PENYUSUNAN
OVERLAP GRAPH
MENGGUNAKAN
SUFFIX
TREE
PADA DNA
SEQUENCE
ADITRIAN RAHIM
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM INSTITUT PERTANIAN BOGOR
PERNYATAAN MENGENAI SKRIPSI DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA
Dengan ini saya menyatakan bahwa skripsi berjudul Penyusunan Overlap Graph menggunakan Suffix Tree pada DNA Sequence adalah benar karya saya dengan arahan dari komisi pembimbing dan belum diajukan dalam bentuk apa pun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir skripsi ini.
Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
ABSTRAK
ADITRIAN RAHIM. Penyusunan Overlap Graph menggunakan Suffix Tree pada DNA Sequence. Dibimbing oleh WISNU ANANTA KUSUMA dan SONY HARTONO WIJAYA.
DNA sequence assembly memiliki peran yang sangat penting dalam analisis genom. Tahap ini bertujuan untuk merekonstruksi fragmen DNA (contig) dari penyatuan fragmen-fragmen pendek menggunakan overlap di antara fragmen tersebut. Metode ini menggunakan overlap graph untuk menyelesaikan masalah. Pada graf ini, node merepresentasikan fragmen dan edge merepresentasikan overlap. Penelitian ini mengembangkan sebuah perangkat lunak untuk menyusun overlap graph menggunakan suffix tree. Evaluasi dilakukan dengan menghitung jumlah node, jumlah edge dan waktu eksekusi dalam beberapa nilai pada minimum overlap. Minimum overlap didefinisikan sebagai panjang minimum dari substring yang diperhitungkan dalam overlap di antara dua fragmen. Hasil evaluasi menunjukkan bahwa jumlah node, jumlah edge dan waktu eksekusi meningkat seiring dengan menurunnya nilai minimum overlap.
Katakunci: Overlap Layout Consensus, DNA sequence assembly, overlap graph, pendeteksian overlap.
ABSTRACT
ADITRIAN RAHIM. Overlap Graph Construction using Suffix Tree on DNA Sequence. Supervised by WISNU ANANTA KUSUMA and SONY HARTONO WIJAYA.
The DNA sequence assembly is very important in genome analysis. This step aims to reconstruct contigous DNA fragment (contigs) from short fragment by concatenating fragments based on the overlap region among them. This method employs overlap graph for solving the problems. In this graph, nodes represent fragments and edges represent overlap. This research developed a software for constructing overlap graph using suffix tree. The evaluation was conducted by measuring the number of nodes, number of edges, and the execution time in many values of minimum overlap. The minimum overlap is defined as the minimum length of substring which is considered as the overlap region between two fragments. The evaluation results showed that the number of nodes, the number of edges, and the execution time were increased with the decreasing of the minimum overlap value.
Skripsi
sebagai salah satu syarat untuk memperoleh gelar Sarjana Komputer
pada
Departemen Ilmu Komputer
PENYUSUNAN
OVERLAP GRAPH
MENGGUNAKAN
SUFFIX
TREE
PADA DNA
SEQUENCE
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM INSTITUT PERTANIAN BOGOR
BOGOR 2013
Judul Skripsi : Penyusunan Overlap Graph menggunakan Suffix Tree pada DNA Sequence
Nama : Aditrian Rahim NIM : G64090035
Disetujui oleh
Dr Wisnu Ananta Kusuma, ST MT Pembimbing I
Sony Hartono Wijaya, SKom MKom Pembimbing II
Diketahui oleh
Dr Ir Agus Buono, MSi MKom Ketua Departemen
Tanggal Lulus:
PRAKATA
Puji dan syukur penulis panjatkan kepada Allah subhanahu wa ta’ala atas segala karunia, rahmat dan ridho-Nya sehingga karya ilmiah ini berhasil diselesaikan. Penelitian ini berfokus pada bidang bioinformatika dengan menitikberatkan pada DNA sequence assembly sebagai salah satu proses untuk mengetahui suatu whole genome pada suatu organisme. Hal yang menjadi motivasi penulis dalam memilih topik ini yaitu bioinformatika sebagai salah satu bidang ilmu yang selalu menawarkan metode-metode baru untuk menyelesaikan masalah-masalah terutama pada bidang biologi molekuler melalui bantuan teknologi informasi.
Terima kasih penulis ucapkan kepada Bapak Dr Wisnu Ananta Kusuma ST, MT selaku pembimbing pertama yang senantiasa selalu membimbing, mengawasi dan mengingatkan penulis pada penelitian ini, serta penulis juga ucapkan terima kasih kepada Bapak Sony Hartono Wijaya SKom, MKom selaku pembimbing kedua yang telah memberikan masukan dan kritik yang sangat bermanfaat untuk memperbaiki penelitian ini. Tak lupa pula penulis menyampaikan terima kasih kepada ayah, ibu dan kakak yang selalu mendukung dan mendoakan selama penelitian ini berlangsung. Ungkapan terima kasih juga disampaikan kepada teman-teman satu bimbingan yaitu Novaldo, Gary, Albert, Aries serta rekan-rekan satu angkatan Ilmu Komputer angkatan 46 yang secara langsung dan tidak langsung membantu penulis pada penelitian ini.
Semoga karya ilmiah ini bermanfaat bagi perkembangan ilmu pengetahuan secara umum dan ilmu komputer pada khususnya.
DAFTAR ISI
DAFTAR TABEL vi
DAFTAR GAMBAR vi
DAFTAR LAMPIRAN vi
PENDAHULUAN 1
Latar Belakang 1
Perumusan Masalah 2
Tujuan Penelitian 2
Manfaat Penelitian 2
Ruang Lingkup Penelitian 2
METODE 2
Mengumpulkan dan Menyiapkan Data DNA Sequence 2
Membentuk Suffix Tree dari Data DNA Sequence 5
Menyusun Overlap Graph 7
Evaluasi 7
HASIL DAN PEMBAHASAN 7
Mengumpulkan dan Menyiapkan Data DNA Sequence 7
Membentuk Suffix Tree dari Data DNA Sequence 9
Menyusun Overlap Graph 9
Evaluasi Jumlah Node dan Edge 11
Evaluasi Waktu Eksekusi 13
Kompleksitas 14
SIMPULAN DAN SARAN 14
Simpulan 14
Saran 14
DAFTAR PUSTAKA 15
DAFTAR TABEL
1 Daftar nukleotida penyusun DNA 3
2 Informasi organisme yang digunakan 8
3 Paramater data DNA sequence menggunakan MetaSim 8
DAFTAR GAMBAR
1 Database organisme hasil unduhan pada MetaSim 3
2 Tampilan database organisme pada MetaSim 4
3 Konfigurasi parameter pada MetaSim 4
4 Tampilan simulasi data DNA sequence 5
5 Suffix tree untuk string 'AACGT' 6
6 Relaxed Suffix Tree untuk string‘AACGT’ 6
7 Ilustrasi penyusunan overlap graph 7
8 Staphylococcus aureus dalam fail FASTA 8
9 Pseudocode pembentukan suffix tree 9
10 Pseudocode pembentukan overlap graph 10
11 Ilustrasi pendeteksian overlap antarfragmen 10
12 Overlap graph R1 dan R2 11
13 Grafik jumlah edge dengan minimum overlap 12
14 Grafik waktu eksekusi dengan minimum overlap 13
DAFTAR LAMPIRAN
1 Source Code 16
2 Screen Shoot Program 20
1
PENDAHULUAN
Latar Belakang
Pemanfaatan teknologi komputer telah mengalami perkembangan pesat terutama pada ilmu biologi. Telah banyak aplikasi-aplikasi komputer yang dikembangkan untuk membantu para peneliti biologi dalam memecahkan masalah, seperti membandingkan DNA sequence, meneliti struktur protein dan perakitan DNA (DNA assembly) (Maftuhah 2007).
Salah satu permasalahan dalam DNA sequencing adalah belum adanya teknologi sequencer yang mampu menghasilkan whole genome dalam satu kali eksekusi. Oleh karena itu, diperkenalkanlah teknik Shotgun sequencing dan DNA sequence assembly. Teknik Shotgun sequencing yaitu teknik memotong DNA secara acak yang menghasilkan fragmen-fragmen (reads) yang saling overlap. DNA sequence assembly merupakan proses penyusunan kembali fragmen-fragmen tersebut menjadi kesatuan DNA sequence yang utuh.
Teknologi sequencer yang selama ini dikenal adalah Sanger sequencer dan sequncer generasi kedua. Sanger sequencer menghasilkan panjang fragmen jauh lebih panjang (500 bp) dibandingkan dengan sequencer generasi kedua (200 bp dan 35-50 bp), namun Sanger menghasilkan jumlah fragmen lebih sedikit dibandingkan dengan yang dihasilkan oleh generasi kedua (Kusuma et al. 2011). Selain itu, sequencer generasi kedua memiliki waktu eksekusi untuk menghasilkan fragmen jauh lebih cepat dibandingkan dengan Sanger. Hal inilah yang menjadi pertimbangan bahwa sequencer generasi kedua lebih banyak digunakan oleh para peneliti biologi.
Salah satu metode untuk melakukan DNA sequence assembly adalah dengan pendekatan Hamiltonian path menggunakan overlap graph. Hamiltonian path adalah jalur terpendek untuk pengunjungan tepat satu kali pada setiap node dalam suatu graf (Abegunde 2010). Overlap graph merupakan representasi berbentuk graf dari fragmen-fragmen yang dihasilkan oleh sequencer. Fragmen direpresentasikan sebagai node dan overlap direpresentasikan sebagai edge.
Pendeteksian overlap antarfragmen sangat sulit dilakukan jika tanpa proses indexing sebelumnya. Selain itu, pendeteksian overlap membutuhkan waktu yang cukup lama. Salah satu cara untuk mendeteksi overlap antarfragmen yaitu dengan membangun suffix tree. Dengan menggunakan suffix tree, pendeteksian dapat dilakukan dengan lebih cepat jika dibandingkan tanpa proses indexing sebelumnya. Selain itu, suffix tree merupakan representasi tree yang dapat menghasilkan suatu algoritme yang efisien untuk mendeteksi overlap pada suatu string dengan kompleksitas linear (Gusfield 1997).
Penelitian sebelumnya yang dilakukan oleh Hernandez (2008) yaitu membangun sebuah perangkat lunak atau assembler dengan mengimplementasikan prefix array. Assembler tersebut memberikan informasi jumlah node dan jumlah edge dalam suatu data DNA sequence.
2
Perumusan Masalah Perumusan masalah pada penelitian ini adalah:
1 bagaimana cara untuk mengimplementasikan suffix tree untuk mendeteksi overlap antarfragmen DNA?
2 bagaimana membentuk graf yang merepresentasikan node sebagai fragmen dan edge sebagai overlap?
Tujuan Penelitian
Tujuan dari penelitian ini adalah membangun perangkat lunak untuk mendeteksi overlap antarfragmen DNA dengan mengimplementasikan suffix tree dan memberikan informasi grafberupa node dan edge.
Manfaat Penelitian
Penelitian ini diharapkan dapat menjadi dasar untuk pengembangan perangkat lunak perakitan DNA yang memiliki kompleksitas algoritme dan waktu eksekusi yang lebih baik.
Ruang Lingkup Penelitian Ruang lingkup penelitian ini antara lain adalah:
1 penelitian ini hanya berfokus pada penyusunan overlap graph yang menampilkan informasi jumlah node dan edge pada suatu data DNA sequence
2 data DNA sequence merupakan data yang diunduh pada website National Center for Biotechnology Information (NCBI) dan dipilih tiga organisme 3 data DNA sequence yang digunakan pada penelitian ini adalah data error
free hasil simulasi menggunakan perangkat lunak MetaSim (Richter et al. 2008)
4 bahasa pemrograman yang digunakan adalah C++ dengan library tambahan seqan (Döring 2008).
METODE
Penelitian ini dilakukan dalam beberapa tahap agar ketercapaian tujuan dapat terlaksana. Terdapat empat tahapan penelitian, di antaranya mengumpulkan dan menyiapkan data DNA sequence, membentuk suffix tree dari data DNA sequence, membentuk overlap graph, evaluasi jumlah node dan edge pada graf dan evaluasi waktu eksekusi.
Mengumpulkan dan Menyiapkan Data DNA Sequence
3 penyimpanan informasi jangka panjang. Setiap makhluk hidup terdiri dari DNA genom. Suatu DNA genom merupakan representasi cetak biru (blue print) yang memiliki instruksi-instruksi untuk membangun suatu komponen lain dari sel, seperti molekul RNA bahkan protein. Bagian dari DNA yang membawa informasi genetis disebut dengan gen (Maftuhah 2007).
DNA terbentuk dari susunan unsur pokok yang disebut dengan nukleotida. Sebuah nukleotida terhubung dengan salah satu dari empat jenis nitrogen organik yang disimbolkan pada empat huruf seperti pada Tabel 1.
Pada DNA terdapat rangkaian komplementer yaitu adenine (A)berpasangan dengan thymine (T) (berlaku sebaliknya) dan cytosine (C) berpasangan dengan guanine (G) (berlaku sebaliknya). Apabila diketahui urutan nukleotida pada salah satu rangkaian, maka dapat ditentukan nukleotida pasangannya. Karena sifat DNA tersebut maka untuk menyatakan ukuran suatu DNA digunakanlah satuan base pair (bp) (Maftuhah 2007).
Data yang digunakan pada penelitian ini adalah data yang diunduh dari
website NCBI pada alamat web
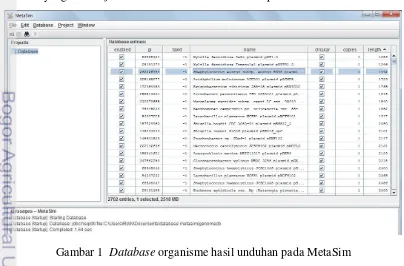
ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/all.fna.tar.gz. Gambar 1 merupakan basis data organisme hasil unduhan yang telah berhasil dimasukan pada MetaSim. Data hasil unduhan tersebut adalah hasil sequencing dari makhluk hidup tertentu. Data sequence ini terdiri atas fragmen-fragmen DNA yang memiliki panjang tertentu yang akan dijadikan failmasukkan untuk diimplementasikan.
Tabel 1 Daftar nukleotida penyusun DNA
Kode 1 huruf Nama Nukleotida Kategori
A Adenine Purine
C Cytosine Pyrimidine
G Guanine Purine
T Thymine Pyrimidine
4
Data DNA sequence diperoleh dari simulasi menggunakan perangkat lunak yang bernama MetaSim. Dengan menggunakan MetaSim, keberadaan error pada suatu DNA sequence dapat dibangkitkan ataupun tidak. Seperti yang diketahui bahwa data masukkan dari sebuah assembler yang menggunakan pendekatan graf sangat sensitif terhadap eror. Oleh karena itu, data DNA sequence pada penelitian ini perlu disimulasikan agar data sequence tersebut tidak memiliki kesalahan (error free). Pemilihan organisme yang akan disimulasikan menggunakan MetaSim tersaji pada Gambar 2.
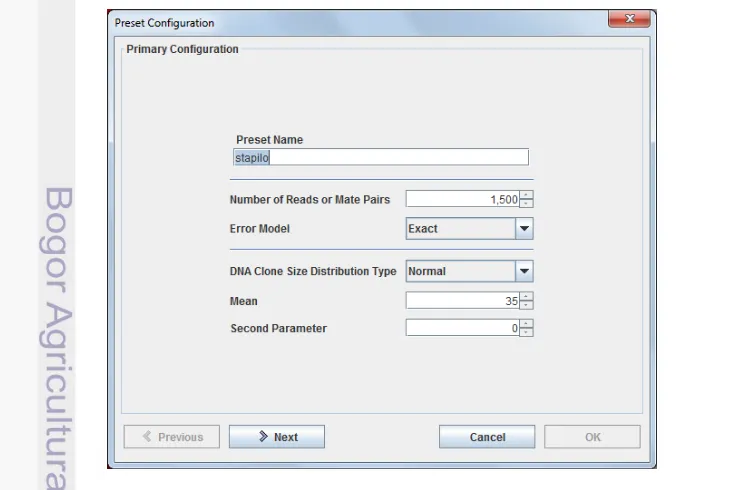
Konfigurasi MetaSim yang tersaji pada Gambar 3 terdapat beberapa
parameter yang harus dipilih dan diisi, “Preset Name” diisi untuk memberikan nama dari konfigurasi itu sendiri, lalu “Number of Reads or Mate Pairs” yaitu
jumlah fragmen yang ingin kita bangkitkan pada suatu DNA sequence. “Error
Model” terdiri atas 454, Empirical, Sanger dan Exact, untuk mendapatkan data sequence yang tidak memiliki kesalahan maka harus dipilih “Error Model” Exact.
“DNA Clone Size Distribution Type” terdiri atas Uniform, Normal, dan Empirical, pada penelitian ini tipe distribusi yang dipilih yaitu Normal, sedangkan “Mean” yaitu panjang sequence setiap fragmennya, penelitian ini dipilih “Mean” dengan
panjang 35 bp dan “Second Parameter” dipilih 0 (nol). Konfigurasi ini menghasilkan fail yang berformat .mprf.
Gambar 2 Tampilan database organisme pada MetaSim
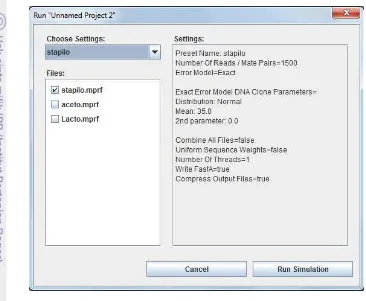
5 Setelah memilih organisme pada basis data dan membentuk konfigurasi pada MetaSim, selanjutnya adalah membuat simulasi data sequence seperti pada Gambar 4. “Choose Settings:” memilih konfigurasi yang telah dibuat sebelumnya. Setelah itu pilih organisme yang ingin disimulasikan pada “Files” berformat .mprf.
selanjutnya pilih “Run Simulation”. Pada akhir proses simulasi didapatkan data sequence yang menjadi data masukan bagi perangkat lunak pada penelitian ini.
Membentuk Suffix Tree dari Data DNA Sequence
Suffix tree adalah struktur data yang merepresentasikan sufiks dari string sedemikian sehingga memudahkan implementasi tertentu dengan cepat untuk berbagai operasi pada string. Suffix tree dari string S adalah pohon yang sisinya (edge) diberikan label string yang merupakan sufiks dari S dan sesuai dengan tepat satu jalur dari root ke leaf. Dalam membangun pohon untuk string S membutuhkan waktu dan ruang linear tergantung panjang S. Jika sudah terbentuk, beberapa operasi dapat dilakukan lebih cepat, misalnya mencari substring di S.
6
Suffix tree untuk string S yang panjangnya n didefinisikan sebagai suatu pohon sedemikian sehingga:
a lintasan dari root ke leaf memiliki hubungan satu-satu dengan sufiks dari S.
b setiap edge diisi string tak kosong.
c semua simpul dalam (internal node) kecuali root memliki paling sedikit dua anak (child).
Di dalam suffix tree, string S selalu ditambahkan dengan sebuah terminal symbol yang tidak termasuk di dalam substring S, biasanya dilambangkan dengan $ seperti pada Gambar 5 (Maftuhah 2007).
Dalam implementasinya suffix tree yang digunakan adalah suffix tree yang lebih sederhana. Suffix tree tersebut tidak menggunakan terminal symbol. Suffix tree ini disebut dengan relaxed suffix tree, seperti pada Gambar 6. Hal ini untuk menghemat ruang memori untuk string yang sangat panjang pada data sequence. Penghilangan terminal symbol ini sangat berpengaruh jika data DNA sequence yang menjadi data masukkan bagi perangkat lunak memiliki panjang sequence yang sangat panjang. Untuk itulah terminal symbol ini dihilangkan.
Relaxed suffix tree ini digunakan untuk menyimpan string data sequence dan nantinya akan digunakan sebagai pendeteksian overlap pada setiap fragmen data sequence.
Gambar 5 Suffix tree untuk string 'AACGT'
7 Menyusun Overlap Graph
Pada tahapan penelitian ini dibentuk suatu graf. Setiap fragmen yang saling overlap akan dijadikan sebuah graf. Setiap node merepresentasikan fragmen dan setiap edge merepresentasikan overlap di antara dua fragmen tersebut. Overlap graph dapat dibentuk jika suffix tree pada masing-masing fragmen telah terbentuk. Setelah suffix tree terbentuk, akhiran pada suatu fragmen dibandingkan dengan awalan fragmen lainnya. Jika akhiran fragmen yang satu sama dengan akhiran fragmen lainnya, kedua fragmen tersebut saling overlap satu sama lain. Salah satu contoh penyusunan overlap graph dapat dilihat pada Gambar 7.
Evaluasi
Tahapan akhir pada penelitian ini adalah evaluasi. Setelah overlap graph terbentuk, selanjutnya adalah menganalisis data keluaran yang dihasilkan oleh perangkat lunak pada penelitian kali ini. Hal-hal yang dianalisis adalah keterhubungan antara jumlah node dan jumlah edge dengan minimum overlap, setelah itu membandingkan waktu eksekusi dengan minimum overlap. Kompleksitas perangkat lunak dianalisis dari kode program dan psedocode yang telah dibentuk.
HASIL DAN PEMBAHASAN
Mengumpulkan dan Menyiapkan Data DNA Sequence
Penelitian ini menggunakan tiga organisme yang dipilih pada basis data MetaSim. Tiga organisme ini dipilih karena panjang sequence yang dimiliki organisme tersebut relatif lebih pendek dengan yang lainnya agar waktu eksekusi yang didapatkan tidak terlalu lama. Informasi organisme tersebut dapat dilihat pada Tabel 2.
8
Dari setiap organisme tersebut akan dilakukan simulasi masing-masing satu kali. Setiap satu kali simulasi menghasilkan sebuah fail berformat FASTA (.fna). Dari hasil simulasi tersebut nantinya didapatkan tiga fail yang menjadi data masukkan pada penelitian ini. Dapat dijamin bahwa tiga fail tersebut merupakan data DNA sequence yang tidak memiliki error. Maksud dari error ini adalah setiap nukleotida pada fragmen atau reads tidak ada yang diganti dengan nukleotida lainnya, namun simulasi ini tidak dapat menghilangkan fragmen yang saling redundant dengan fragmen lainnya.
Untuk mendapatkan data DNA sequence yang tidak memiliki kesalahan, maka error model yang dipilih adalah exact dengan panjang setiap fragmen (Mean) yaitu 35 bp. Informasi parameter yang digunakan untuk setiap organisme pada setiap simulasi dapat dilihat pada Tabel 3.
Gambar 8 merupakan dua baris pertama dari suatu data DNA sequence yang telah disimulasikan menggunakan perangkat lunak MetaSim.
Tabel 2 Informasi organisme yang digunakan
No. Nama Organisme Panjang
Sequence
Asli (bp)
Gi
1 Staphylococcus aureus subsp. aureus ED98 plasmid pAVY
1442 262225764
2 Acetobacter pasteurianus IFO 3283-01 plasmid pAPA01-060
1815 258513334
3 Lactobacillus plantarum WCFS1 plasmid pWCFS102
1917 54307232
Tabel 3 Paramater data DNA sequence menggunakan MetaSim
No. Nama Organisme Number of Read /
9
Dalam fail FASTA tanda lebih besar “>” bermakna bahwa satu fragmen
mulai terbaca dan akhir dari urutan fragmen tersebut akan ditandai lagi dengan
tanda “>” pada baris yang lainnya.
Membentuk Suffix Tree dari Data DNA Sequence
Setelah data DNA sequence didapatkan, langkah selanjutnya adalah membentuk suffix tree dari setiap fragmen-fragmen yang ada pada data DNA sequence. Perangkat lunak yang dikembangkan pada penelitian kali ini akan menerima data masukkan berupa fail dengan format FASTA (.fna) pada suatu path fail. Selanjutnya perangkat lunak membaca satu per satu fragmen yang ada pada fail yang berformat .fna tersebut. Setiap satu fragmen selesai dibaca maka perangkat lunak langsung membentuk suffix tree. Node-node dari tree tersebut disimpan dalam suatu memori yang nantinya akan diakses kembali untuk pendeteksian overlap pada fragmen yang lainnya. Proses ini akan terus dilakukan sampai semua fragmen yang ada pada data masukkan DNA sequence dibaca seluruhnya oleh perangkat lunak dan node-node dari setiap suffix tree pada setiap fragmen disimpan ke dalam memori. Pseudocode untuk pembentukan suffix tree pada setiap fragmen dapat dilihat pada Gambar 9.
Pembentukan suffix tree ini membutuhkan waktu yang sangat lama jika fragmen yang dibaca memiliki panjang string yang sangat panjang. Namun sebaliknya, jika fragmen yang dibaca memiliki panjang string yang relatif pendek, maka pembentukan suffix tree pun cepat. Pada penelitian ini panjang setiap fragmen relatif pendek yaitu hanya 35 bp atau dengan kata lain hanya memiliki 35 karakter nukleotida untuk setiap fragmennya, tetapi jumlah fragmen yang dibaca yaitu 1500 fragmen dan 2000 fragmen. Oleh karena itu, suffix tree yang terbentuk berjumlah 1500 dan 2000.
Menyusun Overlap Graph
Pada proses ini, penyusunan overlap graph dapat dilakukan jika semua fragmen pada data DNA sequence sudah ter-index ke dalam memori. Setelah proses pembentukan suffix tree selesai, penyusunan ataupun pendeteksian overlap graph pada masing-masing fragmen pun lebih mudah dilakukan. Hal ini dikarenakan setiap string yang mewakili akhiran (suffix) pada setiap fragmen akan dibandingkan dengan awalan (prefix) dari fragmen lainnya. Jika akhiran pada fragmen yang satu cocok atau match dengan awalan pada fragmen lainnya, dua fragmen tersebut saling overlap.
for all reads R in data sequence do
generate suffix tree in each reads;
while (!all node generated)
new node; insert node;
endwhile
endfor
10
Pseudocode dan Ilustrasi pendeteksian overlap pada masing-masing fragmen tersaji pada Gambar 10 dan Gambar 11.
Seperti pada Gambar 11, terdapat dua buah fragmen, yaitu R1 dan R2. R1 berisi string ‘TTCGAT’ dan R2 ‘GATAAT’. R1 memiliki substring yang saling overlap dengan R2 yaitu pada substring‘GAT’, sebaliknya R2 memiliki substring yang saling overlap dengan R1 di ‘T’. Substring yang saling overlap ini dengan mudah terlihat jika R1 dan R2 memiliki panjang string yang pendek, namun jika R1 dan R2 tersebut memiliki string yang panjang dan terdiri atas banyak fragmen, maka akan sulit dilakukan pendeteksian overlap. Oleh karena itu suffix tree membantu untuk pendeteksian tersebut.
R1 memiliki suffix tree yang terdiri atas ‘TTCGAT’, ‘TCGAT’, ‘CGAT’,
‘GAT’, ‘AT’ dan ‘T’, sedangkan R2 memiliki suffix tree ‘GATAAT’, ‘ATAAT’,
‘TAAT’, ’AAT’, ‘AT’ dan ‘T’. Terlebih dahulu R1 mendeteksi apakah R1 memiliki string yang overlap dengan R2. Perangkat lunak mengakses semua kemungkinan node pada suffix tree R1 untuk dilakukan proses matching dengan R2, dari proses penelusuran node pada R1 maka terdapat node yang berisi substringdari R1 yaitu ‘GAT’ cocok dengan awalan pada fragmen R2. Perangkat lunak mencatat bahwa R1 saling overlap dengan R2 pada subtring ‘GAT’. Proses ini dilakukan terus-menerus sampai semua fragmen selesai mendeteksi overlap dengan fragmen yang lainnya.
Setelah itu penyusunan overlap graph dilakukan, yaitu dengan mencatat informasi semua fragmen yang saling overlap. Seperti pada Gambar 12, jika R1
Gambar 11 Ilustrasi pendeteksian overlap antarfragmen
for all suffixTree.nodes in data sequence do
while (!all node compared)
for all reads in data sequence do
comparing suffixTree.nodes with reads.prefix;
11 dan R2 saling overlap pada substring‘GAT’ maka R1 dan R2 sebagai node, dan
‘GAT’ sebagai edge. Begitu pula R2 saling overlap dengan R1, R2 dan R1 sebagai node dan substring‘T’ sebagai edge.
Pada perangkat lunak yang dikembangkan dimasukkan satu parameter tambahan, yaitu minimum overlap yang terdeteksi. Hal ini dilakukan karena jika tidak ada minimum overlap maka program akan mengeksekusi sangat lama data masukkan yang diberikan, karena data masukkan berupa fail sequence yang sangat besar. Parameter minimum overlap pada penelitian ini yaitu 10, 15, 20 dan 25.
Data keluaran dari perangkat lunak ini ialah jumlah node, jumlah edge, waktu eksekusi dan fail informasi overlap graph yang berformat .txt. Informasi data keluaran perangkat lunak dapat dilihat pada Lampiran 1.
Evaluasi Jumlah Node dan Edge
Pada evaluasi ini dibandingkan hubungan antara jumlah edge dan minimum overlap. Gambar 13 memperlihatkan bahwa dengan semakin besarnya minimum overlap yang diberikan, jumlah edge pada graf yang dihasilkan semakin kecil. Sebaliknya, jika diberikan minimum overlap yang kecil menghasilkan jumlah egde pada graf semakin besar. Dengan minimum overlap yang lebih sedikit, peluang untuk terjadinya overlap antarfragmen pada data DNA sequence semakin besar, hal inilah yang menyebabkan edge yang dihasilkan pada graf akan lebih banyak.
Pada penelitian ini tidak dibandingkan jumlah node pada graf yang dihasilkan karena jumlah node sama dengan jumlah fragmen pada data DNA sequence. Hal ini bermakna bahwa setiap fragmen pada data DNA sequence saling mengalami overlap.
Pada Gambar 13, bakteri Staphylococcus aureus memiliki jumlah fragmen yang lebih sedikit dibandingkan dengan jumlah fragmen 2 bakteri lainnya, yaitu 1500 fragmen. Sedangkan untuk bakteri Acetobacter pasteurianus dan Lactobacillus plantarum memiliki jumlah fragmen yang sama yaitu 2000 fragmen. Bakteri Staphylococcus aureus memiliki jumlah edge yang paling sedikit karena jumlah fragmen yang menjadi data masukan perangkat lunak merupakan jumlah fragmen yang paling sedikit pula. Berbeda halnya dengan bakteri Acetobacter pasteurianus dan Lactobacillus plantarum, dengan jumlah fragmen yang sama, didapatkan jumlah edge yang dihasilkan bakteri Acetobacter pasteurianus lebih banyak dibandingkan dengan jumlah edge yang dihasilkan oleh bakteri Lactobacillus plantarum.
12
Hal ini dikarenakan data redundant pada bakteri Acetobacter pasteurianus lebih banyak dibandingkan dengan data redundant yang dimiliki oleh bakteri Lactobacillus plantarum. Data redundant merupakan fragmen pada data sequence yang memiliki nukleotida-nukleotida yang sama dengan nukleotida pada fragmen lain. Dengan kata lain suatu fragmen merupakan salinan dari fragmen lainnya. Hal ini yang menyebabkan graf semakin kompleks karena dengan adanya data redundant akan terbentuk dua buah node yang sama dengan edge yaitu node itu sendiri. Sebagai contoh R1 : ATTGCTA dan R2 : ATTGCTA, akan terbentuk suatu node dari R1 ke R2 dan sebaliknya dengan edge AATGCTA. Hal inilah yang seharusnya dihilangkan pada tahap pra proses yaitu menghilangkan salah satu data redundant dengan menghapus R1 saja atau R2 saja. Dengan begitu akan didapatkan graf yang lebih sederhana.
Menurut Zerbino (2009), connectivity pada suatu graf akan meningkat jika nilai parameter k relatif kecil dan connectivity pada graf akan menurun jika nilai parameter k relatif besar. Nilai parameter k ini adalah nilai yang menjadi jumlah karakter yang menjadi node dan edge pada suatu graf. Hal ini memiliki relevansi terhadap jumlah minimum overlap yang ada pada penelitian ini. Jika nilai minimum overlap kecil maka jumlah node dan jumlah edge yang dihasilkan pun akan semakin banyak dan kompleks yang menyebabkan connectivity antarnode semakin besar.
13 Hal ini berbanding terbalik dalam hal specificity. Jika nilai k semakin besar, specificity pada graf pun akan semakin besar pula. Node-node pada graf akan semakin spesifik untuk saling berhubungan karena jumlah node dan edge yang dihasilkan lebih sedikit. Hal ini juga relevan terhadap nilai minimum overlap pada overlap graph pada penelitian kali ini.
Evaluasi Waktu Eksekusi
Seperti yang diketahui sebelumnya, hasil yang diberikan oleh perangkat lunak pada penelitian kali ini yaitu informasi jumlah node, jumlah edge, waktu eksekusi dan suatu fail .txt yang memberikan hasil print out fragmen mana saja yang saling overlap pada data sequence.
Pada evaluasi kali ini, eksekusi untuk setiap bakteri pada masing-masing minimum overlap dilakukan sebanyak 5 kali. Data yang tersaji pada grafik di Gambar 14 merupakan data yang diperoleh dari rata-rata yang dilakukan pencatatan waktu eksekusi sebanyak 5 kali. Hal ini dilakukan untuk mendapatkan rataan karena setiap kali eksekusi didapatkan waktu eksekusi yang berbeda.
Dari grafik pada Gambar 14, memperlihatkan bahwa parameter minimum overlap yang diberikan yaitu 10, 15, 20 dan 25 saling berbanding terbalik dengan hasil eksekusi yang ada. Hal ini membuktikan bahwa semakin besar minimum
14
overlap yang diberikan, waktu eksekusi program pun akan relatif semakin cepat. Namun sebaliknya, jika minimum overlap yang diberikan kecil, maka waktu eksekusi program akan semakin lama. Jika minimum overlap kecil, semua kemungkinan overlap akan semakin banyak dan program akan membutuhkan waktu yang lebih lama untuk mendeteksi serta mencatat overlap pada antarfragmen.
Kompleksitas
Kompleksitas dalam membangun suffix tree dan pendeteksian overlap serta membentuk overlap graph adalah O(n3). Kompleksitas ini didapatkan dari hasil yang ada pada pseudocode yang disajikan pada Gambar 9 dan Gambar 10. Gambar 9 merupakan pseudocode pembentukan suffix tree dengan kompleksitas O(n2) yang memiliki dua buah nested loop di dalamnya. Akan tetapi, worst case secara keseluruhan perangkat lunak yang dibangun pada penelitian kali ini yaitu O(n3). Kompleksitas seperti ini memiliki performa yang buruk untuk ukuran data DNA sequence yang sangat besar, karena waktu eksekusi program terhadap data masukan akan sangat lama.
SIMPULAN DAN SARAN
Simpulan
Penelitian ini telah mampu menghasilkan perangkat lunak yang mampu mendeteksi dan membentuk overlap graph pada suatu data DNA sequence menggunakan suffix tree. Perangkat lunak ini pun telah berhasil memberikan informasi jumlah node dan jumlah edge.
Penelitian ini juga telah berhasil membuktikan bahwa jumlah node dan edge dipengaruhi oleh panjang minimum overlap yang didefinisikan pada setiap fragmen. Kompleksitas yang terbentuk pada perangkat lunak tergolong buruk, karena perangkat lunak ini memiliki kompleksitas O(n3).
Saran Saran untuk penelitian selanjutnya:
1 melakukan tahap pra proses untuk menghapus data redundant pada data DNA sequence agar menghasilkan node dan edge yang lebih sederhana menggunakan metode prefix tree
15
DAFTAR PUSTAKA
Abegunde T, 2010. Comparison of DNA sequence assembly algorithms using mixed data sources [tesis]. Saskatoon (CAN): University of Saskatchewan Döring A, Weese D, Rausch T, Reinert K. 2008. SeqAn an efficient, generic C++
library for sequence analysis. BMC Bioinformatics. 9:11.doi:10.1186/1471-2105-9-11.
Gusfield D. 1997. Algorithms on Strings, Trees, and Sequences. University of California (US) : Cambridge University Press.
Hernandez D, Francois P, Farinelli L, Osteras M, Schrenzel J. 2008. De novo bacterial genome sequencing: Millions of very short reads assembled on a desktop computer. IPSJ Transactions on Bioinformatics. 18:802-809. doi: 10.1101/gr.072033.107.
Hu JW, 2003. An ACGT-Words tree for efficient data access in genomic databases [tesis]. Taiwan (CHN): National Sun Yat-sen University.
Kusuma WA, Ishida T, Akiyama Y. 2011. A combined approach for de novo DNA sequence assembly of very short reads. IPSJ Transactions on Bioinformatics. 4(10):21-33. doi: 10.2197/ipsjtbio.4.21.
Maftuhah DN, 2007. Pencarian string dengan menggunakan metode indexing pada datagenomic [skripsi]. Depok (ID): Universitas Indonesia.
Richter DC, Ott F, Auch AF, Huson DH, Schmid R. 2008. MetaSim – A sequencing simulator for genomics and metagenomics. Plos One. 3(10):e3373. doi:10.1371/journal.pone.0003373.
16
Lampiran 1 Source Code
#include <iostream>
#include <seqan/index.h>
#include <seqan/file.h> #include <seqan/seq_io.h>
#include <string>
using namespace seqan; using namespace std;
typedef Iterator<StringSet<DnaString> >::Type TStringSetIterator; typedef Iterator<StringSet<String<int>> >::Type TIntSetIterator; typedef Index< String<char>, IndexWotd<> > TMyIndex;
static double diffclock(clock_t clock1,clock_t clock2) {
double diffticks=clock1-clock2;
double diffms=(diffticks)/(CLOCKS_PER_SEC/1000); return diffms;
}
StringSet<DnaString> bacaSekuens(char* filename)
{
StringSet<DnaString> bacaFile(char* filename)
17 Lampiran 1 Lanjutan
vector<TMyIndex> getSuffixTree(StringSet<DnaString> & sekuens) {
vector<TMyIndex> indexVector; StringSet<DnaString> suffixtree;
for (TStringSetIterator it = begin(sekuens); it != end(sekuens);
++it) {
TMyIndex myIndex(value(it)); //membuat index suffix tree
Iterator<TMyIndex, TopDown<ParentLinks<Preorder> > > ::
Type myIterator(myIndex); //membuat iterator suffix tree
while (!atEnd(myIterator)) {
//cout << representative(myIterator) << "\n"; //print suffix tree
Dna getRevCompl(Dna const & nucleotide)
{
int start_time, end_time, inputan;
start_time = clock();
vector<TMyIndex> indexBuffer, indexBuffer2; StringSet<DnaString> sekuens, revSequence;
cout << endl << "Masukkan minimum overlap : ";
cin >> inputan; cout << endl;
cout << "Membaca file.." << endl;
//sekuens = bacaSekuens("sekuens.txt"); //pembacaan sekuens dari file
sekuens = bacaFile("input/Lacto.fna");
/*for(TStringSetIterator it = begin(sekuens); it != end(sekuens); ++it){
DnaString buffer = value(it); reverseComplement(buffer);
18
Lampiran 1 Lanjutan
cout << "Membuat suffix tree.." << endl << endl;
indexBuffer = getSuffixTree(sekuens);
ofstream myfile ("output/Lacto-out-TEST.txt");
cout << "Membuat overlap graph.." << endl;
vector<int> nodes;
int indeksTree = 1, edges = 0, nomor = 1;
for(vector<TMyIndex>::iterator it = indexBuffer.begin(); it !=
indexBuffer.end(); ++it) {
TMyIndex tree = *it;
Iterator< TMyIndex, TopDown< ParentLinks<Preorder> > >::Type myIterator(tree);
int indeksTreeElement = 0;
while (!atEnd (myIterator)) {
DnaString str = representative(myIterator);
int indeksSeq = 1;
for (TStringSetIterator it2 = begin(sekuens); it2 !=
end(sekuens); ++it2) {
DnaString seq = value(it2);
Prefix<String<Dna> >::Type pre = prefix(seq, length(str));
//bandingkan str dengan seq
if (indeksTree != indeksSeq)
{
//cout << str << " " << pre << endl;
if((str == pre) && (length(pre) != 0)
&& (length(pre) >= inputan))
{
//apa yang mau dilakukan disini?
19 Lampiran 1 Lanjutan
//memasukkan indeksTree ke dalam vector
if(std::find(nodes.begin(),
nodes.end(), indeksTree) != nodes.end()) {
/* v contains x */
nodes.end(), indeksSeq) != nodes.end()) {
/* v contains x */
myfile << "Reads ke-" << indeksTree;
myfile << " overlap dengan Reads ke-" << indeksSeq;
myfile << " dengan edge : " << str << endl;
cout << "Jumlah node : " << length(nodes) << endl;
cout << "Jumlah edge : " << edges << endl;
myfile.close();
end_time = clock();
cout << endl << "Waktu eksekusi : " << diffclock(end_time,
start_time) << " ms" << endl;
20
21
21 Lampiran 3 Hasil keluaran perangkat lunak
Nama Organisme
Minimum
overlap
Jumlah
Reads
Jumlah
Node
Jumlah
Edge Waktu Eksekusi (ms)
Staphylococcus aureus
10 1500 1500 20414 152551
15 1500 1500 16290 146257
20 1500 1500 12409 139531
25 1500 1500 8606 139295
Acetobacter pasteurianus
10 2000 2000 30062 239532
15 2000 2000 24168 223921
20 2000 2000 18404 210293
25 2000 2000 12465 207022
Lactobacillus plantarum
10 2000 2000 22125 240485
15 2000 2000 17797 237037
20 2000 2000 13567 233616
22
22
RIWAYAT HIDUP
Penulis lahir di Jakarta pada tanggal 22 Mei 1991. Penulis merupakan anak bungsu dari tiga bersaudara dari pasangan Bapak Pramudia G.S dan Ibu Ratna Setiati. Pada tahun 2009, penulis lulus dari SMA Negeri 5 Bogor dan pada tahun itu pula penulis lulus masuk Institut Pertanian Bogor melalui jalur USMI (Undangan Seleksi Masuk IPB) dan diterima di jurusan Ilmu Komputer, Fakultas Matematika dan Ilmu Pengetahuan Alam.