PENGARUH PEMBERIAN AIR BARKARBONASI TERHADAP PROFIL FARMAKOKINETIKA PARASETAMOL PADA TIKUS PUTIH JANTAN
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Ilmu Farmasi
Oleh:
Agatha Devi Mirakel NIM : 038114040
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
2007
PENGARUH PEMBERIAN AIR BARKARBONASI TERHADAP PROFIL FARMAKOKINETIKA PARASETAMOL PADA TIKUS PUTIH JANTAN
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Ilmu Farmasi
Oleh:
Agatha Devi Mirakel NIM : 038114040
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
2007
Liebe: Bapa-ku tercinta di surga, “Sang Guru” kehidupan yang selalu menggenapi janjiNya menjadi indah pada waktunya Mama bundit-ku atas kasih dan doa yang selalu membuatku merasa istimewa Papa dan keluarga atas support, cinta dan segala hal yang belum bisa terucapkan Yosi, gadis kecilku yang setegar batu karang yang membuatku belajar memandang dunia dari sisi yang berbeda Fritas, another miracle in my journey for every single amazing moment we’ve shared.
Untuk semua orang
yang mempercayai keajaiban
v
…………Dan ketika seluruh dunia
berpaling darimu, engkau tau bahwa Dia
selalu hadir untukmu, mengulurkan
tanganNya dan membawa serta keajaiban
cinta. Bahkan ketika semua himpitan beban
dunia membuatmu terluka, disanalah Dia
bekerja menjadikanmu pribadi yang
sempurna.
LEMBAR PERNYATAAN PERSETUJUAN
PUBLIKASI KARYA ILMIAH UNTUK KEPENTINGAN AKADEMIS
Yang bertanda tangan di bawah ini, saya mahasiswa Universitas Sanata Dharma : Nama : Agatha Devi Mirakel
Nomor Mahasiswa : 038114040
Demi pengembangan ilmu pengetahuan, saya memberikan kepada Perpustakaan Universitas Sanata Dharma karya ilmiah saya yang berjudul :
Pengaruh Pemberian Air Berkarbonasi Terhadap Profil Farmakokinetika Parsetamol pada Tikus Putih jantan
beserta perangkat yang diperlukan (bila ada). Dengan demikian saya memberikan kepada Perpustakaan Universitas Sanata Dharma hak untuk menyimpan, me-ngalihkan dalam bentuk media lain, mengelolanya dalam bentuk pangkalan data, mendistribusikan secara terbatas, dan mempublikasikannya di Internet atau media lain untuk kepentingan akademis tanpa perlu meminta ijin dari saya maupun memberikan royalti kepada saya selama tetap mencantumkan nama saya sebagai penulis.
Demikian pernyataan ini yang saya buat dengan sebenarnya. Dibuat di Yogyakarta
Pada tanggal : 1 Februari 2008
Yang menyatakan
( Agatha Devi Mirakel )
PRAKATA
Puji syukur kepada Allah Bapa di Surga, karena oleh berkat, keajaiban dan kasih-Nyalah maka skripsi ini dapat diselesaikan oleh penulis. Skripsi ini disusun untuk memenuhi salah satu syarat memperoleh gelar Sarjana Farmasi (S. Farm.) di Fakultas Farmasi Universitas Sanata Dharma Yogyakarta.
Selama penyusunan skripsi ini, banyak pihak yang telah membantu penulis dalam menyelesaikannya, maka pada kesempatan ini penulis mengucapkan terima kasih kepada :
1. Ibu Rita Suhadi, M. Si., Apt. selaku Dekan Fakultas Farmasi Universitas Sanata Dharma Yogyakarta.
2. Bapak Drs.Mulyono, Apt. selaku pembimbing atas dorongan semangat, diskusi dan pengarahan, peminjaman buku-buku serta kesabaran dan inspirasi dalam membimbing penulis menyelesaikan skripsi.
3. Bapak Yosef Wijoyo,M.Si.,Apt. selaku dosen penguji untuk arahan, dan diskusi, untuk pinjaman buku-buku serta dorongan semangat selama proses penyusunan skripsi.
4. Ibu Christine Patramurti, M.Si., Apt. selaku dosen penguji yang telah memberikan dukungan berupa diskusi-diskusi, kritik dan saran selama penyusunan skripsi ini.
5. Seluruh staf laboratorium: Mas Heru Purwanto, Mas Parjiman, Mas Kayat, Mas Wagiran, Pak Mukmin, Pak Prapto, Mas Parlan, Mas Kunto dan Mas Otok yang telah membantu penulis selama penelitian di laboratorium.
6. Segenap staf administrasi: Pak Tatmo, Mas Narto, Mbak Sari dan Pak Mukmin untuk keramahan, kesabaran dan pelayanan selama masa-masa perkuliahan.
7. Sahabat-sahabatku Angger, Ana Rosa, Dita, Vera, Sari, Sakundita, dan Monika untuk perbedaan, sharing, tawa dan tangisan, persaudaraan, semangat, keceriaan, serta persahabatan yang mengagumkan, juga untuk dukungan selama masa-masa kuliah.
8. Veronika Sulistiawati patner tak terduga dari kelompok A 2003 untuk semua kebersamaan, perdebatan dan dukungan, diskusi, doa dan seluruh harapan yang amat besar selama di laboratorium dan keseluruhan proses penyusunan skripsi ini.
9. Teman-teman selama di laboratorium Angga, Surya dan Galaeh untuk setiap doa sebelum bekerja, untuk penguatan, keceriaan, diskusi, foto-foto, di Laboratorium. Juga Madya, Nia, Agnes, Supri, Eka, Siska, dan Shinta Dewi. 10.Sahabat-sahabatku Diah, Nuning, dan Dewi atas inspirasi selama
bertahun-tahun, doa-doa, kebersaman dan dukungan moral yang sangat besar.
11.Lanny dan Lia tetangga kost dan teman yang baik hati dan pintar, teman sharing dan diskusi dan teman”laporan praktikum” yang selalu ada pada saat yang tepat.
12.Teman-teman kost Sariayu: terutama Yanti dan Vivi yang memberi semangat dan menemani dalam kebersamaan.
13.Teman-teman Seluruh angkatan 2003, khususnya kelas A dan terutama kelompok praktikum B, untuk jiwa yang selalu muda yang membuat hidup
lebih berwarna, kebersamaan dan kekompakan dalam setiap tahun yang telah dilalui.
14.Setiap orang yang tidak dapat disebutkan satu-persatu namun memberi kontribusi yang amat berharga dalam tiap tahap hingga saat penulis menyelesaikan skripsi ini.
Penulis menyadari bahwa skripsi yang disusun ini masih banyak memiliki kekurangan. Oleh karena itu penulis mengharapkan kritik dan saran untuk perbaikan dan perkembangan selanjutnya. Akhir kata, semoga skripsi ini berguna bagi kemajuan ilmu pengetahuan.
Penulis
INTISARI
Absorpsi obat yang diberikan peroral sebagai contoh parasetamol, dipengaruhi berbagai faktor fisiologis termasuk adanya makanan dan minuman dalam saluran cerna. Minuman berkarbonasi dengan kandungan utama air berkarbonasi sering dikonsumsi masyarakat dan terdapat kemungkinan suatu saat minuman itu dikonsumsi bersama dengan parasetamol. Tujuan penelitian ini adalah untuk mengetahui pengaruh air berkarbonasi terhadap profil farmakokinetika parasetamol, parameter farmakokinetika yang dipengaruhi serta seberapa besarnya, juga hal yang diakibatkan dari pengaruh tersebut.
Penelitian ini merupakan penelitian eksperimental murni, rancangan acak lengkap pola satu arah. Digunakan sepuluh ekor tikus putih jantan galur wistar sebagai subyek uji. Lima ekor sebagai kelompok kontrol dan lima ekor sebagai kelompok perlakuan. Subyek diberi parasetamol peroral dosis 300 mg/kgBB, dilanjutkan pemberian air barkarbonasi 5,8115 mg/kgBB (kelompok perlakuan) atau air dengan volume setara air barkarbonasi (kelompok kontrol). Penetapan kadar parasetamol dilakukan dengan metode HPLC oleh Howie et al.(1977) yang dimodifikasi oleh Wijoyo (2001). Hasilnya diolah menggunakan program STRIPE (Johnston & Woolard, 1983 yang dimodifikasi oleh Jung) dan dianalisis statistik dengan paired t-tes (taraf kepercayaan 95%).
Hasilnya, air berkarbonasi memberikan perbedaan bermakna terhadap profil farmakokinetika fase absorbsi dan eliminasi parasetamol: ka(+131,61%);
Cmaks(+27,74%); tmaks(-29,42%); AUC0-~(+28,35%); ClT(-21,62%); β(-15,00%);
k13(-13,04%); t½eliminasi(+42,55%); MRT(+18,08%). Perantaranya diduga adalah
peningkatan kecepatan pengosongan lambung, penurunan biotransformasi dan atau ekskresi parasetamol. Akibatnya mungkin berupa peningkatan daya analgesik dan resiko hepatotoksisitas(pada pemakaian dosis berganda).
Kata kunci : farmakokinetika, interaksi, parasetamol, air barkarbonasi.
ABSTRACT
Absorbtion for the drug given orally for example paracetamol, influenced by many physiologic factor, include the presence of foods and beverages in gastrointestinal track. Carbonated soft drink which contain carbonated water often to be consumed and it’s possible that once people consume it concomitanly with paracetamol. The aim of this research was to know wheather carbonated water influence paracetamol’s pharmacokinetics profile or not, include the parameters that were affected, the amount also the effect.
This was a pure experimental research, completely randomized one way variance. Ten white male Wistar strain rats used as the subjects. Five rats as the control group and others as the treatment group. Subjects were given paracetamol orally (300 mg/kgBB), continued with carbonated water 5,8115 mg/kgBB (treatment group) and pure water with the same volume (control group). HPLC method by Howie et al., modified by Wijoyo (2001) was used to determine paracetamol concentration in the blood. The results were proceed by STRIPE (Johnston & Woolard, 1983 modified by Jung) and statistically analized by paired t-test with (95% confidence intervals).
The results showed that carbonated water affected paracetamol’s pharmacokinetics profile on absorbtion and elimination phase: ka(+131,61%);
Cmax(+27,74%); tmax(-29,42%); AUC0-~(+28,35%); ClT(-21,62%); β(-15,00%);
k13(-13,04%);t½elimination(+42,55%); MRT(+18,08%). The mediator was presume
as the acceleration of gastric emptying and the decrease biotransformatin and or excretion of paracetamol. The posibble results will be the increase of analgetic capacity and hepototoxcicity risk (in multiple dose administration).
Key words: pharmacokinetics, interaction, paracetamol, carbonated water
DAFTAR ISI
HALAMAN JUDUL ... ii
HALAMAN PERSETUJUAN PEMBIMBING ... iii
HALAMAN PENGESAHAN ... iv
HALAMAN PERSEMBAHAN ... v
PRAKATA... vii
PERNYATAAN KEASLIAN KARYA ... x
INTISARI ... xi
ABSTRACT... xii
DAFTAR ISI... xiii
DAFTAR TABEL... xv
DAFTAR GAMBAR ... xx
DAFTAR LAMPIRAN... xxiii
BAB I. PENGANTAR... 1
A. Latar Belakang ... 1
1. Permasalahan ... 4
2. Keaslian penelitian... 4
3. Manfaat penelitian ... 4
B. Tujuan Penelitian ... 5
BAB II. PENELAAHAN PUSTAKA ... 6
A. Farmakokinetika ... 6
1. Pengertian ... 6
2. Analisis Farmakokinetika ... 7
3. Parameter farmakokinetika ... 14
4. Strategi Penelitian farmakokinetika ... 25
B. Interaksi farmakokinetika ... 33
1. Pengertian ... 33
2. Mekanisme Interaksi Farmakokinetika ... 34
3. Akibat... 42
4. Perantara ... 43
5. Penyebab ... 43
6. Penafsiran... 43
C. Parasetamol ... 44
1. Terapetik ... 44
2. Kimia... 45
3. Farmakokinetika ... 46
4. Hepatotoksisitas ... 50
D. Air Berkarbonasi ... 52
E. Metode Penetapan Kadar Parasetamol Dalam Darah ... 54
1. Metode Gas Liquid Chromatography (GLC) ... 55
2. Metode Spektrofotometri-Diferensial ... 56
3. Metode oleh Micelli dkk ... 56
4. Metode Cafetz dkk ... 56
5. Metode High Performance Liquid Chromatograpy (HPLC) ... 57
F. Landasan Teori... 59
G. Hipotesis ... 62
BAB III. METODOLOGI PENELITIAN ... 63
A. Jenis dan Rancangan Penelitian ... 63
B. Variabel-variabel Penelitian... 63
1. Variabel Bebas ... 63
2. Variabel Tergantung ... 64
3. Variabel Pengacau... 65
C. Bahan Penelitian ... 65
D. Alat Penelitian... 66
E. Tata Cara Penelitian ... 67
1. Optimasi penetapan kadar parasetamol ... 67
2. Penelitian lanjutan... 69
F. Analisis Hasil ... 71
1. Perhitungan parameter farmakokinetika ... 71
2. Analisis statistik ... 71
BAB IV. HASIL DAN PEMBAHASAN ... 73
A. Optimasi metode ... 73
1. Pembuatan dan penetapan kurva baku ... 76
2. Penetapan harga perolehan kembali, kesalahan acak, dan kesalahan sistemik. ... 80
3. Uji stabilitas parasetamol ... 84
B. Penelitian lanjutan... 85
1. Penetapan dosis parasetamol... 85
2. Penetapan dosis air berkarbonasi ... 86
3. Penetapan waktu pengambilan cuplikan ... 86
C. Analisis Hasil ... 87
1. Profil absorbsi parasetamol... 91
2. Profil distribusi parasetamol ... 96
3. Profil eliminasi parasetamol ... 98
BAB V. KESIMPULAN DAN SARAN ... 101
A. Kesimpulan ... 101
B. Saran ... 102
DAFTAR PUSTAKA ... 103
LAMPIRAN... 107
BIOGRAFI PENULIS ... 158
DAFTAR TABEL
Tabel I. Sifat dari model satu kompartemen terbuka ... 10
Tabel II. Sifat dari model dua kompartemen terbuka ... 11
Tabel III. Rangkuman model kompartemen, rute pemberian dan persamaan kadar dalam darah, serum dan urin ... 13
Tabel IV. Ketergantungan parameter farmakokinetika primer terhadap variabel fisiologi ... 15
Tabel V. Ketergantungan parameter famakokinetika sekunder dan turunan terhadap parameter farmakokinetika primer... 25
Tabel VI. Pembersihan parasetamol yang diperoleh isolated perfusea liver... 50
Tabel VII. Parameter farmakokinetika & farmakodinamika parasetamol pada manusia ... 52
Tabel VIII. Asam bikarbonat ... 52
Tabel IX. Karbon dioksida... 53
Tabel X. Faktor yang mempengaruhi pengosongan lambung ... 61
Tabel XI. Parameter farmakokinetika model 2 kompartemen terbuka... 70
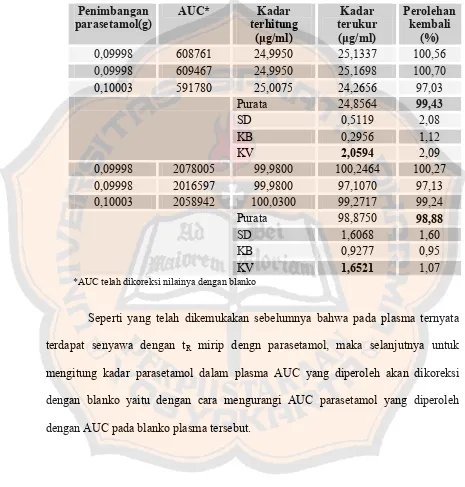
Tabel XII. Harga perolehan kembali, kesalahan sistemik, kesalahan acak penetapan kadar parasetamol dalam plasma dengan HPLC-Intraday... 82
Tabel XIII. Harga perolehan kembali, kesalahan sistemik, kesalahan acak penetapan kadar parasetamol dalam plasma dengan
HPLC- Interday... 83
Tabel XIV. Peruraian parasetamol dalam plasma setelah disimpan pada suhu 0oC ... 84
Tabel XV. Data kadar parasetamol dalam plasma setelah pemberian parasetamol dalam plasma dosis 300 mg/kgBB ... 86
Tabel XVI. Kenaikan kadar parasetamol dalam plasma setelah pemberian parasetamol oral 300 mg/kg BB pada tikus akibat pemberian air berkarbonasi ... 89
Tabel XVII. Pengaruh pemberian air berkarbonasi terhadap profil farmakokinetika parasetamol pada tikus putih jantan... 90
Tabel XVIII. Seri kadar larutan intermediet kurva baku parasetamol... 108
Tabel XIX. Data persamaan kuva baku ... 109
Tabel XX. Contoh perhitungan kadar larutan parasetamol pada penentuan nilai perolehan kembali, kesalahan sistemik dan kesalahan acak (intraday dan interday) ... 110
Tabel XXI. Konversi perhitungan dosis antar jenis subyek... 131
Tabel XXII. Data kadar parasetamol dalam plasma dalam berbagai waktu... 132
Tabel XXIII. Hasil pengolahan STRIPE untuk kelompok kontrol 1... 139
Tabel XXIV. Hasil pengolahan STRIPE untuk kelompok kontrol 2... 140
Tabel XXV. Hasil pengolahan STRIPE untuk kelompok kontrol 3... 141
Tabel XXVI. Hasil pengolahan STRIPE untuk kelompok kontrol 4... 142
Tabel XXVII. Hasil pengolahan STRIPE untuk kelompok kontrol 5... 143
Tabel XXVIII. Hasil pengolahan STRIPE untuk kelompok perlakuan 1... 144
Tabel XXIX. Hasil pengolahan STRIPE untuk kelompok perlakuan 2... 145
Tabel XXX. Hasil pengolahan STRIPE untuk kelompok perlakuan 3... 146
Tabel XXXI. Hasil pengolahan STRIPE untuk kelompok perlakuan 4... 147
Tabel XXXII. Hasil pengolahan STRIPE untuk kelompok perlakuan 5... 148
Tabel XXXIII. Rangkuman parameter farmakokinetika parasetamol pada tiap-tiap subyek uji... 154
DAFTAR GAMBAR
Gambar 1. Tahap analisis farmakokinetika ... 7
Gambar 2. Tahapan aksi hayati / biologi obat dalam tubuh ... 26
Gambar 3. Contoh struktur protein ... 29
Gambar 4. Rangkuman prinsip penafsiran dan penilaian interaksi farmakokinetika serta akibat kinetika farmakologi, toksikologi dan klinisnya ... 43
Gambar 5. Struktur parasetamol ... 45
Gambar 6. Metabolisme parasetamol ... 49
Gambar 7. Struktur asam bikarbonat ... 52
Gambar 8. Gambaran denaturasi protein ... 75
Gambar 9. Gugus kromofor dan auksokrom parasetamol ... 76
Gambar 10. Kromatogram blangko plasma ... 77
Gambar 11. Kromatogram parasetamol dalam plasma dengan kadar 100 µg/ml ... 77
Gambar 12. Disosiasi parasetamol... 78
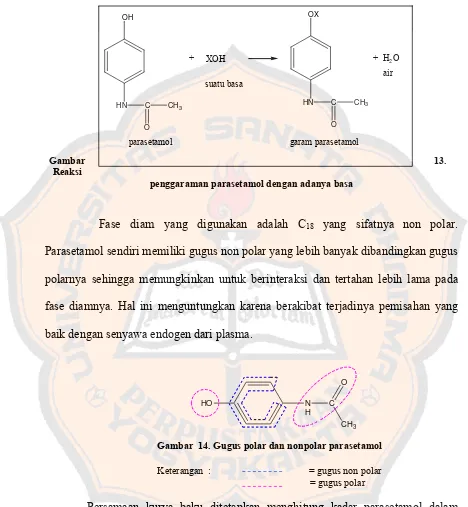
Gambar 13. Reaksi penggaraman parasetamol dengan adanya basa ... 79
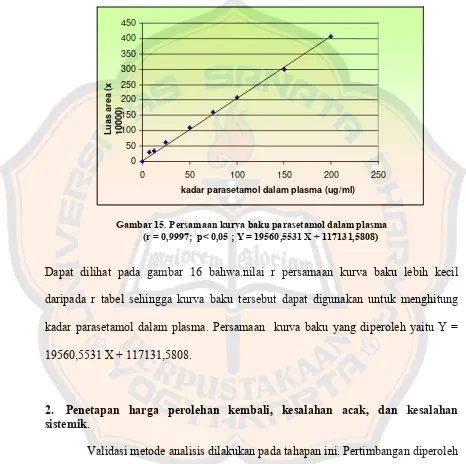
Gambar 14. Gugus polar dan nonpolar parasetamol... 79
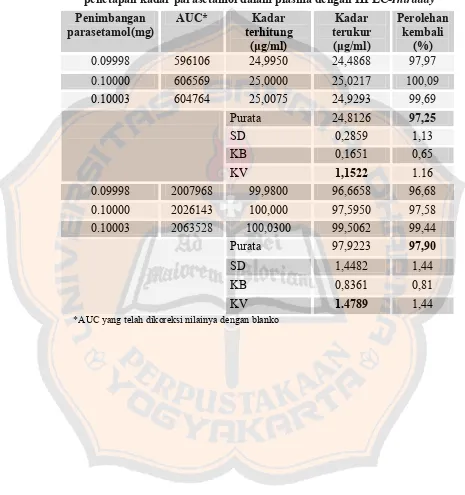
Gambar 15. Persamaan kurva baku parasetamol dalam plasma ... 80
Gambar 16. Kromatogram kontrol 3, menit ke 20... 85
Gambar 17. Kurva kekerabatan kadar parasetamol lawan waktu pada tikus jantan akibat pemberian parasetamol oral dosis 300 mg/kg BB ... 87 Gambar 18. Kurva kekerabatan ln kadar parasetamol lawan waktu pada
tikus jantan akibat pemberian parasetamol oral dosis 300 mg/kg BB ... 87 Gambar 19. Perubahan liku kenaikan kadar parasetamol lawan waktu
pada tikus jantan setelah pemberian parasetamol oral dosis 300 mg/kg.BB dengan dan tanpa air berkarbonasi ... 88 Gambar 20. Perubahan liku kenaikan ln kadar parasetamol lawan waktu
pada tikus jantan setelah pemberian parasetamol oral dosis 300 mg/kg.BB dengan dan tanpa air berkarbonasi ... 88 Gambar 21. Kurva kadar parasetamol dalam plasma lawan waktu pada
kelompok kontrol (A) dan kelompok perlakuan (B)... 133 Gambar 22. Kurva ln kadar parasetamol dalam plasma lawan waktu
pada kelompok kontrol (A) dan kelompok perlakuan (B) ... 134 Gambar 23a. Kurva kadar parasetamol dalam plasma lawan waktu pada
kelompok kontrol 1- perlakuan 1 ... 135 Gambar 23b. Kurva kadar parasetamol dalam plasma lawan waktu pada
kelompok kontrol 2- perlakuan 2 ... 135 Gambar 23c. Kurva kadar parasetamol dalam plasma lawan waktu pada
kelompok kontrol 3 – perlakuan 3 ... 135
Gambar 23d. Kurva kadar parasetamol dalam plasma lawan waktu pada kelompok kontrol 4 – perlakuan 4 ... 136 Gambar 23e. Kurva kadar parasetamol dalam plasma lawan waktu pada
kelompok kontrol 5 – perlakuan 5 ... 136 Gambar 24a. Kurva ln kadar parasetamol dalam plasma lawan waktu
pada kelompok kontrol 1- perlakuan 1 ... 137 Gambar 24b. Kurva ln kadar parasetamol dalam plasma lawan waktu
pada kelompok kontrol 1- perlakuan 1 ... 137 Gambar 24c. Kurva ln kadar parasetamol dalam plasma lawan waktu
pada kelompok kontrol 1- perlakuan 1 ... 137 Gambar 24d. Kurva ln kadar parasetamol dalam plasma lawan waktu
pada kelompok kontrol 1- perlakuan 1 ... 138 Gambar 24e. Kurva ln kadar parasetamol dalam plasma lawan waktu
pada kelompok kontrol 1- perlakuan 1 ... 138
DAFTAR LAMPIRAN
Lampiran 1. Perhitungan dan penimbangan pembuatan kurva baku parasetamol ... 108 Lampiran 2. Contoh data dan perhitungan pembuatan larutan
parasetamol pada penentuan nilai perolehan kembali, kesalahan sistemik dan kesalahan acak (intraday dan interday ... 110 Lampiran 3. Contoh perhitungan dosis awal untuk orientasi dosis... 111 Lampiran 4. Kromatogram kurva baku parasetamol dalam plasma ... 112 Lampiran 5. Contoh kromatogram kelompok kontrol... 116 Lampiran 6. Contoh kromatogram kelompok perlakuan ... 123 Lampiran 7. Contoh perhitungan dosis dan volume air berkarbonasi ... 131 Lampiran 8. Data kadar parasetamol dalam plasma pada berbagai
waktu ... 132 Lampiran 9. Kurva kadar parasetamol dalam plasma lawan waktu ... 133 Lampiran 10. Kurva ln kadar parasetamol dalam plasma lawan waktu... 134 Lampiran 11. Kurva kadar parasetamol dalam plasma lawan waktu
untuk tiap– tiap pasang subyek uji... 135 Lampiran 12. Kurva ln kadar parasetamol dalam plasma vs waktu untuk
tiap – tiap pasang kontrol-perlakuan ... 137 Lampiran 13. Hasil pengolahan STRIPE untuk kelompok kontrol... 139 Lampiran 14. Hasil pengolahan STRIPE untuk kelompok perlakuan... 144
Lampiran 15. Contoh perhitungan parameter farmakokinetika parasetamol ... 149 Lampiran 16. Rangkuman parameter farmakokinetika parasetamol pada
tiap-tiap subyek uji... 154 Lampiran 17. Analisis stastistik SPSS (12.00) data ka... 156
Lampiran 18 Sertifikat analisis parasetamol ... 169
BAB I PENGANTAR
A. Latar Belakang
Obat-obat yang diberikan dengan jalur pemberian ekstravaskular seperti
peroral contohnya parasetamol, mengalami suatu tahapan yang disebut absorpsi
yaitu absorbsi pada saluran cerna (gastrointestinal absorbtion). Pada proses
absorbsi ini, obat terlebih dahulu harus melewati membran pada tempat absorbsi
seperti dinding pembuluh kapiler pada saluran cerna agar dapat tersedia dalam
saluran sistemik dan siap memberikan aksi. Tahap absorbsi tidak akan terjadi pada
obat-obat yang diberikan dengan jalur pemberian intravaskular (misalnya secara
intravena, intraarterial, intraspinal dan intraserebral), karena obat tidak perlu
menembus suatu membran agar dapat tersedia pada saluran sistemik. Proses
absorbsi obat pada saluran cerna ini dapat dipengaruhi oleh beberapa faktor
fisiologi seperti waktu tinggal dalam saluran cerna (transit time); kecepatan
pengosongan lambung; tempat absorpsi (lambung, usus); keefektifan luas
permukaan pada tiap tempat absorpsi; pH pada saluran cerna; aliran darah pada
tempat absorpsi; ada dan tidaknya makanan dalam saluran cerna serta masih
banyak lagi (Wagner, 1979). Memperhatikan faktor-faktor fisiologi diatas, maka
sangat mungkin bila suatu saat makanan atau minuman yang dikonsumsi bersama
dengan pemberian obat peroral mempengaruhi proses absorbsi obat tersebut
dengan cara mengubah satu atau lebih faktor-faktor fisiologi.
Salah satu minuman yang mungkin dikonsumsi bersama dengan obat
adalah minuman berkarbonasi. Di Indonesia jenis minuman berkarbonasi ini
sudah lama dikenal. Rasa segar yang menjadi keistimewaan jenis minuman ini
disebabkan oleh kandungan air berkarbonasi yang menjadi komponen utamanya.
Sensasi ”bubling” yang ditimbulkan olah kandungan gas yang terlarut,
menjadikannya unik dan digemari (Anonim, 2002). Air barkarbonasi sering dijual
dalam bentuk kombinasi dengan bahan tambahan seperti gula dan bahan perasa
seperti yang terdapat dalam minuman ringan berkarbonasi (carbonatedsoft drink)
namun ada juga yang dijual dalam bentuk air barkarbonasi tanpa campuran
apapun yang sering disebut sebagai air soda. Minuman jenis ini dapat dikonsumsi
kapan saja mulai dari anak-anak, dewasa, hingga orang tua. Contoh yang dapat
dijumpai mengenai konsumsi minuman berkarbonasi bersama dengan obat adalah
konsumsi salah satu merk minuman berkarbonasi bersama dengan parasetamol
(Hermansaksono, 2005).
Parasetamol adalah suatu metabolit aktif dari fenasetin yang berkhasiat
sebagai analgesik-antipiretik. yang juga sudah lazim digunakan sejak tahun 1893
(AMA, 1994; Hardam, Gilman & Limbird, 1996). Di Indonesia parasetamol telah
banyak beredar sebagai obat bebas dengan berbagai nama dagang. Depkes RI
menganjurkan parasetamol sebagai pilihan utama untuk pengobatan demam
(www.depkes.go.id.). Meskipun tergolong obat yang sudah lama digunakan,
namun karena efek sampingnya yang relatif sedikit, sifatnya yang tidak
mengiritasi lambung dan aman untuk digunakan pada anak-anak, membuat obat
Saat mengkonsumsi air berkarbonasi, kandungan gas yang terlarut
didalamnya memiliki kecenderungan memenuhi lambung (peningkatan volume)
dan menyebabkan tekanan pada lambung (gastric distention). Kedua hal tersebut
termasuk stimulus dalam pengosongan lambung (Mayersohn, 2002). Maka bagi
obat seperti parasetamol yang melalui tahapan absorpsi di usus halus, faktor
fisiologi berupa kecepatan pengosongan lambung akan mempengaruhi efektifitas
absorpsinya (Whitehouse, 1981) dan mungkin juga terhadap profil
farmakokinetika parasetamol yang lain (distribusi dan eliminasi). Untuk itulah
penelitian ini dilakukan untuk membuktikan apakah terdapat interaksi air
barkarbonasi-parasetamol yang berpengaruh terhadap profil farmakokinetika dari
parasetamol.
Penelitian mengenai pengaruh air barkarbonasi terhadap profil
farmakokinetika parasetamol ini menggunakan HPLC (High Liquid Performance
Chromatography) untukmengukur kadar parasetamol utuh dalam plasma. Metode
yang digunakan mengacu pada metode yang dikembangkan oleh Howie,
Adriaensenss & Prescott, (1977) dengan modifikasi yang dilakukan oleh Wijoyo
(2001). Metode ini dipilih dengan pertimbangan bahwa metode ini memenuhi
parameter senstivitas, selektivitas, ketepatan dan ketelitian serta dapat mengatasi
masalah volume cairan biologis (darah) yang terbatas pada subyek uji (tikus)
1. Permasalahan
a. Apakah pemberian air barkarbonasi mempengaruhi profil farmakokinetika
parasetamol?
b. Parameter farmakokinetika apa yang dipengaruhi dan berapa besarnya
pengaruh tersebut?
c. Hal apa yang mungkin terjadi akibat perubahan profil farmakokinetika
tersebut?
2. Keaslian Penelitian
Sejauh penelusuran pustaka di USD, penelitian mengenai pengaruh air
barkarbonasi terhadap profil farmakokinetika parasetamol belum pernah
dilakukan. Penelitian tentang parasetamol yang pernah ada sebelumnya adalah
antaraksi farmakokinetika jamu merit dengan parasetamol (Kristianto, 2000);
antaraksi parasetamol dengan vegeta (Delima, 2004) serta antaraksi parasetamol
dengan jamu antangin (Sulistyowati,2005).
3. Manfaat
a. Manfaat teoritis yang didapatkan dari penelitian ini adalah memberikan
informasi tentang pengaruh pemberian air barkarbonasi terhadap profil
farmakokinetika parasetamol.
b. Manfaat praktis yang didapatkan dari penelitian ini adalah memberikan
barkarbonasi dengan parasetamol pada kinerja farmakologi-toksikologi
parasetamol.
B. Tujuan Penelitian
Penelitian ini bertujuan untuk:
1. Penelitian ini bertujuan untuk membuktikan kebenaran bahwa air berkarbonasi
dapat mempengaruhi profil farmakokinetika parasetamol.
2. Mengetahui parameter farmakokinetika apa saja yang dipengaruhi dan berapa
besarnya pengaruh tersebut.
3. Mengetahui hal yang mungkin terjadi akibat perubahan profil farmakokinetika
BAB II
PENELAAHAN PUSTAKA
Sehubungan dengan maksud penelitian ini maka di dalam bab ini akan ditelaah lebih lanjut mengenai farmakokinetika, analisis farmakokinetika, interaksi farmakokinetika, parasetamol dan air berkarbonasi.
A. Farmakokinetika
1. Pengertian
Farmakokinetika adalah suatu cabang dari ilmu farmakologi. Farmakologi didefinisikan sebagai ilmu yang mempelajari interaksi obat dengan organisme hidup dan segala aspek dari interaksi tersebut. Berarti, baik obat maupun organisme hidup dapat saling mempengaruhi. Bagian farmakokinetika dikhususkan untuk mempelajari bagian tentang pengaruh obat terhadap organisme hidup. Oleh Makoid dan Cobby (2002) farmakokinetika didefinisikan sebagai suatu perhitungan matematika dari waktu proses absorsi, distribusi, metabolisme dan ekskresi (ADME) dari obat didalam tubuh. Faktor biologi, psikologi dan fisika-kimia yang dapat mempengaruhi proses perpindahan obat di dalam tubuh juga dapat mempengaruhi tingkat dan kecepatan ADME obat tersebut di dalam tubuh. Sejauh ini aksi farmakologi banyak berhubungan dengan kadar obat di dalam plasma, begitu pula dengan aksi toksikologi.
2. Analisis farmakokinetika
Analisis farmakokinetika dilakukan dengan tujuan untuk mendapatkan parameter farmakokinetika. Pada tahap selanjutnya parameter-parameter tersebut dapat digunakan untuk berbagai macam tujuan misalnya menentukan laju absorpsi, metabolisme dan ekskresi melalui urin: memperhitungkan ketersedian hayati (bioavailabilitas) suatu produk; menghubungkan respon farmakologi dengan konsentrasi obat di dalam plasma, cairan tubuh lain atau jaringan; memprediksi kadar obat dalam darah setelah pemberian dosis ganda; mengoptimalkan aturan dosis untuk obat-obat tertentu dan masih banyak lagi. Dalam mempelajari analisis farmakokinetika terlebih dahulu harus dipahami tetang model kompartemen, ordo kinetika, strategi penelitian dan teknik analisis obat dalam cairan biologis.
Gambar 1. Tahap analisis farmakokinetika
(Wagner, 1975 dengan revisi) Pemberian obat
dengan dosis tertentu kepada subyek
Pencuplikan sampel melalui cairan biologis (misal darah atau urin ) atau jaringan
Penetapan kadar obat utuh dan atau metabolinya terhadap fungsi waktu
Data
Penetapan model kompartemen farmakokinetika
Aplikasi model Penjabaran model kompartemen
a. Analisis model kompartemen adalah tahapan yang pertama dilakukan setelah didapat data kadar obat tak berubah atau metabolitnya dalam darah atau urin (cairan biologis yang paling sering digunakan). Tahap ini penting untuk mencocokkan data hasil uji dengan rumus perhitungan parameter farmakokinetika. Setelah berada di dalam badan (sirkulasi sitemik) obat akan terdistribusi dengan cepat ke berbagai organ dengan sifat beragam. Badan dianggap suatu kumpulan kompartemen (multi kompartemen) yang terpisah satu sama lain, untuk menyederhanakannya badan dianggap sebagai suatu sistem satu atau dua kompertemen terbuka. hal tersebut didasarkan pada asumsi bahwa proses perpindahan (distribusi) obat antar kompertemen bersifat bolak-balik antara darah disatu pihak dan tempat distribusi di pihak lain. Cara pengerjaannya adalah dengan mengikuti metode plot semilogaritma kadar obat lawan laktu dengan perhitungan matematika.
1) Model satu kompartemen terbuka. Diasumsikan bahwa badan adalah kompertemen tunggal, seluruh kompertemen yang ada dianggap sebagai sentral. Kompartemen sentral didefinisikan sebagai jumlah seluruh bagian badan (organ atau jaringan) dimana kadar obat didalamnya segera berada dalam kesetimbangan dengan kadar obat dalam darah atau plasma (Ritschel, 1992). Pada model ini seolah-olah tidak terdapat fase distribusi. Adanya fase distribusi hanya digambarkan dengan Vd. Kurva semilogaritma hanya menunjukkan
Tabel I. Sifat dari model satu kompartemen terbuka
Aplikasi
Intravaskular
(Intravena, intrakardiak, intra-arterial)
Ekstravaskular
(oral, peroral, rectal,intramuscular, subkutan, intrakutan)
Tidak ada absorpsi, semua obat yang diinjeksikan berada dalam sirkulasi sitemik, distribusi yang cepat antara aliran darah dan jaringan; kesetimbangan (steady state) langsung tercapai; penurunan kadar obat tergantung pada ekskresi dan metabolisme.
Absorpsi berjalan seturut pelepasan obat dan mekanisme absorpsi; pada waktu 0 tidak terdapat obat pada sirkulasi sistemik; selama terjadi proses absorpsi, konsentrasi obat meningkat sampai puncak (peak) dan kemudian menurun sejalan dengan eliminasi (metabolisme dan ekskresi); tidak semua obat terabsorpsi.
(Ritschel, 1992) Sifat
Model
D = dosis yang diberikan Vd = volume distribusi C = kadar obat dalam plasma kel = tetapan laju eliminasi
D ka kel
D = dosis yang diberikan Vd = volume distribusi C = kadar obat dalam plasma Ka = tetapan laju absorpsi kel = tetapan laju eliminasi
BLOOD LEVEL (pada kertas semi logaritma) Log Kadar waktu Log Kadar waktu BODY
Vd C
D kel
kel
BODY
Tabel II. Sifat dari model dua kompartemen terbuka
Aplikasi
Intravaskular
(Intravena, intrakardiak, intra-arterial)
Ekstravaskular
(oral, peroral, rectal, intramuscular, subkutan, intrakutan)
Sifat
Tidak ada absorpsi, semua obat yang diinjeksikan berada dalam sirkulasi sitemik; distribusi yang lambat antara aliran darah dan jaringan; kesetimbangan (steady state) tercapai beberapa saat setelah pemberian; penurunan kadar pada tahap pertama kurva kadar obat terjadi karena distribusi; penurunan kadar obat pada bagian kedua tergantung pada pendistribusian kembali (back distribution) obat dari jaringan ke dalam darah, ekskresi dan metabolisme.
Absorpsi berjalan seturut pelepasan obat dan mekanisme absorpsi; pada waktu 0 tidak terdapat obat pada sirkulasi sistemik; selama terjadi absorpsi, konsentrasi obat meningkat sampai puncak (peak) diikuti penurunan dikarenakan distribusi lambat sampai tercapai
kesetimbangan; penurunan monoeksponen tergantung pada pendistribusian kembali (back distribution) obat dari jaringan ke darah, ekskresi dan metabolisme
Model
k13
D
D = dosis yang diberikan CC = kompartemen sentral PC = kompartemen perifer k12, k21 = tetapan distribusi k13 = tetapan eliminasi dari
komp.sentral
Vc = volume distribusi komp. sentral
C = kadar obat dalam plasma
β = tetapan eliminasi tota
ka k13
D.f
D = dosis yang diberikan CC = kompartemen sentral PC = kompartemen perifer Ka = tetapan absorpsi F = fraksi obat terabsorpsi k12, k21 = tetapan distribusi k13 = tetapan eliminasi dari
komp.sentral
Vc = volume distribusi komp. sentral
C = kadar obat dalam plasma
β = tetapan eliminasi total
CC
Vc C PC
CC
Lanjutan tabel II
(Rischel,1992)
BLOOD LEVEL (pada kertas semi logaritma)
Log Kadar
waktu
Log Kadar
waktu ka > α
Log Kadar
waktu
Tabel III. Rangkuman model kompartemen, rute pemberian dan persamaan kadar dalam darah, serum dan urin
BLOOD LEVEL Rute
pemberian
Model kompartemen
Persamaan kadar obat (µg/ml) (pada kertas semi logaritma)
Log Kad Waktu Intravaskuler -Intravena -intrakardiak -intra-arterial Satu kompartemen terbuka
C(t) = C(0) e–kel..t
Log Kadar Waktu Ekstravaskular -oral -peroral -rektal -intramuskular -subkutan -intrakutan Satu kompartemen terbuka
C(t) = M e–kel. t – N e–ka.t
M= Intersep dari hasil
back extrapolation
slope persamaan monoeksponen eliminasi dengan ordinat (µg/ml) N = intersep persamaan
monoeksponen absorbsi dengan ordinat (µg/ml) Log Kadar Waktu Intravascular -Intravena -intrakardiak -intra-arterial dua kompartemen terbuka
C(t) = B e –β.t + L e -α t
Log Kadar
Waktu
ka > α
Log Kadar
waktu
ka < α
Ekstravaskular -oral -peroral -rektal -intramuskular -subkutan -intrakutan dua kompartemen terbuka
C(t) = M e–β.t + L e-α t - N e–kel.t
M = intersep dari hasil back ekstrapolation slope persamaan monoeksponen eliminasi dengan ordinat (µg/ml) L = intersep slope
persaman distribusi dengan ordinat (µg/ml)
N= kadar hipotetik obat pada t (0) yang diperoleh dari penjumlahan nilai L dan M (µg/ml)
(Ritschel, 1992)
β
3. Parameter farmakokinetika
Parameter farmakokinetika didefinisikan sebagai besaran yang diturunkan secara matematik dari hasil pengukuran kadar obat atau metabolitnya didalam darah atau urin (Suryawati & Donatus, 1998), dimana parameter ini akan menjadi acuan bagi keefektifan perubahan fisiologi pada tahap farmakokinetika. Didasarkan pada hubungannya dengan perubahan fisiologis, parameter farmakokinetika dapat dikelompokkan menjadi tiga yaitu :
• Parameter farmakokinetika primer yaitu parameter yang nilainya dipengaruhi secara langsung oleh perubahan fisiologi. Termasuk dalam parameter ini adalah tetapan laju absorpsi (ka); fraksi obat yang diabsorpsi (f); volume
distribusi (Vd); sedangkan pembersihan renal (ClR) dan pembersihan hepatik
(ClH).
• Parameter farmakokinetika sekunder adalah parameter yang nilainya tergantung pada parameter farmakokinetika primer. Tetapan laju eliminasi (kel),
waktu paruh eliminasi (t½ eliminasi) dan fraksi obat yang diekskresikan dalam
bentuk utuh (fe) adalah contoh dari parameter farmakokinetika sekunder ini (Rowland & Tozer, 1995)
• Parameter farmakokinetika turunan, nilai parameter ini tidak hanya tergantung pada parameter farmakokinetika primer tetapi juga pada dosis seperti pada dijumpai pada kadar obat dalam plasma dalam kondisi tunak (Css) dan luas
Tabel IV. Ketergantungan parameter farmakokinetika primer terhadap variabel fisiologi
PARAMETER FARMAKOKINETIKA PRIMER
FAKTOR FISIOLOGI
Tetapan laju absorpsi (ka) Aliran darah pada tempat absorpsi, pengosongan lambung (oral), motilitas usus (oral)
Bioavailabilitas Pengosongan lambung, sekresi asam lambung dan enzim hidrolitik pada empedu dan motilitas usus.
Pembersihan Hepatik (ClH); bioavailabilitasa
Aliran darah hepatik, keterikatan dalam darah,aktivitas hepatoselular.
Pembersihan renal (ClR) Aliran darah ginjal, keterikatan dalam darah, sekresi aktif, reabsorpsi aktif, filtrasi glomerular, pH urin,aliran urin
Volume distribusi (Vd);
a
Eliminasi hepatik diasumsikan sebagai satu-satunya penyebab penurunan bioavailabilitas. Keterikatan dalam darah, keterikatan dengan jaringan, partisi pada lemak, komposisi tubuh, ukuran tubuh
(Rowland&Tozer,1995)
a. Luas Area di Bawah Kurva (Area Under the Curve/AUC). AUC total (AUC 0-~) menggambarkan jumlah obat yang terukur dalam
darah pada wakru nol sampai tak hingga. Diperoleh dari hasil penjumlahan nilai AUC0-tn dengan AUCtn- ∝( Ritschel, 1992)
=AUC ∝
-0
AUC 0-tn + AUC tn- ∝ (1)
Keterangan :
n
t -0
AUC = Luas area dibawah kurva kadar obat di dalam darah lawan waktu dari waktu nol hingga waktu n
∞ -tn
AUC = Luas area dibawah kurva kadar obat di dalam darah lawan waktu dari waktu nol hingga waktu tak hingga
∞ -0
Besarnya AUC0-tn menggambarkan jumlah obat yang terukur dalam darah
pada rentang waktu tertentu. Nilainya dapat diperkirakan dengan aturan trapezoid, metode ini akurat bila terdapat cukup titik-titik data pengukuran kadar obat di dalam darah (Shargel, Wu-Pong, & Yu, 2005). Area diantara tiap titik dijumlahkan sebagai :
) t (t 2 Cn C
AUC 0-tn = n+1 + n− n+1 (2)
Keterangan :
n
t -0
AUC = Luas area dibawah kurva kadar obat di dalam darah lawan waktu dari waktu nol hingga waktu n
tn+1 = waktu saat n+1(menit)
tn = waktu saat n (menit)
Cn = konsentrasi pada waktu tn (µg/ml)
Cn+1 = konsentrasi pada waktu tn+1 (µg/ml)
tn -0
AUC menggambarkan AUC dari waktu nol sampai dengan waktu terakhir pengukuran kadar obat di dalam darah. Selanjutnya area yang tersisa dihitung dengan membagi kadar obat di dalam darah dengan kel
atau β ( Ritschel, 1992)
β atau k C AUC el n
-tn ∞ = (3)
Keterangan :
∞ -tn
AUC = Luas area dibawah kurva kadar obat di dalam darah lawan waktu dari waktu nol hingga waktu tak hingga
kel = tetapan laju eliminasi obat (menit)
Cn = kadar obat pada titik terakhir pengambilan sample (µg/ml)
Karena persamaan kadar obat dalam darah pada model dua kompartemen terbuka adalah
C(t) = Me -β t + Le-α t + Ne-ka t (4)
maka nilai AUC0- ∝ dapat dihitung dengan persamaan berikut.
a -0 k N α L β M
AUC ∞= + − (5)
Keterangan :
∝ -0
AUC = Luas area dibawah kurva kadar obat di dalam darah lawan waktu dari waktu nol hingga waktu tak hingga
M = Intersep dari hasil back extrapolation slope persamaan monoeksponen eliminasi dengan ordinat (µg/ml)
β = slope (tetapan laju) eliminasi total (disposisi lambat) [menit-1] L = intersep dari slope persamaan distribusi dengan ordinat (µg/ml)
α = slope (tetapan laju) distribusi (disposisis cepat) [menit-1] ka = slope (tetapan laju) absorpsi (µg/ml)
N = kadar hipotetik obat pada t (0) yang diperoleh dari penjumlahan nilai L dan M (µg/ml)
( Ritschel, 1992)
b. Tetapan laju absorpsi (ka), adalah fraksi obat yang diabsorpsi
tiap satuan waktu, karenanya tetapan ini menentukan jumlah obat yang dipindahkan dari tempat absorpsinya ke dalam darah tiap satuan waktu (Notari dkk, 1975; Ritschel, 1992)
absorbsi t 0.693 k 1 a 2
= (6)
Keterangan :
ka = slope (tetapan laju) absorpsi (menit-1)
c. Cmaks, didefinisikan sebagai kadar maksimum yang terdapat
dalam plasma setelah pemberian oral. Waktu yang dibutuhkan untuk mencapainya dinamakan tmaks. Nilai tmaks tidak tergantung pada dosis
namun tergantung pada tetapan laju absorpsi (ka) dan tetapan laju distibusi
(α). Cmaks sering disebut juga kadar puncak dimana laju obat yang
diabsorpsi sebanding dengan laju obat yang dieliminasi. Nilai dapat diperoleh dari persamaan kadar obat dalam tubuh
( )( ) ( )( ) ( )( ) ⎥⎦⎤ ⎢ ⎣ ⎡ ⎭ ⎬ ⎫ ⎩ ⎨ ⎧ − − − + ⎭ ⎬ ⎫ ⎩ ⎨ ⎧ − − − + ⎭ ⎬ ⎫ ⎩ ⎨ ⎧ − − −
= − − −αtmaks
a a a 21 βtmaks a 21 αtmaks a 21 a maks e k β k α k k e β α β k β k e α β α k α k Vc f D k
C (7)
Keterangan :
Cmaks = konsentrasi puncak (peak) kadar obat dalam darah (µg/ml)
ka = slope (tetapan laju) absorpsi (menit-1)
D = dosis obat yang diberikan
f = fraksi obat terabsorpsi
Vc = volume distribusi kompartemen sentral (ml)
α = slope (tetapan laju) distribusi (disposisis cepat) [menit-1]
β = slope (tetapan laju) eliminasi total (disposisi lambat) [menit-1] k21 = tetapan laju distribusi untuk perpindahan obat dari kompartemen
perifer ke kompartemen sentral (menit)
d. Volume distribusi (Vdss).Volume distribusi (Vd) adalah suatu
volume distribusi yang berkaitan dengan jumlah obat yang terdistribusi dalam cairan tubuh pada kondisi kesetimbangan/tunak (steady-state). Bentuk lain untuk menyatakan Vd antara lain: Vdexstrap (volume distribusi
dengan metode ekstrapolasi); Vdarea (volume distribusi dengan metode
area); Vdβ ( volume distribusi selama fase eliminasi). Vdss sifatnya tidak
tergantung pada laju eliminasi sehingga dapat digunakan untuk mengkorelasikan data dari suatu individu ke individu yang lain.
Besarnya Vd tergantung pada faktor fisiologi seperti laju aliran darah pada berbagai jaringan, kelarutan dalam lemak, koefisien partisi dan perbedaan tipe jaringan serta pH. Pada pemberian secara intravena (i.v.) dengan model satu kompartemen Vd dapat ditetapkan segera setelah tejadi kesetimbangan dengan membagi dosis yang diberikan (D) dengan konsentrasi obat mula-mula [C(0)]. Pada pemberian ekstravaskular (e.v.), prosedur ini diperbolehkan bila dosis yang diberikan dikalikan dengan fraksi dosis yang sebenarnya terabsorpsi. Tujuan penetapan Vd adalah untuk menghubungkan kadar obat dalam plasma dengan total jumlah obat yang terdapat dalam darah pada berbagai waktu.
(
)
(
a)(
a)
a 21 a 21 21 12 ss k α k β N k k D f k Vc dimana , Vc x k k k Vd − − − = +
= (8)
Keterangan :
Vdss = volume distribusi pada kondisi tunak / steady-state(ml)
Vc = volume distribusi kompartemen sentral / plasma (ml)
k12 = tetapan laju distribsi untuk perpindahan obat dari kompartemen sentral ke kompartemen perifer (menit-1)
ka = slope (tetapan laju) absorpsi (menit-1)
f = fraksi obat terabsorpsi (fraksi dari 1)
N = kadar hipotetik obat mula-mula pada t = 0 dalam modol dua kompertemen pada pemebrian ekstravaskuler (µg/ml)
D = dosis obat yang diberikan (µg)
Nilai dari (Vdss/Vc) – 1 adalah pengukuran langsung terhadap k12/k21. Nilai
(Vdss/Vc) – 1 semakin besar menggambarkan jumlah obat lebih besar
terdistribusi pada kompartemen perifer namun bila nilainya semakin kecil maka menggambarkan semakin besarnya obat yang terdistribusi pada kompartemen sentral.
e. Tetapan laju distribusi (α), tetapan laju distribusi (disposisi cepat yang sering disimbolkan dengan α, sifatnya adalah campuran (hybrid). Nilianya dapat diperoleh dari persamaan garis monoeksponen
distribusi:
(
2 21 13)
k 4k b b 1/2
α= + − atau (9)
eliminasi 2 1
t 0,693
α= (10)
Keterangan :
α = slope (tetapan laju) distribusi (disposisis cepat) [menit-1]
k21 = tetapan laju distribsi untuk perpindahan obat dari kompartemen perifer ke kompartemen sentral (menit-1)
k31 = tetapan laju eliminasi obat dari kompartemen sentral (menit-1)
b = k12 + k21 + k13
t½ eliminasi = waktu paruh eliminasi (menit)
f. Tetapan laju distribusi sentral-perifer (k12), didefinisikan sebagai
tetapan laju disribusi dari kompartemen sentral ke kompartemen perifer.
13 k 21 k β α 12
k = + − − (11)
Keterangan :
k12 = tetapan laju distribusi untuk perpindahan obat dari kompartemen sentral
ke kompartemen perifer (menit-1)
k21 = tetapan laju distribusi untuk perpindahan obat dari kompartemen perifer
ke kompartemen sentral (menit-1)
k13 = tetepan laju eliminasi obat dari kompartemen sentral (menit) α = slope (tetapan laju) distribusi (disposisis cepat) [menit-1]
β = slope (tetapan laju) eliminasi total (disposisi lambat) [menit-1]
C(0) = kadar hipotetik obat pada t (0) yang diperoleh dari penjumlahan nilai A dan B (µg/ml)
g. Tetapan laju distribusi perifer-sentral (k21) menyatakan tetapan
laju disribusi dari kompartemen perifer ke kompartemen sentral. Sama seperti k12 sifat k21 juga merupakan tetapan laju distribusi yang tunggal.
(
k α)
M(
ka β)
L β α N k α M k β L k a a a 21 − + − + += (12)
Keterangan :
k21 = tetapan laju distribusi untuk perpindahan obat dari kompartemen perifer
ke kompartemen sentral (menit-1) ka = slope (tetapan laju) absorpsi (menit-1)
L = intersep dari slope persamaan distribusi dengan ordinat (µg/ml)
M = Intersep dari hasil back extrapolation slope persamaan monoeksponen eliminasi dengan ordinat (µg/ml)
α = slope (tetapan laju) distribusi (disposisis cepat) [menit-1]
β = slope (tetapan laju) eliminasi total (disposisi lambat) [menit-1]
N = kadar hipotetik obat pada t (0) yang diperoleh dari penjumlahan nilai L dan M (µg/ml)
h. Kliren total (ClT), Kliren menggambarkan volume darah atau
juga semua jalur ekskresi termasuk metabolisme. Obat dapat dibersihkan dari tubuh melalui berbagai jalur. Dua organ penting adalah ginjal (ClR)
dan hati(ClH). Jalur selain ginjal dan hati (paru-paru, kulit, saliva, air susu,
dll) biasanya diabaikan. Kliren dapat diperoleh dari data dosis (D), bioavailabilitas absolut (f) dan AUC0−∝.
∝ −
=
0 T
AUC f x D
Cl (13)
Atau dapat pula dihitung dari Vc dan tetapan eliminasi dari kompartemen sentral
13 c
T V xk
Cl = (14)
Karena kliren total merupakan penjumlahan dari kliren organ-organ maka:
ClT = ClH + ClR +Clx (15)
Keterangan :
ClT = kliren total (ml/menit)
ClH = kliren hepatik (ml/menit)
ClR = kliren ginjal (ml/menit)
Clx = kliren hati (ml/menit)
D = dosis (µg)
k13 = tetapan laju eliminasi dari kompertemen sentral (menit)
Vc = volume distribusi kompartemen sentral (ml)
∝ -0
AUC = Luas area dibawah kurva kadar obat di dalam darah lawan waktu dari waktu nol hingga waktu tak hingga
f = fraksi obat terabsorpsi
i. Tatapan laju eliminasi (disposisi lambat) total (β), diperoleh dari slope persamaan garis monoeksponen eliminasi dan merupakan suatu tetapan hibrida (hybrid constant)
12 21 2
k k b b
β = − − − (16)
Keterangan :
β = slope (tetapan laju) eliminasi total (disposisi lambat) [menit-1] b = k21 + k12 + k13
k21 = tetapan laju distribsi untuk perpindahan obat dari kompartemen perifer ke kompartemen sentral (menit-1)
k13 = tetapan laju elimani untuk perpindahan obat kompartemen sentral (menit-1)
j. Tetapan laju eliminasi kompartemen sentral (k13),
menggambarkan tetapan laju eliminasi obat dari kompartemen sentral.
21 13
k
β α
k = (17)
k. Waktu paruh eliminasi (t½ eliminasi), adalah waktu yang
diperlukan agar kadar obat dalam darah menjadi setengahnya.
T ss eliminasi
2 1
Cl Vd x 0,693
t = (18)
∝ ∝
=
0-AUC AUMC
MRT (19)
Keterangan :
MRT = waktu tinggal rata-rata obat di dalam tubuh (menit-1)
∝ − 0
AUMC = first moments dari kurva kadar obat dalam plasma, diperoleh dengan mengkalikan persamaan konsentrasi obat dalam plasma dengan waktu.
∝ − 0
AUC = Luas area dibawah kurva kadar obat di dalam darah lawan waktu dari waktu nol hingga waktu tak hingga
Berkaitan dengan profil proses absorpsi distribusi dan eliminasi terutama pada pemberian obat ekstravaskular masing-masing parameter dapat digunakan untuk mengkaji proses proses tersebut. Seperti profil absorpsi dapat dikaji melalui parameter ka, Cpmaks, tmaks, fa dan AUC0−∝ Profil distribusi dikaji dengan
parameter seperti tetapan kecepatan distribusi (α), tetapan kecepatan perpindahan obat dari kompertemen sentral ke perifer (k12), tetapan kecepatan perpindahan
obat dari kompertemen perifer ke sentral (k21), setelah dicapai keseimbangan
distribusi. Parameter disposisi (β) dan k13 digunakan untuk mengkaji profil
eliminasi suatu obat (Donatus, 1998).
Tabel V. Ketergantungan parameter famakokinetika sekunder dan
turunan terhadap parameter farmakokinetika primer
Persamaan
Parameter Farmakokinetika Sekunder
Waktu paruh eliminasi(t1/2 el)
Tetapan laju eliminasi(Kel/β)
Fraksi obat yang diekskresikan dalam bentuk utuh (f)
total R Cl Cl Vd Cl Cl Vd 0.693 x Parameter Turunan AUC (oral)
Kadar obat dalam plasma dalam kondisi tunak(Css)
(
)
(
)
(
intervalpemberian)
total Cl oral ilitas Bioavailab Dosis total Cl oral ilitas Bioavailab Dosis
( Rowland &Tozer, 1995)
4. Strategi penelitian farmakokinetika
farmakodinamika terjadi pada saat obat berinteraksi dengan reseptor pada jaringan sasaran sehingga dapat menimbulkan efek (Arieens & Simonis, 1982)
-disintegrasi sediaan obat - disolusi zat aktif
- absorpsi - distribusi - metabolisme - ekskresi -disintegrasi sediaan obat - disolusi zat aktif
- absorpsi - distribusi - metaboli •
•
Gambar 2. Tahapan aksi hayati / biologi obat dalam tubuh
(Arieens & Simonis, 1982; Bowman & Rand,1990)
Subyek yang digunakan dalam penelitian farmakokinetika adalah makluk hidup, maka harus dipahami dengan baik variabel yang menentukan kesahihan penelitian. Hal ini penting karena subyek hidup seringkali sulit dikendalikan. Maka dari itu strategi penelitian penting ditetapkan sebelumnya, agar hasil penelitian dapat diandalkan. Tahapannya strategi penelitian umumnya sebagai berikut.
a. Pemilihan rancangan uji coba. Dalam memilih rancangan perlu dipertimbangkan variabel yang terdapat pada subyek uji maupun sistem penelitian.
sme - ekskresi I. Tahap farmasetika
Obat tersedia
Dosis untuk diabsorpsi
Ketersediaan farmasetis
II. Tahap farmakokinetika
Obat tersedia untuk beraksi
Ketersediaan hayati /bioavailability
III. Tahap farmakodinamika
Efek
interaksi obat reseptor dalam jaringan sasaran -disintegrasi sediaan obat - disolusi zat aktif
1) Variabilitas antar subyek (umur, daya tahan, berat badan, kemampuan metabolisme)
2) Variabilitas perlakuan (dosis & formulasi yang berbeda)
3) Variabilitas waktu (perubahan lingkungan, kelelahan, efek sisa perlakuan lainnya)
4) Variabilitas (dalam subyek)
5) Variabilitas residual, yakni yang tidak dapat diidentifikasi (misalnya kesalahan penetapan kadar dll.)
b. Pemilihan subyek uji dan jumlahnya. Subyek uji meliputi hewan (uji pra klinis) dan manusia (uji klinis). Pada penggunaan hewan sebagai subyek uji ada hal-hal yang harus dipertimbangkan seperti bentuk sediaan, cara pemberian, kemudahan penanganan, kemudahan pengosongan lambung, kemudahan pengambilan cuplikan hayati (cairan biologis), dan volume maksimal yang dapat diterima oleh hewan uji. Yang tidak kalah penting adalah kemiripan mekanisme absorpsi, distribusi, metabolisme dan ekskresi dengan diri manusia. Hewan yang mungkin digunakan meliputi anjing, kera, babi, kelinci , mencit dan tikus. Jumlah subyek uji dapat ditentukan dari variabilitas antar subyek bagi obat. Variabilitas AUC (luas daerah di bawah kurva) dapat digunakan sebagai tolak ukurnya. Semakin kecil variabilitas antar subyek maka jumlah subyek uji yang diperlukan relatif semakin sedikit.
merupakan tempat yang paling cepat dicapai oleh obat dan paling logis digunakan untuk penetapan kadar obat dalam tubuh. Alasannya karena darah mengambil obat dari tempat absorpsi, mendistribusikan pada jaringan sasaran dan menghantarkan ke organ eliminasi. Alasan lain karena pada sebagian besar obat, bentuk utuh adalah bentuk yang aktif secara farmakologi (Tozer, 1979).
1) Darah adalah bagian kompleks dari cairan tubuh. Darah memiliki
volume kurang lebih seperduabelas berat badan, 55% adalah bagian cair (plasma terbuffer yang mengandung protein dam lemak terlarut) dan 45% bagian padat(sel darah tersusupensi). Beruntung bahan penyusun utama yaitu sel darah merah atau eritrosit dapat dipisahkan dari plasma dengan sentrifugasi sederhana. Nemun perlakuannya harus hati-hati karena sel darah merah dapat pecah dan menyulitkan pemisahan komponen yang tidak diinginkan (Chamberlain, 1995; Pearce, 2002). Jika darah dibiarkan mengendap tanpa penambahan antikoagulan, sel darah merah biasanya akan menggumpal (membeku) dan menghasilkan cairan yang disebut serum yang dapat didekantasi. Serum dalam banyak hal serupa dengan plasma kecuali bahwa serum tidak lagi mengandung faktor pembekuan karena sudah digunakan pada proses pembekuan (Murray, Granner, Mayes & Rodwell, 2000; Chamberlain, 1995).
dengan ultrafiltrasi ataupun dialisis dapat ikut menghilangkan pula sebagian besar fraksi obat. Meskipun ada pendapat bahwa obat dalam bentuk bebaslah yang penting secara fisiologi, namun karena obat-obatan tersebut biasanya terikat protein sehingga kadar dalam bentuk bebas sangat rendah, maka adalah hal yang biasa mengukur total obat dalam plasma atau serum (Chamberlain, 1995).
Metode denaturasi adalah bagian dari deproteinisasi plasma guna menyiapkan plasma untuk dianalisis. Denaturasi dilakukan untuk memecah ikatan obat-protein sehingga protein dapat mengendap dan filtratnya diisolasi. Pada denaturasi terjadi proses modifikasi struktur sekunder, tersier dan kuartener protein sehingga dapat digunakan untuk merusak kemampuan berikatan dengan obat dan mengendapkan protein (Bruice 1998; Chamberlain, 1995).
A B
C D D
Gambar 3. Contoh struktur protein: (A) primer pada ribonuklease; (B) sekunder berbentuk lembaran berlipat pararel; (C) tersier dengan berbagai macam ikatan; (D) kuartener pada
protein globular yang kompleks
Metode denaturasi protein diantaranya sebagi berikut. a) Mengubah pH dengan asam trikloroasetat (TCA), asam perklorat dan asam tungstat biasa adalah asam-asam kuat yang bisa ditambahkan untuk denaturasi. Penambahan asam kuat akan menggangu muatan anion-kation pada ikatan protein sehingga terjadi gangguan elektrostatik serta rusaknya ikatan hidrogen pada protein.
b) Pemakaian reagen khusus yang dapat menciptakan suatu ikatan hidrogen yang lebih kuat yang akan menggangu ikatan antar protein dalam molekul.
c) Pemakaian pelarut organik seperti metanol, etanol dan asetionitril yang mengganggu ikatan hidrofobik pada bagian gugus-gugus non-polar dengn cara berikatan dengan gugus tersebut.
d) Penggunaan enzym proteolitik seperti subtilisin, tripsin, proteinase, papain dan ketodase untuk menghindari kerusakan analit karena denaturasi dengan bahan kimia. Prosedur tersebut berhasil untuk preparasi obat dari jaringan, namun enzim subtilisin sukses untuk digesti protein plasma.
e) Penggunaan panas pada suhu 90°C selama 5-15 menit dapat dipakai untuk menganggu gaya tarik antar molekul, berakibat pada denaturasi.
ditetapkan dengan tepat. Penggunaan urin akan lebih baik daripada darah bila obat diekskresikan secara sempurna dalam bentuk tak berubah di dalam urin. Keterbatasan urin adalah pengosongan kandung kemih sulit dilakukan, dekomposisi selama penyimpanan dan kemungkinan terhidrolisisnya konjugat metabolit tak stabil dalam urin.
a. Pemilihan metode penetapan kadar. Metode dikatakan memenuhi syarat digunakan dalam penelitian farmakokinetik bila memenuhi syarat berikut.
1) Selektivitas atau spesifitas menempati prioritas utama karena betuk
obat yang ditetapkan adalah bentuk utuh atau metabolitnya, sehingga dengan metode tersebut harus dapat dibedakan antara obat utuh dengan metabolitnya (Mulja & Hanwar, 2003)
2) Sensitivitas berkaitan dengan kemampuan metode untuk
mengidentifikasi perbedaan yang kecil antar analit (Mulja & Hanwar, 2003) faktor yang menjadi pertimbangan sensitivitas adalah slope (kemiringan) kurva baku dan presisi. Jika terdapat dua metode dengan presisi yang sama maka metode yang memiliki slope lebih curam bersifat lebih sensitif (Skoog, 1985)
3) Ketelitian (Akurasi) didefinisikan sebagai kedekatan nilai hasil
4) Ketepatan (Presisi) adalah tingkat kesamaan/kesesuain bila
pengukuran dilakukan berulang kali pada cuplikan hayati yang sama (Mulja & Hanwar, 2003). Parameternya adalah standar deviasi absolut (absolut standart deviation = SD), koevisien variasi = KV (coefficient of variation = CV) atau sering juga disebut sebagai standar deviasi relatif
(relative standart deviation = RSD) (Skoog et al., 1998) Untuk bioanalisis KV sebesar 15-20% masih dapat diterima (Mulja & Hanwar, 2003).
5) Cepat. Kecepatan pengukuran kada obat dengan suatu metode
analisis juga harus dipertimbangkan mengingat sampel yang dianalisis dalam jumlah banyak ( Donatus, 1998).
b. Pemilihan takaran dosis. Dosis yang diberikan harus menjamin dapat terukurnya kadar obat sampai rentang waktu tertentu sampai didapat data yang mencukupi unutuk analisis farmakokinetika. Harus diperhatikan pula adanya kinetika tergantung dosis yaitu berubahnya parameter farmakokinetika suatu obat bila dosis yang diberikan berubah. Bila hal tersebut terjadi maka obat diasumsikan mengikuti kinetika ordo nol. Pemilihan takaran dosis dapat dipertimbangkan dari harga ED50 dan LD50 (Kaplan, 1998)
c. Pemilihan lama dan banyaknya waktu pengambilan cuplikan. Darah dan urin paling sering digunakan sebagai cuplikan hayati dalam analisis farmakokinetika. Jika digunakan darah maka lama pengambilan adalah 3-5 x t½el obat yang diuji. Sedang frekuensi berkaitan dengan model
setidaknya harus dilakukan 3 kali pencuplikan pada masing-masing tahap yaitu absorpsi, distribusi dan eliminasi (Donatus, 1989)
d. Analisis dan evaluasi hasil merupakan tahapan akhir dari analisis farmakokinetika. Tahapan ini meliputi analisi model kompertemen, analisis data uji coba dan perhitungan parameter farmakokinetika, analisis statistika dan evaluasi hasil.
A. Interaksi farmakokinetika 1. Pengertian
farmakokinetika terdapat pula interaksi farmakodinamika yang terjadi bila efek suatu obat berubah karena kehadiran obat atau agen kimia lain pada tempat aksinya. Hasil interaksi bisa menyebabkan hasil efek yang tetap sama, lebih kuat atau lebih lemah. Secara klinis bermakna namun dapat juga tidak bermakna. Selanjutnya dalam penelitian ini akan lebih dibahas tentang interaksi farmakokinetika.
2. Mekanisme interaksi farmakokinetika
Beberapa obat mengalami interaksi dengan cara yang unik. Banyak sekali obat yang berinteraksi tidak hanya dengan satu macam mekanisme saja, namun bisa dua atau lebih. Oleh karena itu untuk lebih jelasnya disini akan dibahas tentang berbagai mekanisme interaksi, khususnya interaksi farmakokinetika. Berdasarkan fase terjadinya, interaksi farmakokinetik dapat di golongkan sebagai berikut.
1) Efek perubahan pH saluran cerna (gastrointestinal). Perpindahan dengan mekanisme difusi pasif seperti pada membran mukosa tergantung pada jumlah obat dalam bentuk molekul tak-terion (non-ionized), bentuk yang larut-lemak (lipid-soluble). Karena itu pKa,
kelarutan dalam lemak , pH usus dan berbagai parameter terkait formulasi farmasetika menentukan absorpsi obat. Peningkatan pH karena H2-bloker dan antasida dapan mempengaruhi kelarutan ketokonazol dan
mengurangi absorpsinya. Absorpsi asam salisilat lebih tinggi pada pH yang rendah dibanding pH tinggi. Secara teoritis hal ini mungkin disebabkan perubahan pH lambung, namun dalam praktek keluarannya (outcome) sering tidak pasti karena beberapa mekanisme seperti pembentukan khelat dan perubahan motilitas usus juga dapat mempengaruhi.
dalam susu, antasida atau bahan-bahan berzat besi. Selain itu kompleks yang terbentuk tersebut mengurangi efek antibakteri dari antibiotik (Mustchler & Darendorf, 1995; Stockely,1994).
3) Perubahan motilitas saluran cerna. Usus halus bagian atas adalah tempat utama bagi absorpsi sebagian besar obat. Hal-hal yang dapat mengubah kecepatan pengosongan lambung dapat mempengaruhi absorpsi obat. Contohnya propanthelin menunda pengosongan lambung yang berakibat pada pengurangan laju absorpsi parasetamol (asetaminofen) sedang metklopramid berefek sebaliknya, namun demikian jumlah obat yang terabsorpsi tidak berubah. Obat antikolinergik mengurangi motilitas usus, antidepresan trisiklik tersebut dapat meningkatkan absorpsi dikumarol kemungkinan dengan meningkatkan waktu untuk berdisolusi dan diabsorpsi (Stockely,1994). 4) Malabsorpsi yang disebabkan oleh obat. Neomisin menyebabkan sindrom malabsorpsi yang serupa dengan malaria non-tropik. Efeknya adalah perubahan absorpsi beberapa obat termasuk digoxin dan penisilin V (Stockely,1994)
secara farmakologi sedang yang terikat bersifat inaktif atau sering diistilahkan “restricrive”drug. Meski demikian ikatan dengan protein plasma ini berifat
reversibel sehingga pada akhirnya molekul obat yang semula inaktif tersebut menjadi aktif dan selanjutnya mengalami metabolisme dan ekskresi seperti molekul bebas lainnya (Stockely,1994).
Dua buah obat dapat dapat saling bersaing dan saling mendesak satu sama lain dalam berikatan pada protein plasma yang sama. Interaksi ini sangat umum dijumpai namun hanya bermakna klinis bila obat terikat dengan protein plasma dalam jumlah yang bersar, indeks terapi sempit dan volume distribusi (Vd) relatif kecil (Mustchler & Darendorf, 1995). Obat seperti itu misalnya
sufonilurea seperti tolbutamid (terikat 96%, Vd 10 L). Antikoagulan oral
seperti warfarin (terikat 99%, Vd 9 L) dan fenitoin (terikat 90%, Vd 35 L).
Contoh lain diazoxide, fenilbutazon dan sulfanilamid (Stockely,1994).
1) Induksi enzim. “Toleransi” adalah fenomena umum yang
berkembang pada beberapa obat. Contohnya pada penggunaan babiturat, sejalan dengan waktu dosis harus ditingkatkan untuk memperoleh efek hipnotik yang sama. Hal ini dikarenakan barbiturat meningkatkan aktivitas enzim mikrosomal (“induksi”enzim) sehingga metabolisme dan ekskresinya sendiri ditingkatkan. Fenomena juga terjadi pada kehadiran obat lain yang dimetabolisme dengan enzim yang sama. Misalnya pada antikoagulan oral seperti warfarin, metabolisme enzimatiknya meningkat dan dibutuhkan dosis yang lebih dengan kehadiran diklorafenazon (agen penginduksi enzim) (Stockely,1994).
Jumlah induksi enzim tergantung pada obat dan dosisnya., namun prosesnya memerlukan beberapa hari atau minggu, dan bertahan dalam waktu yang sama setelah penggunaan agan penginduksi tersebut dihentikan. Efek ini tidak hanya disebabkan oleh obat namun juga pestisida hidrokarbon terklorinasi seperti lindane dan dichopane, juga setelah merokok. Hal yang harus diperhatikan adalah ketika agen penginduksi dihentikan penggunaanya, obat obyek yang semula ditingkatkan dosisnya harus dikurangi kembali, bila tidak akan timbul overdosis (Stockely,1994).
penginhibisi ditingkatkan. Inhibisi enzim tidak memerlukan waktu yang lama bila dibandingkan induksi enzim, hanya dua-tiga hari saja, dan toksisitas terjadi dengan cepat (Stockely,1994).
Pada pasien epilepsi dengan terapi fenitoin, kehadiran kloramfenikol menyebabkan akumulasi fenitoin yang tidak terdeteksi sampai pasien mulai menampakkan maniefestasi keracunan. Makna klinis inhibisi enzim tergantung pada peningkatan kadar obat dalam serum. Jika tetap berada dalam kisaran terapetik, interaksi ini dianggap bermanfaat dan dianggap berbahaya jika sampai atau melebihi batas minimun ketoksikan (Stockely,1994).
3) Perubahan aliran darah yang melalui hati. Setalah absorpsi,
sirkulasi portal akan membawa obat langsung menuju hati sebelum didistribusikan oleh aliran darah keselurah bagian tubuh lainnya. Obat yang cukup larut lemak mengalami metabolisme yang cukup signifikan melalui first pass effect ini. Simetidin (namun tidak dengan ranitidin) mengurangi aliran darah hepatik sehingga ketersediaan hayati propanolol meningkat. Propanolol juga mengurangi klirennya sendiri juga obat lain seperti lidokain. Beberapa obat yang lain memiliki efek meningkatkan aliran darah hepatik sehingga metabolismenya meningkat (Stockely,1994) .
dalam glomerolus dari tubulus dimana molekul-molekul kecil yang dapat melewati pori dari membran glomerular ( misal: air, garam, beberapa obat-obatan) disaring ke dalam lumen dari tubulus. Sementara molekul yang lebih besar, seperti protein plasma, dan sel darah ditahan. Aliran darah kemudian membawa bagian yang tersisa pada tubulus ginjal dan digunakan transport aktif untuk memindahkan obat dan metabolitnya dari darah dan mensekresinya ke dalam filtrat tubular. Sel tubulus memiliki sistem transpor aktif maupun pasif untuk mereabsorsi obat. Gangguan oleh obat pada pH cairan tubulus dengan sistem transpor aktif dan dengan aliran darah menuju ginjal dapat mengubah ekskresi obat lain (Stockely,1994).
1) Perubahan pH urin. Reabsorpsi pasif obat tergantung jumlah obat
yang terdapat pada bentuk tidak terion, bentuk larut lemak yang dalam hal ini tergantung pada pKa dan pH urin. Perubahan pH yang
mengurangi jumlah obat tidak terion (urin basa untuk obat asam dan urin asam untuk obat basa) meningkatkan ekskresi obat. Makna klinis dari interaksi ini kecil karena sebagian besar obat baik asam lemah maupun basa lemah dimetabolisme oleh hati menjadi bentuk inaktif dan sedikit yang diekskresi dalam bentuk utuh. Prakteknya hanya sedikit obat yang mengalami interaksi ini (perkecualian termasuk pada perubahan ekskresi quinidin dan salisilat karena perubahan pH urin oleh antasida) dalam kasus overdosis perubahan pH urin digunakan untuk meningkatkan ekskresi obat seperti fenobarbital dan salisilat
2) Perubahan sekresi tubuler aktif dari ginjal. Obat-obat dengan
mekanisme transpor aktif yang sama dalam tubulus ginjal dapat saling berkompetisi dalam ekskresi. Probenesid mengurangi ekskresi penisilin dan obat-obat lain dengan berkompetisi pada mekanisme ekskresi dimana penisilin akan tertahan. Meski demikian probenesid akhirnya juga tertahan karena mengalami reabsorsi pasif sepanjang tubulus ginjal (Stockely,1994).
3) Perubah