Possible Predictor for Response to Electroconvulsive
Therapy in Depressed Psychotic Inpatients
Rachel Maayan, Yana Yagorowski, Daniel Grupper, Mordechai Weiss,
Biana Shtaif, Mahmoud Abou Kaoud, and Abraham Weizman
Background: Dehydroepiandrosterone (DHEA) and its
sulfate derivative DHEAS are neuroactive steroids. In the
brain, they interact with
g
-aminobutyric acid (GABA
A)
receptors, which are involved in the regulation of anxiety
and mood. The relevance of circulatory neurosteroids to
psychiatric
disorders
and
biological
treatment
is
unknown.
Methods: Basal plasma levels of cortisol, DHEA, and
DHEAS and the DHEAS–DHEA ratio were determined in
17 psychiatric inpatients before and after six
electrocon-vulsive (ECT) therapy sessions, and all changes were
statistically analyzed. For baseline values, 25 healthy
individuals served as control subjects. Severity of
depres-sion and psychosis in the patients was measured with the
Hamilton Depression Rating Scale (HDRS) and the Brief
Psychiatric Rating Scale, respectively.
Results: Both basal and post-ECT levels of cortisol,
DHEA, and DHEAS were significantly higher in the
patients than in the control subjects. DHEAS levels in
responding patients were higher at completion of
treat-ment than at baseline. Patients defined as ECT
nonre-sponders (change in HDRS
,
30% from before
treat-ments) exhibited elevated basal DHEAS levels compared
with ECT responders.
Conclusions: Markedly elevated basal DHEAS levels
(mean
1
2 SD of control value) are associated with
resistance to ECT and may serve as a potential predictive
marker of nonresponsiveness to ECT in depressed
pa-tients. Biol Psychiatry 2000;48:693–701 © 2000 Society
of Biological Psychiatry
Key Words: Dehydroepiandrosterone (DHEA), DHEAS,
neurosteroids, electroconvulsive therapy (ECT),
depres-sion, GABA
Areceptor
Introduction
D
ehydroepiandrosterone (DHEA) and
dehydroepi-androsterone sulfate (DHEAS) are neuroactive
ste-roids that are synthesized in situ within the brain,
inde-pendent of their synthesis in the steroidogenic organs
(Baulieu 1991, 1992, 1997; Baulieu and Robel 1990;
Mellon 1994; Regelson and Kalimi 1994; Robel and
Baulieu 1995a, 1995b; Robel et al 1995). Some of the
neuroactive steroids have been found to interact with the
g
-aminobutyric acid (GABA)-gated chloride ion channels
and may represent an important class of neuromodulators
that can rapidly alter central nervous system excitability
via nongenomic mechanisms (Baulieu and Robel 1996;
Deutsch et al 1992; Majewska 1992, 1995; Majewska et al
1986; Majewska and Schwartz 1987; McEwen 1991).
DHEA, but not DHEAS, is associated with antiaggressive
effects (Haug et al 1988). Researchers speculate that the
sedative effects of some neurosteroids are achieved via
direct or indirect enhancement of GABA-mediated
chlo-ride ion conductance (Majewska 1995; Regelson and
Kalimi 1994; Robel and Baulieu 1995a, 1995b; Young et
al 1996). They may act either by decreasing the level of
the GABA
Aantagonistlike pregnenolone sulfate (Robel
and Baulieu 1995b) or by increasing the level of the
GABA
Aagonistlike metabolites of progesterone (such as
di- and tetrahydro- progesterone) (Young et al 1996) or the
level of the DHEA metabolite androsterone (Majewska
1995).
In contrast to the GABA agonists, the neurosteroids
DHEA and DHEAS display allosteric antagonistic
prop-erties at GABA
Areceptors (Akiva et al 1991; Demirgoren
et al 1991; Majewska 1995; Majewska et al 1990; Spivak
1994); DHEAS appears to be the more potent (Baulieu and
Robel 1998). Alterations in the synthesis of the
neuros-teroids may result in GABA-mediated behavioral changes.
Neuroactive steroids were reported to be altered during
major depression in humans and to play a putative role in
the treatment of depression with antidepressants. A
signif-icant decrease in the plasma concentrations of 5
a
-preg-From the Laboratory of Biological Psychiatry, Felsenstein Medical Research Center, Tel Aviv (RM, BS, MAK, AW), Research Unit, Geha Psychiatric Hospital, Beilinson Campus, Petah Tiqva (AW), Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv (AW), and Be’er Yaakov Mental Health Center, Be’er Yaakov (YY, DG, MW), Israel.
Address reprint requests to Abraham Weizman, M.D., Geha Psychiatric Hospital, PO Box 102, Petah Tiqva 49100, Israel.
Received August 27, 1999; revised February 9, 2000; accepted February 15, 2000.
© 2000 Society of Biological Psychiatry 0006-3223/00/$20.00
nan-3
a
-ol-20-one (3
a
,5
a
-THP) and 5
b
-pregnan-3
a
-ol-20-one
(3
a
,5
b
-THP),
which
are
positive
allosteric
modulators of the GABA
Areceptor, and a concomitant
increase in 5
a
-pregnan-3
b
-ol-20-one (3
b
,5
a
-THP), which
may act as a functional antagonist for GABA-agonistic
steroids, were reported in patients with major depression.
These alterations in the composition of neuroactive
ste-roids were corrected following successful treatment with
various antidepressants. Pregnenolone, DHEA,
progester-one, and 5
a
-pregnan-3,20-dione (5
a
-DHP) plasma
con-centrations were not altered by the antidepressants
(Ro-meo et al 1998). The authors suggested that progesterone
metabolism is dysregulated during depression, possibly
resulting in a decreased GABA-ergic neurotransmission.
Electroconvulsive therapy (ECT) is one of the most
effective treatments for major depression (Avery and
Winokur 1977; Barton 1977; Lambourn and Gill 1978).
ECT is especially recommended for patients with
psy-chotic depression and refractory major depression, as well
as for patients with schizophrenia with affective symptoms
(Avery 1993; Solan et al 1988). Usually six ECT
treat-ments lead to a significant improvement. ECT, as a major
stressor, causes a rise in the stress hormones (Purdy et al
1991; Weizman et al 1987), which may be followed by a
corresponding increase in the steroids active at the
GABA
Areceptors. It is possible that in the brain, the in
situ glial synthesis of allopregnanolone and
allotetrahydro-deoxycorticosterone, which are GABA-positive steroids,
may be heightened following ECT, and there may also be
changes in the levels of the GABA-negative steroids, such
as DHEAS and pregnenolone sulfate (for a review of brain
neurosteroids see Deutsch et al 1992). DHEA may be
involved in the termination of the stress response through
its putative anxiolytic activity (Melchior and Ritzmann
1994).
The role of neurosteroids in neuropsychiatric disorders
is still unknown. One can speculate that changes in the
levels of the neuroactive steroids and the ratio between
them and their sulfate derivatives (which have more potent
antagonistic activity) may be involved in the
pathophysi-ology of stress states such as anxiety and depression.
Interestingly, Ferguson et al (1964) demonstrated 25 years
ago that ECT is associated with increased urinary DHEAS
excretion. The aims of our study were 1) to investigate the
influence of six sequential ECT sessions in drug-resistant
psychiatric patients on the plasma levels of DHEA and
DHEAS and the ratio between the free steroid and its
sulfate derivative (the corresponding weak and potent
GABA
Aantagonists) and 2) to assess whether one of these
parameters may predict clinical responsiveness to ECT.
We hypothesized that ECT will be associated with
in-creases in plasma DHEA and/or DHEAS and cortisol
levels and that pretreatment high neurosteroid levels will
interfere with the response to ECT.
Methods and Materials
Study Population
The study population consisted of 17 hospitalized psychiatric patients, seven men and 10 women, of mean age 40.4 6 3.1 years. Two patients had major depression with psychotic fea-tures, 10 had schizophrenia with comorbid depression (Siris 1995), and five were schizoaffective patients with current de-pressive signs and symptoms. The diagnoses of schizophrenia, schizoaffective disorder, and major depression were established according to the DSM-IV criteria using the Structured Clinical Interview for DSM-IV—Patient Version (SCID-P; First et al 1995). The duration of the psychiatric disorders was 14.868.4 years. All were physically healthy with no infection or history of drug or alcohol abuse. Six patients were receiving zuclo-penthixol, three fluphenazine, four haloperidol, and four clothia-pine; three patients also were being treated with carbamazepine, three with clomipramine, and one with maprotiline. Patients were maintained on the same drug treatment for at least 4 weeks, and treatment regimen was not altered during the entire study period. An independent psychiatrist recommended ECT according to clinical judgment because of the patients’ drug resistance. For the patients with psychotic depression, drug resistance was defined as failure to respond to adequate trials with antipsychotics combined with antidepressants (a heterocyclic antidepressant, a selective serotonin reuptake inhibitor, or another class of antide-pressant medication) before initiation of ECT (Prudic et al 1996). The schizophrenic and schizoaffective patients were defined as drug resistant if no satisfactory response was obtained following at least three adequate trials with antipsychotics (Meltzer et al 1990) combined with antidepressants, without or with carbam-azepine. For baseline comparison, 25 healthy age- and gender-matched control subjects (10 men, 15 women; mean age 38.56
2.7 years) were used. The control subjects were assessed by a clinical interview according to the SCID guidelines (First et al 1995) and medical examination. The rates of cigarette smoking were similar: 14 of 17 of the patients and 22 of 25 of the control subjects were smokers. None of the patients or control subjects used hormones. Both patients and control subjects were sampled during the summer (June-August) to avoid seasonal variations (Deslypere et al 1983).
The study was approved by the Be’er Yaakov Review Board for Clinical Research, and informed consent was obtained from all participants.
ECT Procedure
and succinylcholine 0.5–1.0 mg/kg IV. Seizure duration was monitored with a single-channel electroencephalograph (includ-ing electrodes placed over the contralateral hemisphere) and cuff technique. Treatment was given twice weekly for a total of six sessions. The starting charge of the ECT stimulus dosage (pulse width3frequency (2)3train duration3current) was 48 mc; this was increased gradually during the ECT course according to the rise in seizure threshold, as described previously (Coffey et al 1995; Fink 1992; Sackheim et al 1987).
Clinical Assessments
The severity of the depressive, anxiety, and psychotic symptoms was measured with the Hamilton Depression Rating Scale (HDRS; Hamilton 1960), the Hamilton Anxiety Rating Scale (HARS; Hamilton 1959), and the Brief Psychiatric Rating Scale (BPRS; Overall and Gorham 1962). Clinical assessments were performed 1 day before initiation of the ECT course and 1 day after the sixth treatment. The rater (YY) was blind to the laboratory measures, and the laboratory staff was blind to the clinical data.
Hormone Determinations
Blood samples were obtained before and 20 min after the first and sixth ECT session (ECT-1, ECT-6). Morning (7:00 to 8:00 AM) basal samples were collected concomitantly from the control subjects. All hormone determinations were performed in the same run to avoid interassay variability.
The levels of DHEA, DHEAS, and cortisol were determined by commercial radioimmunoassay (RIA) kits, as follows:
• DHEA-DSL 9000 Active DHEA coated tube RIA kit
(Diagnostic Systems Laboratories, Webster, TX): sensitiv-ity 0.02 ng/mL; specificsensitiv-ity-cross-reactivsensitiv-ity with DHEAS 0.88%, all others negligible; assay variability 10.2% be-tween runs, 5.6 –10.6% within runs, according to level.
• DHEA-S-DSL-3500 Active DHEAS coated tube RIA kit (Diagnostic Systems Laboratories): sensitivity 17 ng/mL; specificity-cross-reactivity with DHEA 41%; assay vari-ability 10% between runs, 6.3–9.4% within runs, according to level.
• Cortisol-TKCO1 Coat-A-Count kit (Diagnostic Products
Corporation, Los Angeles): sensitivity 0.2mg%; specifici-ty-cross-reactivity: with prednisolone 76%, 11-deoxycorti-sol 11.4%, prednisone 2.3%, cortisone and corticosterone 1%, all others#0.3%; assay variability 4.0 – 6.4% between runs, 3– 4.8% within runs, according to level.
Statistical Analysis
Analyses of variance (ANOVAs) with or without repeated measures followed by the Student–Newman–Keuls post hoc test were used for the evaluation of the hormonal data. The changes in the HDRS, HARS, and BPRS scores were analyzed by two-tailed Student’s paired t test. Correlation was analyzed by Pearson’s correlation test. All results are expressed as means6
SEMs.
Results
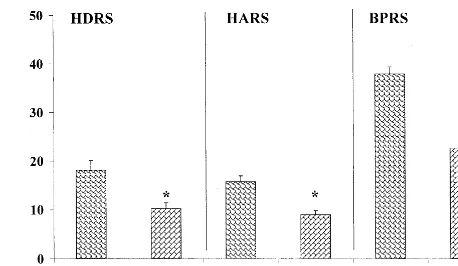
Clinical Effects of ECT
As expected, the HDRS, HARS, and BPRS scores for the
whole group significantly decreased after completion of
the six ECT sessions: p
,
.0001 for all (Figure 1).
Hormone Effects of ECT
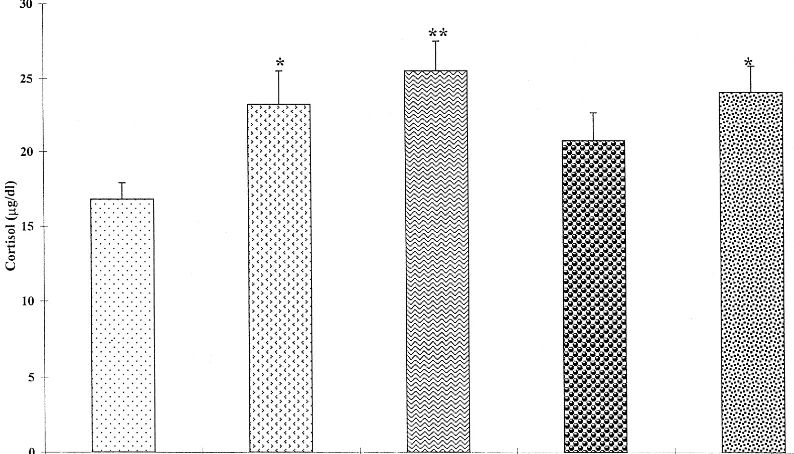
PATIENTS VERSUS CONTROL SUBJECTS.
The
pa-tients had significantly higher levels of cortisol, DHEA,
and DHEAS than did the healthy control subjects both at
baseline and after the last session [F(4,90)
5
4.44, p
5
.0025; F(4,90)
5
3.16, p
5
.017; and F(4,90)
5
5.51, p
5
.005, respectively; Figures 2– 4]. No significant difference
was found in the DHEAS–DHEA ratio [F(4,90)
5
1.38,
p
5
.24, ns; Figure 5]. The patients did not have elevated
cortisol and DHEA compared with the controls subjects at
the pre–ECT-6 time point, and the DHEAS–DHEA ratio
did not differ significantly at the post–ECT-6 time point.
No correlation was found between the pretreatment level
of depression and DHEAS or DHEA levels (r
5 2
.11 and
r
5 2
.28, respectively, both ns).
PATIENT LEVELS BEFORE, DURING, AND AFTER ECT.
Cortisol and DHEA plasma levels remained unaltered
during the study period [F(67)
5
2.19, p
5
.1, ns, and
F(67)
5
0.30, p
5
.8, ns, respectively; Figures 2 and 3],
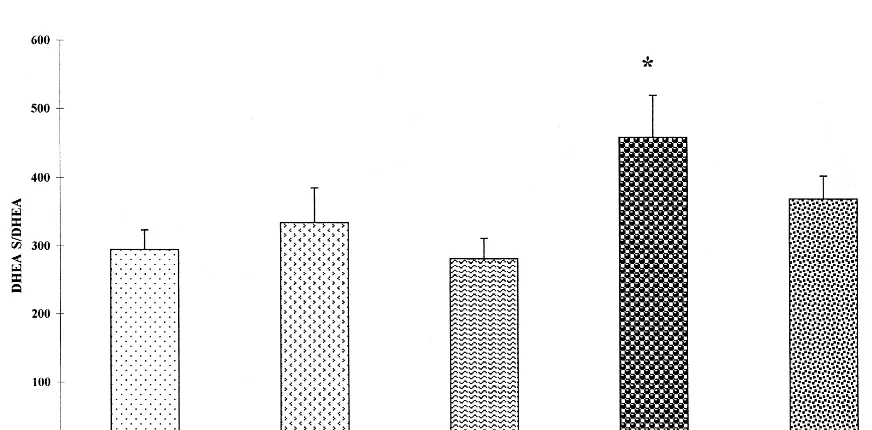
but DHEAS levels rose significantly from the beginning to
the end of treatment. Both pre– and post–ECT-6 DHEAS
levels were significantly higher than the pre– and post–
ECT-1 levels [F(67)
5
5.11, p
5
.003; Figure 4]. A
significant difference was also found in the DHEAS–
DHEA ratio [pre–ECT-6 vs. post–ECT-1, F(67)
5
3.44,
p
5
.02; Figure 5]. Because DHEA and/or DHEAS may
act as a cortisol antagonist (Hechter et al 1997), we
assessed also possible alterations in DHEAS– cortisol and
DHEA– cortisol ratios, which were not affected
signifi-cantly by the repeated ECT [F(67)
5
2.13, p
5
.11, ns,
and F(67)
5
2.36, p
5
.08, ns, respectively].
Relationship between Clinical Response and
Alterations in DHEAS
No statistically significant correlations were found
be-tween the changes in the severity of depressive, anxiety,
and psychotic symptoms (post–ECT-6 minus pre–ECT-1)
and the corresponding changes in DHEAS, DHEA, and
cortisol levels (correlations were
2
.28, .29, and .11 for
depression;
2
.14, .17, and .13 for anxiety and
2
.11, .38,
and .29 for psychosis, all ns).
Because DHEAS was the only hormone altered
signif-icantly by ECT, we decided (post hoc) to evaluate the
possibility that pre-ECT DHEAS levels can predict a
positive clinical response to ECT. Patients were divided
Figure 2. Levels of serum cortisol before, during, and after electroconvulsive therapy (ECT). *p, .05 vs. control subjects. **p,
.01 vs. control subjects.
into two groups by basal DHEAS levels. A cutoff point of
elevated DHEAS level was defined as a level higher than
the control mean
1
2 SD (
.
2200 ng/mL). Eight patients
were found to have a basal DHEAS above this cut-off
(5171
6
407 ng/mL), and nine patients a nonelevated basal
DHEAS level (1691
6
407 ng/mL). A positive clinical
response of the depressive, anxiety, and psychotic symptoms
to ECT was defined as a reduction of at least 30% from
baseline in the HDRS scores a day after ECT-6. This cutoff
for clinical response was chosen a priori because most of the
patients were chronic (mean duration of the psychiatric
disorders: 14.8
6
8.4 years), drug-resistant psychotic patients
with depression who are difficult to treat. Accordingly, eight
of the nine patients with a nonelevated basal DHEAS level
responded to the ECT treatment as opposed to only one of the
eight patients with an elevated level (Fisher’s exact test: p
,
.005, odds ratio
5
56, 95% confidence interval 2.9 –1072.4).
Such a relationship was not found for either anxiety or
psychotic symptom response to ECT (Fisher’s exact test:
HARS, p
5
.13, ns; BPRS, p
5
.29, ns). The patient
subgroups with high and normal baseline DHEAS levels did
not differ significantly in their ECT-induced changes in
Figure 4. Levels of serum dehydro-epiandrosterone sulfate (DHEAS) before, during, and after electrocon-vulsive therapy (ECT). *p,.05 vs control subjects. **p,.01 vs. con-trol subjects. #p , .05 vs. pre– ECT-1. ##p, .01 vs. post–ECT-1 within “total.” 1p, .05 vs. post– ECT-1.
DHEAS levels [1188
6
449 vs. 662
6
485 ng/mL; t(15)
5
0.77, p
5
.45, ns].
Discussion
The major findings of this study are that ECT induces an
elevation in DHEAS plasma levels and that nonelevated
basal DHEAS levels are associated with a clinically
relevant response (improvement
.
30%) of depressive
symptoms (as assessed by the HDRS) to ECT, at least
following six repeated ECT treatments. In addition, we
demonstrated that cortisol, DHEAS, and DHEA basal
levels are higher in drug-resistant psychotic depressed
patients than in healthy control subjects.
Our finding of elevated basal plasma levels of DHEA
and DHEAS in psychiatric patients is in accordance with
earlier studies. Hansen et al (1982) reported high basal
DHEA levels in psychotic depressed patients, and
Tollef-son et al (1990) found elevated 24-hour urinary DHEAS
levels in nonpsychotic patients with major depression. In a
more recent study, however, Romeo et al (1998) failed to
demonstrate altered DHEA in 11 hospitalized patients
with severe major depression. Furthermore, other
re-searchers reported on reduced DHEA and/or DHEAS
levels or DHEA and/or DHEAS– cortisol ratios in
de-pressed patients (Berr et al 1996; Goodyer et al 1998;
Herbert 1998; Osran et al 1993). Our results regarding the
effect of ECT are in agreement with Ferguson et al (1964)
who demonstrated that ECT increased urinary DHEAS
levels. It is of note that DHEA and/or DHEAS show
increased concentrations both in blood and in the brain
following acute stress (Zinder and Dar 1999). Thus, the
pretreatment elevated DHEA and/or DHEAS levels in the
patients may be related to stress of hospitalization, and the
increase obtained immediately after ECT may be related to
the stress of the iatrogenic convulsions or to ECT
premedication.
The increase in neuroactive steroid levels during
de-pression may act to attenuate the dede-pression-associated
overactivity of the hypothalamic-pituitary-adrenal (HPA)
axis (Wolf et al 1997), as well as the severity of the
depressive symptoms (Wolkowitz et al 1995, 1997b). It is
of note that DHEA has been successfully tested as an
antidepressant (Reus 1997; Wolkowitz et al 1997a, 1997b,
1999) and may act as an endogenous antidepressant.
Furthermore, the antidepressant responses to DHEA
treat-ment were directly correlated with treattreat-ment-induced
in-creases in plasma DHEAS (Wolkowitz et al 1997b).
Previous studies have demonstrated that depression is
associated with a decrease in cerebrospinal fluid (CSF)
and plasma levels of the GABA-agonistic neurosteroids
3
a
,5
a
-tetrahydroprogesterone (THP, allopregnanolone),
and 3
a
,5
b
-THP (Romeo et al 1998; Uzunova et al 1998),
which occurs concomitantly with an increase in plasma
levels of 3
b
,5
a
-THP, but not DHEA. Furthermore, in one
study, CSF levels of allopregnanolone correlated
nega-tively with the severity of depression as assessed by the
HDRS, and successful antidepressant therapy led to
nor-malization of both the CSF and plasma content of the
GABA-active neurosteroids (Romeo et al 1998; Uzunova
et al 1998). Based on preclinical and clinical studies, these
authors suggested that the antidysphoric and anxiolytic
profiles of selective serotonin reuptake inhibitors may be
related to their ability to increase brain allopregnanolone
availability (Guidotti and Costa 1998; Uzunov et al 1996).
Unfortunately the role of the balance between
neuros-teroids with GABA-agonistic and antagonistic activity
were not assessed in nonpsychotic and psychotic
de-pressed patients.
was found in our study population between the changes in
DHEAS levels and changes in depression scores.
None-theless, a recent study (Heinz et al 1999) has demonstrated
in abstinent alcoholics negative correlations between
DHEAS plasma concentrations and both self-rated
depres-sion and observer-rated depresdepres-sion scores. In addition, the
ratio of DHEAS to cortisol concentrations was also
neg-atively correlated with depression scores.
In conclusion, depression in drug-resistant psychotic
inpatients is associated with high levels of the putative
allosteric GABA
Areceptor antagonist DHEAS as
com-pared with healthy controls, and a markedly elevated
(mean
1
2 SD of control levels) basal level of this
neurosteroid is a predictor of resistance to ECT. The
relationship between elevated DHEAS levels, other
GABA-active neurosteroids, depression, anxiety and
psy-chosis, as well as the clinical response to antidepressants,
anxiolytics, and ECT, merit further investigation. Our data
should be considered as preliminary pilot data and should
inspire more rigorous studies in homogeneous psychiatric
populations.
We are grateful for the editorial and secretarial help of Gloria Ginzach and Hanni Penn.
References
Abadie JM, Wright B, Correa G, Browne ES, Porter JR, Svec F (1993): Effect of dehydroepiandrosterone on neurotransmitter levels and appetite regulation in the obese Zucker rat. Diabetes 42:662– 669.
Akiva Y, Young J, Kabbadj K, Sanch MHJ, Tucman D, Vourch’ C, et al (1991): Neurosteroids: Biosynthesis, metabolism and function of pregnenolone and dehydroepiandrosterone in the brain. J Steroid Biochem Mol Biol 40:71– 81.
Avery D, Winokur G (1977): The efficacy of electroconvulsive therapy and antidepressants in depression. Biol Psychiatry 12:507–523.
Avery DH (1993): Electroconvulsive therapy. In: Dumer DL, editor. Current Psychiatric Therapy, Vol 84. Philadelphia: Saunders, 524 –525.
Barton JL (1977): ECT in depression: The evidence of controlled studies. Biol Psychiatry 12:687– 695.
Baulieu EE (1991): Neurosteroids: A new function in the brain. Biol Cell 71:3–10.
Baulieu EE (1992): Neurosteroids: An overview. In: Biggio G, Concas A, Costo E, editors. Gabaergic Synaptic Transmis-sion. New York: Raven, 1–167.
Baulieu EE (1997): Neurosteroids: Of the nervous system, by the nervous system, for the nervous system. Recent Prog Horm Res 52:1–32.
Baulieu EE, Robel P (1990): Neurosteroids: A new brain function? J Steroid Biochem Mol Biol 37:305– 403. Baulieu EE, Robel P (1996): Dehydroepiandrosterone and
dehy-droepiandrosterone sulfate as neuroactive steroids. J Endo-crinol 150(suppl):S221–S239.
Baulieu EE, Robel P (1998) Dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEA-S) as neuroac-tive steroids. Proc Natl Acad Sci U S A 95:4089 – 4091. Berr C, Lafont S, Debuire B, Dartigues JF, Baulieu EE (1996):
Relationships of dehydroepiandrosterone sulfate in the el-derly with functional, psychological, and mental status, and short-term mortality: A French community-based study. Proc Natl Acad Sci U S A 93:13410 –13415.
Coffey CE, Lucke J, Weiner RD, Krystal AD, Aque M (1995): Seizure threshold in electroconvulsive therapy (ECT): II. The anticonvulsant effect of ECT. Biol Psychiatry 37:777–788. D’Elia G (1970): Unilateral electroconvulsive therapy. Acta
Psychiatr Scand Suppl 215:5–98.
Demirgoren S, Majewska MK, Spivak CE, London ED (1991): Receptor binding and electrophysiological effects of dehy-droepiandrosterone sulfate, an antagonist of the GABAA receptor. Neuroscience 45:127–135.
Deslypere JP, de Biscop G, Vermeulen A (1983): Seasonal variation of plasma dehydroepiandrosterone sulphate and urinary androgen excretion in post-menopausal women. Clin Endocrinol (Oxf) 18:25–30.
Deutsch SI, Mastropaolo J, Hitri A (1992): GABA active steroids: Endogenous modulators of GABA-gated chloride ion conductance. Clin Neuropharmacol 15:352–364. Ferguson HC, Bartram ACG, Fowlie HC, Cathro DM, Birchall
K, Mitchell FL (1964): A preliminary investigation of steroid excretion in depressed patients before and after electrocon-vulsive therapy. Acta Endocrinol (Copenh) 47:58 – 66. Fink M (1992): Electroconvulsive therapy. In: Paykel ES, editor.
Handbook of Affective Disorders, 2nd ed. Edinburgh: Churchill Livingstone, 359 –367.
First MB, Spitzer RL, Gibbon M, Williams JBW (1995): Structural clinical interview for Axis I DSM-IV disorders— patient edition, SCID-T/P, version 2.0. New York: New York State Psychiatric Institute, Biometrics Research Department. Goodyer IM, Herbert J, Altham PM (1998): Adrenal steroid secretion and major depression in 8- to 16-year-olds, III. Influence of cortisol/DHEA ratio at presentation on subse-quent rates of disappointing life events and persistent major depression. Psychol Med 28:265–273.
Guidotti A, Costa E (1998): Can the antidysphoric and anxiolytic profiles of selective serotonin reuptake inhibitors be related to their ability to increase brain 3a,5a-etrahydroprogesterone (allopregnanolone) availability? Biol Psychiatry 44:865– 873. Hamilton M (1959): The assessment of anxiety states by rating.
Br J Med Psychol 32:50 –55.
Hamilton M (1960): A rating scale for depression. J Neurol Neurosurg Psychiatry 23:56 – 62.
Hansen CR, Kroll F, Mackenzie TB (1982): Dehydroepiandros-terone and affective disorders. Am J Psychiatry 139:386 –387. Haug M, Spetz JF, Ouss-Schlegel ML, Baulieu EE, Robel P (1988): The role of dehydroepiandrosterone and preg-nenolone in the expression of stress behavior towards lactat-ing females in mice. Pathol Biol (Paris) 36:995–1001. Hechter O, Grossman A, Chatterton RT Jr (1997): Relationship
Heinz A, Weingartner H, George D, Hommer D, Wolkowitz OM, Linnoila M (1999): Severity of depression in abstinent alcoholics is associated with monoamine metabolites and dehydroepiandrosterone-sulfate concentrations. Psychiatry Res 89:97–106.
Herbert J (1998): Neurosteroids, brain damage, and mental illness. Exp Gerontol 33:713–727.
Howard JS III (1992): Severe psychosis and the adrenal andro-gens. Integr Physiol Behav Sci 27:209 –215.
Lambourn J, Gill D (1978): A controlled comparison of simu-lated and real ECT. Br J Psychiatry 133:514 –519.
Majewska MD (1992): Neurosteroids: Endogenous bimodal modulators of GABAA receptor. Mechanism of action and physiological significance. Prog Neurobiol 38:379 –395. Majewska MD (1995): Neuronal actions of
dehydroepiandros-terone. Possible roles in the brain development, aging, mem-ory and affect. Ann N Y Acad Sci 774:111–120.
Majewska MD, Demigoren S, Spivak CE, London ED (1990): The neurosteroid dehydroepiandrosterone sulfate is an allo-steric antagonist of the GABAAreceptor. Brain Res 526:143– 146.
Majewska MD, Harrison NL, Schwartz RD, Barker JL, Paul SM (1986): Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science 232:1004 –1007. Majewska MD, Schwartz RD (1987): Pregnenolone-sulfate: An endogenous antagonist of the gamma-aminobutyric acid re-ceptor complex in the brain. Brain Res 404:355–360. Maurice T, Junien JL, Privat A (1997): Dehydroepiandrosterone
sulfate attenuates dizocilpine-induced learning impairment in mice via sigma 1 receptors. Behav Brain Res 83:159 –164. McEwen BS (1991): Nongenomic and genomic effects of
ste-roids on neural activity. Trends Pharmacol Sci 12:141–147. Melchior CL, Ritzmann RF (1994): Dehydroepiandrosterone is an anxiolytic in mice on the plus maze. Pharmacol Biochem Behav 47:437– 441.
Mellon SH (1994): Neurosteroid biochemistry: Modes of action and clinical relevance. J Clin Endocrinol Metab 78:1003– 1009.
Meltzer HY (1990): Defining treatment refractoriness in schizo-phrenia. Schizophr Bull 16:563–565.
Osran H, Reist C, Chen CC, Lifrak ET, Chicz-DeMet A, Parker LN (1993): Adrenal androgens and cortisol in major depres-sion. Am J Psychiatry 150:806 – 809.
Overall JE, Gorham DR (1962): The Brief Psychiatric Rating Scale. Psychol Rep 10:799 – 812.
Paul SM, Purdy RH (1992): Neuroactive steroids. FASEB J 6:2311–2322.
Prince RJ, Simmonds MA (1992): 5b-Pregnan-3b-ol-20-one, a specific antagonist at the neurosteroid site of the GABAA receptor complex. Neurosci Lett 135:273–275.
Prudic J, Haskett RF, Mulsant B, Malone KM, Pettinati HM, Stephens S, et al (1996): Resistance to antidepressant medi-cations and short-term clinical response to ECT. Am J Psychiatry 153:985–992.
Purdy RH, Morrow AL, Moore PH, Paul MS (1991): Stress-induced elevations of gamma-aminobutyric acid type A receptor-active steroids in the rat brain. Proc Natl Acad Sci U S A 8:4553– 4557.
Reddy DS, Kaur G, Kulkami SK (1998): Sigma (sigma1) receptor mediated antidepressant-like effects of neurosteroids in the Porsolt forced swim test. Neuroreport 9:3069 –3073. Reddy DS, Kulkarni SK (1997): Differential anxiolytic effects of
neurosteroids in the mirrored chamber behavior test in mice. Brain Res 752:61–71.
Regelson W, Kalimi M (1994): Dehydroepiandrosterone (DHEA)—the multifunctional steroid. Ann N Y Acad Sci 719:564 –575.
Reus VI, Wolkowitz OM, Frederick S (1997): Antiglucocortico-steroid treatments in psychiatry. Psychoneuroendocrinology 22(suppl 1):S121–S124.
Robel P, Baulieu EE (1995a): Dehydroepiandrosterone (DHEA) is a neuroactive neurosteroid. Ann N Y Acad Sci 774:82–110. Robel P, Bauleiu EE (1995b): Neurosteroids: Biosynthesis and
function. Crit Rev Neurobiol 9:383–394.
Robel P, Young J, Corpechot C, Mayo W, Perche F, Hugh M, et al (1995): Biosynthesis and assay of neurosteroids in rats and mice: Functional correlates. J Steroid Biochem Mol Biol 53:355–360.
Romeo E, Stro¨hle A, Spolletta G, di Michele F, Hermann B, Holsboer F, et al (1998): Effects of antidepressant treatment on neuroactive steroids in major depression. Am J Psychiatry 155:910 –913.
Sackheim HA, Decina P, Prohovnik I, Maliz S (1987): Seizure threshold in electroconvulsive therapy. Arch Gen Psychiatry 44:355–360.
Sands D (1954): Further studies on endocrine treatment in adolescence and early adult life. J Ment Sci 100:211–219. Siris SG (1995): Depression and schizophrenia. In: Hirsch SR,
Weinberger DR, editors. Schizophrenia. Oxford, UK: Black-well Science, 128 –145.
Skolnick P, Layer RT, Popik P, Nowak G, Paul IA, Trullas R (1996): Adaptation of N-methyl-D-aspartate (NMDA) recep-tors following antidepressant treatment: Implications for the pharmacotherapy of depression. Pharmacopsychiatry 29:23– 26.
Solan WJ, Khan A, Avery DH (1988): Psychotic and nonpsy-chotic depression: Comparison of response to ECT. J Clin Psychiatry 49:L97–L99.
Spivak CE (1994): Desensitisation and noncompetitive blockade of GABAA receptors in central midbrain neurons by a neurosteroid dehydroepiandrosterone sulfate. Synapse 16: 113–122.
Tollefson GD, Haus E, Garvey MJ, Evans M, Tuason VB (1990): 24-hour urinary dehydroepiandrosterone sulfate in unipolar depressive treatment with cognitive and/or pharmacotherapy. Ann Clin Psychiatry 2:39 – 45.
Uzunov DP, Cooper TB, Costa E, Guidotti A (1996): Fluoxetine-elicited changes in brain neurosteroid content measured by negative ion mass fragmentography. Proc Natl Acad Sci U S A 93:12599 –12604.
Uzunova V, Sheline Y, Davis JM, Rasmusson A, Uzunov DP, Costa E, et al (1998): Increase in the cerebrospinal fluid content of neurosteroids in patients with unipolar major depression who are receiving fluoxetine or fluvoxamine. Proc Natl Acad Sci U S A 95:3239 –3244.
beta-endor-phin, growth hormone, prolactin and cortisol secretion in de-pressed patients. Psychopharmacology 93:122–126.
Wolf OT, Ko¨ster B, Kirschbaum C, Pietrowsky R, Kern W, Hellhammer DH, et al (1997): A single administration of dehydroepiandrosterone does not enhance memory perfor-mance in young healthy adults, but immediately reduces cortisol levels. Biol Psychiatry 42:845– 848.
Wolkowitz OM, Reus VI, Canick J, Levin B, Lupien S (1997a): Gluctocorticoid medication, memory and steroid psychosis in medical illness. Ann N Y Acad Sci 823:81–96.
Wolkowitz OM, Reus VI, Keebler A, Nelson N, Friedland M, Brizendine L, et al (1999): Double-blind treatment of major depression with dehydroepiandrosterone. Am J Psychiatry 156:646 – 649.
Wolkowitz OM, Reus VI, Roberts E, Manfredi F, Chan T, Ormiston S, et al (1995): Antidepressant and cognition-enhancing effects of DHEA in major depression. Dehydro-epiandrosterone and aging. Ann N Y Acad Sci 774:337–339. Wolkowitz OM, Reus VI, Roberts E, Manfredi F, Chan T, Raum WJ, et al (1997b): Dehydroepiandrosterone (DHEA) treat-ment of depression. Biol Psychiatry 41:311–318.
Young J, Corpechot C, Perche F, Eychenne B, Haug M, Baulieu EE, et al (1996): Neurosteroids in the mouse brain: behavioral and pharmacological effects of a 3 beta-hydroxysteroid de-hydrogenase inhibitor. Steroids 61:144 –149.