simpulan
Pada penelitian ini, sudah disajikan pengklasifikasian fragmen metagenome menggunakan metode SVM. Secara keseluruhan penelitian ini sudah menghasilkan akurasi yang baik, bahkan pada panjang fragmen kecil 400 bp yaitu 65.3% untuk takson genus, 72.0% untuk takson order, 78.2% untuk takson kelas, dan 82.1% untuk takson filum. Pada panjang fragmen besar (10 Kbp) akurasi
mencapai ˃ 95% untuk semua eve takson. Dari penggunaan berbagai panjang
fragmen ini disimpulkan bahwa semakin panjang fragmen yang digunakan maka akan semakin besar hasil akurasi yang diperoleh dan sebaliknya.
Penggunaan 4 fungsi kernel yang berbeda pada pemodelan SVM pun telah diterapkan. Dari hasil penelitian ini disimpulkan bahwa fungsi kernel yang diterapkan tidak terlalu berpengaruh terhadap hasil pengklasifikasian karena metode ekstraksi fitur yang digunakan ternyata sudah baik. Metode ekstraksi fitur spaced k-mers dengan variabel w = 3 dan d = 0, 1, 2 telah menghasilkan fitur yang dapat memisahkan data secara linear. Sehingga kondisi linearly separable sudah terpenuhi tanpa perlu menerapakan fungsi kernel apapun pada pembentukan model SVM.
Kemudian untuk hasil pengklasifikasian data uji yang tidak ada modelnya pada data latih menunjukkan hasil yang serupa dengan hasil aplikasi BLAST. Fragmen data uji Burkholderia sp JV3 sebagian besar dikelaskan menjadi Xanthomonas dan Pseudomonas. Ini menunjukkan bahwa kinerja pengklasifikasian pada penelitian ini bergantung pada unsur nukleotida yang dimiliki oleh setiap data, baik data latih yang akan menjadi model maupun data uji yang akan diprediksi kelasnya.
19
Saran
Beberapa saran untuk penelitian selanjutnya yaitu:
1 Menggunakan sequence data 16S rRNA yang dihasilkan dari proses sequencing dan sudah banyak tersedia di genbank dengan panjang fragmen yang mendominasi yaitu 400 bp, sehingga tidak perlu menggunakan data simulasi.
2 Menambah jumlah kelas pada data latih sehingga dapat melakukan prediksi untuk lebih banyak kelas.
3 Menggunakan data riil misal Sargasso Sea atau yang lainnya.
DAFTAR PUSTAKA
Boswell D. 2002. Introduction to support vector machine [Internet]. [diunduh 2012 Des 9]. Tersedia pada: http://www.work.caltech.edu/~boswell/ IntroToSVM.pdf
Hsu CW, Chang CC, Lin CJ. 2003. A practical guide to support vector classification [Internet]. [diunduh 2012 Des 9]. Tersedia pada: http://www.csie.ntu.edu.tw/~cjlin
Hsu CW, Lin CJ. 2002. A comparison of methods for multiclass support vector machine. IEEE Transactions on Neural Networks. 13(2):415–425. doi: 10.1109/72.991427.
Kusuma, WA. 2012. Combined approaches for improving the performance of de novo DNA sequence assembly and metagenomic classification of short fragments from next generation sequencer [disertasi]. Tokyo (JP): Tokyo Institute of Technology.
Kusuma WA, Akiyama Y. 2011. Metagenome fragment binning based on characterization vector. Di dalam: International Conference on Bioinformatics and Biomedical Technology (ICBBT 2011); 2011 Mar 25–27; Sanya, China. Liu L, Ho YK, Yau S. 2006. Clustering DNA sequences by feature vectors.
Molecular Phylogenetics and Evolution. 41(1):64–69. doi: 10.1016/j.ympev. 2006.05.019
McHardy AC, Martín HG, Tsirigos A, Hugenholtz P, Rigoutsos I. 2007. Accurate phylogonetic classification of variabel-length DNA fragments. Nature Methods. 4(1):63–72. doi: 10.1038/nmeth976.
McHardy AC Rigoutsos I. 007. What’s in the mi : phy ogenetic c assification of
metagenome sequence samples. Current Opinion in Microbiology. 10(5):499– 503. doi: 10.1016/j.mib.2007.08.004.
Osuna EE, Freund R, Girosi F. 1997. Support vector machines: training and applications. AI Memo (1602).
Refaeilzadeh P, Tang L, Liu H. 2009. Cross-validation. Di dalam: Liu L, Öszu MT, editor. Encyclopedia of Database Systems. New York (US): Springer. Richter DC, Ott F, Auch AF, Schmid R, Huson DH. 2008. MetaSim: a sequencing
20
21 Lampiran 1 Daftar nama organisme data latih
No Nama Organisme No Nama Organisme
1 Bacillus amyloliquefaciens FZB42
23 Bartonella tribocorum CIP 105476
2 Bacillus anthracis str. 'Ames Ancestor'
24 Bordetella avium 197N chromosome
3 Bacillus anthracis str. Ames chromosome
25 Bordetella bronchiseptica RB50 4 Bacillus anthracis str. Sterne
chromosome
26 Bordetella parapertussis 12822 5 Bacillus cereus ATCC 10987
chromosome
27 Bordetella pertussis Tohama I 6 Bacillus cereus ATCC 14579 28 Bordetella petrii DSM 12804 7 Bacillus cereus E33L 29 Borrelia afzelii PKo
8 Bacillus cereus subsp. cytotoxis NVH 391-98
30 Borrelia duttonii Ly
9 Bacillus clausii KSM-K16 31 Borrelia garinii PBi chromosome chromosome linear
10 Bacillus halodurans C-125 chromosome
32 Borrelia hermsii DAH chromosome
11 Bacillus licheniformis ATCC 14580
33 Borrelia recurrentis A1 12 Bacillus subtilis subsp. subtilis
str. 168 chromosome
34 Borrelia turicatae 91E135 chromosome
13 Bacillus thuringiensis serovar konkukian str. 97-27
chromosome
35 Bradyrhizobium japonicum USDA 110 chromosome
14 Bacillus thuringiensis str. Al Hakam chromosome
36 Bradyrhizobium sp. BTAi1 chromosome
15 Bacillus weihenstephanensis KBAB4
37 Bradyrhizobium sp. ORS278 chromosome
16 Bacteroides fragilis NCTC 9343 chromosome
38 Brucella abortus S19 chromosome 1 17 Bacteroides fragilis YCH46
chromosome
39 Brucella abortus bv. 1 str. 9-941 chromosome chromosome I 18 Bacteroides thetaiotaomicron
VPI-5482 chromosome
40 Brucella canis ATCC 23365 chromosome I
19 Bacteroides vulgatus ATCC 8482 chromosome
41 Brucella melitensis biovar Abortus 2308 chromosome chromosome I
20 Bartonella bacilliformis KC583 42 Brucella melitensis bv. 1 str. 16M chromosome chromosome I 21 Bartonella henselae str.
Houston-1
43 Brucella ovis ATCC 25840 chromosome chromosome I 22 Bartonella quintana str.
Toulouse
22
Lampiran 1 Lanjutan
No Nama Organisme No Nama Organisme
45 Brucella suis ATCC 23445 chromosome I
65 Burkholderia thailandensis E264 chromosome chromosome I 46 Burkholderia ambifaria AMMD
chromosome chromosome 1
66 Burkholderia vietnamiensis G4 chromosome chromosome 1 47 Burkholderia ambifaria MC40-6
chromosome chromosome 1
67 Burkholderia xenovorans LB400 chromosome 1
48 Burkholderia cenocepacia AU 1054 chromosome 3
68 Campylobacter concisus 13826 49 Burkholderia cenocepacia
HI2424 chromosome chromosome 1
69 Campylobacter curvus 525.92 chromosome
50 Burkholderia cenocepacia J2315 chromosome chromosome 1
70 Campylobacter fetus subsp. fetus 82-40
51 Burkholderia cenocepacia MC0-3 chromosome chromosome 1
71 Campylobacter hominis ATCC BAA-381
52 Burkholderia mallei ATCC 23344 chromosome chromosome 1
72 Campylobacter jejuni RM1221
53 Burkholderia mallei NCTC 10229 chromosome I
73 Campylobacter jejuni subsp. doylei 269.97
54 Burkholderia mallei NCTC 10247 chromosome I
74 Campylobacter jejuni subsp. jejuni NCTC 11168 chromosome 55 Burkholderia mallei SAVP1
chromosome I
75 Candidatus Phytoplasma australiense
56 Burkholderia multivorans ATCC 17616 chromosome chromosome 1
76 Candidatus Phytoplasma mali
57 Burkholderia phymatum STM815 chromosome chromosome 1
77 Chlamydophila abortus S26/3 58 Burkholderia phytofirmans PsJN
chromosome chromosome 1
78 Chlamydophila caviae GPIC 59 Burkholderia pseudomallei
1106a chromosome I
79 Chlamydophila felis Fe/C-56 60 Burkholderia pseudomallei
1710b chromosome chromosome I
80 Chlamydophila pneumoniae AR39
61 Burkholderia pseudomallei 668 chromosome I
81 Chlamydophila pneumoniae CWL029
62 Burkholderia pseudomallei K96243 chromosome chromosome 1
82 Chlamydophila pneumoniae J138
63 Burkholderia sp. 383 chromosome 1
83 Chlamydophila pneumoniae TW-183
64 Burkholderia sp. 383 chromosome chromosome 2
23 Lampiran 1 Lanjutan
No Nama Organisme No Nama Organisme
85 Chlorobium limicola DSM 245 chromosome
107 Clostridium tetani E88 chromosome
86 Chlorobium luteolum DSM 273 chromosome
108 Clostridium thermocellum ATCC 27405 chromosome
87 Chlorobium phaeobacteroides BS1 chromosome
109 Corynebacterium diphtheriae NCTC 13129 chromosome 88 Chlorobium phaeobacteroides
DSM 266 chromosome
110 Corynebacterium efficiens YS-314
89 Chlorobium phaeovibrioides DSM 265 chromosome
111 Corynebacterium glutamicum ATCC 13032
90 Chlorobium tepidum TLS 112 Corynebacterium glutamicum R chromosome
91 Clostridium acetobutylicum ATCC 824
113 Corynebacterium jeikeium K411 92 Clostridium beijerinckii NCIMB
8052 chromosome
114 Corynebacterium urealyticum DSM 7109
93 Clostridium botulinum A str. ATCC 19397
115 Cupriavidus metallidurans CH34 chromosome
94 Clostridium botulinum A str. ATCC 3502
116 Cupriavidus necator N-1 chromosome chromosome 1 95 Clostridium botulinum A str.
Hall
117 Cupriavidus taiwanensis LMG 19424 chromosome 1
96 Clostridium botulinum A3 str. Loch Maree
118 Dehalococcoides ethenogenes 195
97 Clostridium botulinum B str. Eklund 17B
119 Dehalococcoides sp. BAV1 98 Clostridium botulinum B1 str.
Okra
120 Dehalococcoides sp. CBDB1 chromosome
99 Clostridium botulinum E3 str. Alaska E43
121 Ehrlichia canis str. Jake 100 Clostridium botulinum F str.
Langeland
122 Ehrlichia chaffeensis str. Arkansas
101 Clostridium difficile 630 chromosome
123 Ehrlichia ruminantium str. Gardel
102 Clostridium kluyveri DSM 555 124 Ehrlichia ruminantium str. Welgevonden
103 Clostridium novyi NT 125 Francisella philomiragia subsp. philomiragia ATCC 25017 chromosome
104 Clostridium perfringens ATCC 13124
126 Francisella tularensis subsp. holarctica FTNF002-00 chromosome
105 Clostridium perfringens str. 13 127 Francisella tularensis subsp. holarctica LVS chromosome 106 Clostridium phytofermentans
ISDg
24
Lampiran 1 Lanjutan
No Nama Organisme No Nama Organisme
129 Francisella tularensis subsp. mediasiatica FSC147
151 Helicobacter pylori 26695 130 Francisella tularensis subsp.
novicida U112
152 Helicobacter pylori G27 chromosome
131 Francisella tularensis subsp. tularensis FSC198
153 Helicobacter pylori HPAG1 chromosome
132 Francisella tularensis subsp. tularensis SCHU S4
154 Helicobacter pylori J99 133 Francisella tularensis subsp.
tularensis WY96-3418
155 Helicobacter pylori P12 chromosome
134 Frankia alni ACN14a chromosome
156 Helicobacter pylori Shi470 chromosome
135 Frankia sp. CcI3 chromosome 157 Lactobacillus acidophilus NCFM chromosome
136 Frankia sp. EAN1pec chromosome
158 Lactobacillus brevis ATCC 367 137 Geobacter bemidjiensis Bem
chromosome
159 Lactobacillus casei ATCC 334 138 Geobacter lovleyi SZ
chromosome
160 Lactobacillus casei BL23 chromosome
139 Geobacter metallireducens GS-15 chromosome
161 Lactobacillus delbrueckii subsp. bulgaricus ATCC 11842
140 Geobacter sulfurreducens PCA chromosome
162 Lactobacillus delbrueckii subsp. bulgaricus ATCC BAA-365 chromosome
141 Geobacter uraniireducens Rf4 chromosome
163 Lactobacillus fermentum IFO 3956
142 Haemophilus ducreyi 35000HP 164 Lactobacillus gasseri ATCC 33323
143 Haemophilus influenzae 86-028NP chromosome
165 Lactobacillus helveticus DPC 4571
144 Haemophilus influenzae PittEE chromosome
166 Lactobacillus johnsonii NCC 533 145 Haemophilus influenzae PittGG
chromosome
167 Lactobacillus plantarum WCFS1 146 Haemophilus influenzae Rd
KW20 chromosome
168 Lactobacillus reuteri DSM 20016 chromosome
147 Haemophilus somnus 129PT chromosome
169 Lactobacillus reuteri JCM 1112 148 Haemophilus somnus 2336
chromosome
170 Lactobacillus sakei subsp. sakei 23K
149 Helicobacter acinonychis str. Sheeba chromosome
171 Lactobacillus salivarius UCC118 150 Helicobacter hepaticus ATCC
51449 chromosome
172 Leptospira biflexa serovar Patoc strain 'Patoc 1 (Ames)'
25 Lampiran 1 Lanjutan
No Nama Organisme No Nama Organisme
173 Leptospira biflexa serovar Patoc strain 'Patoc 1 (Paris)'
chromosome chromosome I
194 Mycobacterium avium subsp. paratuberculosis K-10
174 Leptospira borgpetersenii serovar Hardjo-bovis L550 chromosome 1
195 Mycobacterium bovis AF2122/97
175 Leptospira interrogans serovar Copenhageni str. Fiocruz L1-130 chromosome chromosome I
196 Mycobacterium bovis BCG str. Pasteur 1173P2
176 Leptospira interrogans serovar Lai str. 56601 chromosome chromosome I
197 Mycobacterium gilvum PYR-GCK chromosome
177 Listeria innocua Clip11262 198 Mycobacterium leprae TN chromosome
178 Listeria monocytogenes EGD-e 199 Mycobacterium marinum M 179 Listeria monocytogenes serotype
4b str. F2365 chromosome
200 Mycobacterium smegmatis str. MC2 155
180 Listeria welshimeri serovar 6b str. SLCC5334
201 Mycobacterium sp. JLS chromosome
181 Methanococcus maripaludis C5 chromosome
202 Mycobacterium sp. KMS chromosome
182 Methanococcus maripaludis C6 chromosome
203 Mycobacterium sp. MCS chromosome
183 Methanococcus maripaludis C7 chromosome
204 Mycobacterium tuberculosis CDC1551
184 Methanococcus maripaludis S2 chromosome
205 Mycobacterium tuberculosis F11 185 Methanosarcina acetivorans C2A
chromosome
206 Mycobacterium tuberculosis H37Ra
186 Methanosarcina barkeri str. Fusaro chromosome
207 Mycobacterium tuberculosis H37Rv
187 Methanosarcina mazei Go1 chromosome
208 Mycobacterium ulcerans Agy99 188 Methylobacterium extorquens
PA1 chromosome
209 Mycobacterium vanbaalenii PYR-1 chromosome
189 Methylobacterium populi BJ001 chromosome
210 Mycoplasma agalactiae PG2 190 Methylobacterium radiotolerans
JCM 2831 chromosome
211 Mycoplasma arthritidis 158L3-1 191 Methylobacterium sp. 4-46
chromosome
212 Mycoplasma capricolum subsp. capricolum ATCC 27343 192 Mycobacterium abscessus ATCC
19977 chromosome chromosome 1
213 Mycoplasma gallisepticum str. R(low) chromosome
26
Lampiran 1 Lanjutan
No Nama Organisme No Nama Organisme
215 Mycoplasma hyopneumoniae 232 238 Psychrobacter arcticus 273-4 216 Mycoplasma hyopneumoniae
7448 chromosome
239 Psychrobacter cryohalolentis K5 chromosome
217 Mycoplasma hyopneumoniae J chromosome
240 Psychrobacter sp. PRwf-1 chromosome
218 Mycoplasma mobile 163K 241 Pyrobaculum aerophilum str. IM2 chromosome
219 Mycoplasma mycoides subsp. mycoides SC str. PG1
chromosome
242 Pyrobaculum arsenaticum DSM 13514
220 Mycoplasma penetrans HF-2 243 Pyrobaculum calidifontis JCM 11548 chromosome
221 Mycoplasma pneumoniae M129 244 Pyrobaculum islandicum DSM 4184 chromosome
222 Mycoplasma pulmonis UAB CTIP
245 Pyrococcus abyssi GE5 chromosome
223 Mycoplasma synoviae 53 246 Pyrococcus furiosus DSM 3638 224 Onion yellows phytoplasma
OY-M
247 Pyrococcus horikoshii OT3 chromosome
225 Pseudomonas aeruginosa PA7 248 Rickettsia akari str. Hartford 226 Pseudomonas aeruginosa PAO1
chromosome
249 Rickettsia bellii OSU 85-389 227 Pseudomonas aeruginosa
UCBPP-PA14
250 Rickettsia bellii RML369-C 228 Pseudomonas fluorescens Pf-5
chromosome
251 Rickettsia canadensis str. McKiel 229 Pseudomonas fluorescens Pf0-1
chromosome
252 Rickettsia conorii str. Malish 7 230 Pseudomonas putida F1
chromosome
253 Rickettsia felis URRWXCal2 231 Pseudomonas putida GB-1
chromosome
254 Rickettsia massiliae MTU5 232 Pseudomonas putida KT2440
chromosome
255 Rickettsia prowazekii str. Madrid E chromosome
233 Pseudomonas putida W619 chromosome
256 Rickettsia rickettsii str. 'Sheila Smith'
234 Pseudomonas syringae pv. phaseolicola 1448A chromosome
257 Rickettsia rickettsii str. Iowa chromosome
235 Pseudomonas syringae pv. syringae B728a
258 Rickettsia typhi str. Wilmington 236 Pseudomonas syringae pv.
tomato str. DC3000 chromosome
259 Shewanella amazonensis SB2B chromosome
237 Pseudomonas syringae pv. tomato str. DC3000 plasmid pDC3000A
27 Lampiran 1 Lanjutan
No Nama Organisme No Nama Organisme
261 Shewanella denitrificans OS217 282 Staphylococcus aureus subsp. aureus COL chromosome 262 Shewanella frigidimarina
NCIMB 400
283 Staphylococcus aureus subsp. aureus JH1
263 Shewanella halifaxensis HAW-EB4 chromosome
284 Staphylococcus aureus subsp. aureus JH9
264 Shewanella loihica PV-4 285 Staphylococcus aureus subsp. aureus MRSA252 chromosome 265 Shewanella oneidensis MR-1 286 Staphylococcus aureus subsp.
aureus MSSA476 chromosome 266 Shewanella pealeana ATCC
700345 chromosome
287 Staphylococcus aureus subsp. aureus MW2
267 Shewanella putrefaciens CN-32 chromosome
288 Staphylococcus aureus subsp. aureus Mu3
268 Shewanella sediminis HAW-EB3 289 Staphylococcus aureus subsp. aureus Mu50
269 Shewanella sp. ANA-3 chromosome chromosome 1
290 Staphylococcus aureus subsp. aureus N315
270 Shewanella sp. MR-4 chromosome
291 Staphylococcus aureus subsp. aureus NCTC 8325 chromosome 271 Shewanella sp. MR-7
chromosome
292 Staphylococcus aureus subsp. aureus USA300_FPR3757 chromosome
272 Shewanella sp. W3-18-1 chromosome
293 Staphylococcus aureus subsp. aureus USA300_TCH1516 chromosome
273 Shewanella woodyi ATCC 51908 chromosome
294 Staphylococcus aureus subsp. aureus str. Newman chromosome 274 Shigella boydii CDC 3083-94
chromosome
295 Staphylococcus epidermidis ATCC 12228 chromosome 275 Shigella boydii Sb227 296 Staphylococcus epidermidis
RP62A
276 Shigella dysenteriae Sd197 297 Staphylococcus haemolyticus JCSC1435 chromosome 277 Shigella flexneri 2a str. 2457T 298 Staphylococcus saprophyticus
subsp. saprophyticus ATCC 15305
278 Shigella flexneri 2a str. 301 chromosome
299 Streptococcus agalactiae 2603V/R
279 Shigella flexneri 5 str. 8401 chromosome
300 Streptococcus agalactiae A909 280 Shigella sonnei Ss046
chromosome
301 Streptococcus agalactiae NEM316
28
Lampiran 1 Lanjutan
No Nama Organisme No Nama Organisme
303 Streptococcus gordonii str. Challis substr. CH1
325 Streptococcus suis 05ZYH33 304 Streptococcus mutans UA159
chromosome
326 Streptococcus suis 98HAH33 305 Streptococcus pneumoniae
CGSP14
327 Streptococcus thermophilus CNRZ1066 chromosome 306 Streptococcus pneumoniae D39 328 Streptococcus thermophilus
LMD-9 307 Streptococcus pneumoniae G54
chromosome
329 Streptococcus thermophilus LMG 18311 chromosome
308 Streptococcus pneumoniae Hungary19A-6
330 Streptomyces avermitilis MA-4680
309 Streptococcus pneumoniae R6 331 Streptomyces coelicolor A3(2) chromosome
310 Streptococcus pneumoniae TIGR4 chromosome
332 Streptomyces griseus subsp. griseus NBRC 13350 311 Streptococcus pyogenes M1 GAS
chromosome
333 Sulfolobus acidocaldarius DSM 639 chromosome
312 Streptococcus pyogenes MGAS10270 chromosome
334 Sulfolobus solfataricus P2 chromosome
313 Streptococcus pyogenes MGAS10394 chromosome
335 Sulfolobus tokodaii str. 7 chromosome
314 Streptococcus pyogenes MGAS10750 chromosome
336 Synechococcus elongatus PCC 6301 chromosome
315 Streptococcus pyogenes MGAS2096 chromosome
337 Synechococcus elongatus PCC 7942 chromosome
316 Streptococcus pyogenes MGAS315 chromosome
338 Synechococcus sp. CC9311 317 Streptococcus pyogenes
MGAS5005 chromosome
339 Synechococcus sp. CC9605 318 Streptococcus pyogenes
MGAS6180 chromosome
340 Synechococcus sp. CC9902 chromosome
319 Streptococcus pyogenes MGAS8232 chromosome
341 Synechococcus sp. JA-2-3B'a(2-13)
320 Streptococcus pyogenes MGAS9429 chromosome
342 Synechococcus sp. JA-3-3Ab 321 Streptococcus pyogenes NZ131
chromosome
343 Synechococcus sp. PCC 7002 chromosome
322 Streptococcus pyogenes SSI-1 chromosome
344 Synechococcus sp. RCC307 323 Streptococcus pyogenes str.
Manfredo