Uji kualitatif dan
Uji kualitatif dan
kuantitatif DNA dan
kuantitatif DNA dan
RNA
RNA
Uji Kuantitatif
Uji Kuantitatif
Uji kuantitatif DNA dengan spektrofotometri
UV-Vis, DNA murni dapat menyerap cahaya
ultraviolet karena keberadaan basa-basa
purin dan pirimidin. Pita ganda DNA dapat
menyerap cahaya UV pada
260 nm, sedang
kontaminan protein atau phenol akan
menyerap cahaya pada
280 nm.
Sehingga kemurnian DNA dapat dukur
dengan menghitung nilai absorbansi
260
nm dibagi dengan nilai absorbansi
280
(Å260/Å280), dan nilai kemurnian DNA
berkisar antara 1.8-2.0.
Mengukur Konsentrasi DNA/RNA
Mengukur Konsentrasi DNA/RNA
Serta untuk mengukur konsentrasi DNA
digunakan rumus sebagai berikut:
[DNA] = Å260 x 50 x faktor pengenceran
Å
260= Nilai absorbansi pada
260 nm
50 = larutan dengan nilai absorbansi 1.0
sebanding dengan 50 ug untai ganda DNA
per ml (dsDNA)
[RNA] = Å260 x 40 x faktor pengenceran
Uji Kualitatif
Uji Kualitatif
Metoda standar yang digunakan untuk
memisahkan, mengidentifikasi dan
memurnikan fragmen DNA adalah
elektroforesis gel agorose.
Teknik ini sederhana, cepat terbentuk,
dan mampu memisahkan campuran
potongan DNA sesuai dengan ukurannya
secara akurat, dibanding dengan densitas
gradient sentrifugasi.
Selanjutnya, lokasi DNA dalam gel
tersebut dapat diidentifikasi secara
langsung dengan menggunakan pewarna
berfluorescen.
Electrophoresis for nucleic acid
Electrophoresis for nucleic acid
Agarose Gel Electrophoresis
Purification for Specific Fragment of DNA
-DNA Electro-elution
-Electrophoresis onto DEAE-cellulose
membranes
Polyacrylamide Gels
Pulse-field Gel Electrophoresis (PFGE)
11/23/18 fatchiyah, JB-UB 5
Preparing and Running Standard
Preparing and Running Standard
Agarose DNA Gels
Agarose DNA Gels
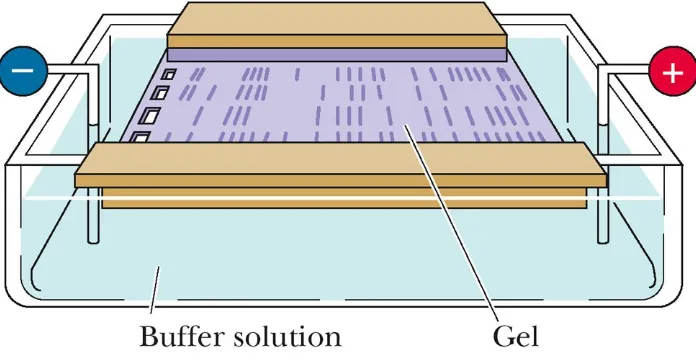
The equipment and supplies necessary for
conducting agarose gel electrophoresis are relatively simple and include:
An electrophoresis chamber and power
supply
Gel casting trays, which are available in a
variety of sizes and composed of
UV-transparent plastic. The open ends of the trays are closed with tape while the gel is being
cast, then removed prior to electrophoresis.
Sample combs, around which molten agarose
is poured to form sample wells in the gel.
Preparing and Running Standard
Preparing and Running Standard
Agarose DNA Gels
Agarose DNA Gels
Electrophoresis buffer, usually
Tris-acetate-EDTA (TAE) or Tris-borate-EDTA
(TBE).
Loading buffer, which contains
something dense (e.g. glycerol) to allow
the sample to "fall" into the sample wells,
and one or two tracking dyes, which
migrate in the gel and allow visual
Preparation of Gel
Preparation of Gel
11/23/18 fatchiyah, JB-UB 8 Ethidium bromide, a fluorescent dye used for
staining nucleic acids. NOTE: Ethidium bromide is a known mutagen and should be handled as a
hazardous chemical - wear gloves while handling.
Transilluminator (an ultraviolet lightbox), which is
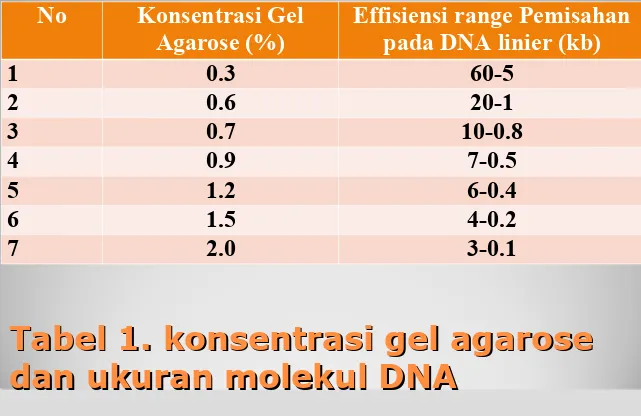
Tabel 1. konsentrasi gel agarose
Tabel 1. konsentrasi gel agarose
dan ukuran molekul DNA
dan ukuran molekul DNA
No Konsentrasi Gel
Agarose (%) Effisiensi range Pemisahan pada DNA linier (kb)
1 0.3 60-5
2 0.6 20-1
3 0.7 10-0.8
4 0.9 7-0.5
5 1.2 6-0.4
6 1.5 4-0.2
DNA and RNA molecules are negatively
charged, thus move in the gel matrix
toward the positive pole (+)
Linear DNA molecules are separated
according to size
The mobility of circular DNA molecules is
affected by their topological structures.
The mobility of the same molecular weight
DNA molecule with different shapes is:
supercoiled> linear> nicked or relaxed
11/23/18 fatchiyah, JB-UB 10
Chemistry of nucleic acids
Migration of DNA Fragments in
Migration of DNA Fragments in
Agarose
Agarose
Fragments of linear DNA migrate through
agarose gels with a mobility that is inversely proportional to the log10 of their molecular weight.
In other words, if you plot the distance from the
well that DNA fragments have migrated against the log10 of either their molecular weights or
number of base pairs, a roughly straight line will appear.
Circular forms of DNA migrate in agarose
distinctly differently from linear DNAs of the
same mass.
Typically, uncut plasmids will appear to
migrate more rapidly than the same plasmid
when linearized. Additionally, most
preparations of uncut plasmid contain at
least two topologically-different forms of
DNA, corresponding to supercoiled forms and
nicked circles.
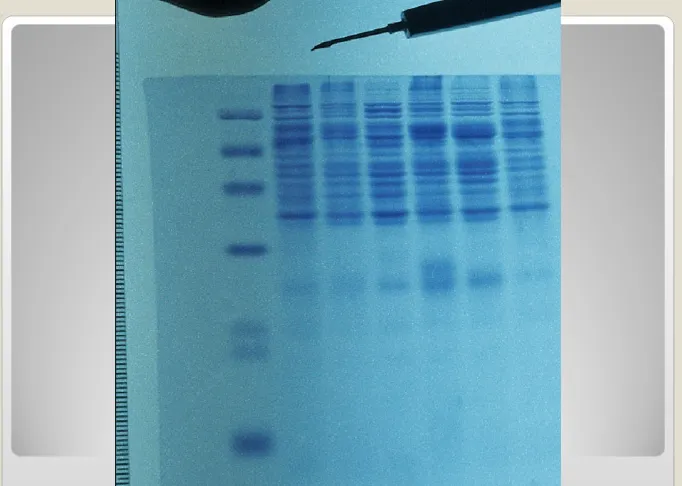
The image to the right shows an
DNA Migration
DNA Migration
11/23/18 fatchiyah, JB-UB 14
large moderate small
Picture of DNA separation by gel electrophoresis
M
M
S
Factors of DNA Migration
Factors of DNA Migration
• Several additional factors have important effects on
the mobility of DNA fragments in agarose gels, and can be used to your advantage in optimizing
separation of DNA fragments.
Chief among these factors are:
Agarose Concentration Voltage
Electrophoresis buffer Effects of Ethidium
Bromide
11/23/18 fatchiyah, JB-UB 15
Purification for Specific
Purification for Specific
Fragment of DNA
Fragment of DNA
In addition to its importance as an analytical tool,
gel electrophoresis is widely used for isolating and then purifying specific fragments of DNA, usually in preparation for subcloning
Several techniques can be used to purify DNA
from agarose gels, and choosing between them is, to some extent, a matter of personal preference. They all start out by excising the desired "band" from an ethidium-stained gel viewed with a UV transilluminator. Because UV light can fragment DNA, it is best to work expeditiously and keep exposure time to a minimum.
DNA Electroelution
DNA Electroelution
Cut out the desired piece of agarose using a
razor blade or scalpel blade, and try to get as little extra agarose as possible.
The block of agarose containing DNA is then
subjected to any of the following. The block of agarose is placed in a piece of dialysis tubing with a small amount of fresh electrophoresis
buffer, the ends sealed with clamps, and the bag placed into an electrophoresis chamber.
Application of current will cause the DNA to
migrate out of the agarose, but it will be trapped within the bag.
DNA Electroelution . .
DNA Electroelution . .
11/23/18 fatchiyah, JB-UB 21 Progress can be monitored using a
transilluminator, as shown below. When the DNA is out of the agarose, the flow of current is
reversed for a few seconds to knock the DNA off of the side of the tubing.
The buffer containing the DNA is then collected
and the DNA precipitated with ethanol.
Electroelution is more time consuming than
22
To separate DNA of different
To separate DNA of different
size ranges
size ranges
Narrow size range of DNA: use
polyacrylamide
Wide size range of DNA: use agarose
gel
Very large DNA(>30-50kb): use
Electrophoresis onto
Electrophoresis onto
DEAE-cellulose membranes
cellulose membranes
At
low concentrations of salt
,
DNA binds avidly to
DEAE-cellulose membranes
.
Fragments of DNA are
electrophoresed in a standard
agarose gel until they resolve
adequately. One then makes a
slit in the gel slightly ahead
of the fragment(s) of interest
and resumes electrophoresis
until all of that fragment has
migrated and stuck onto the
membrane.
Electrophoresis onto
Electrophoresis onto
DEAE-cellulose membranes
cellulose membranes
The membrane is then removed, washed free
of agarose in low salt buffer (150 mM NaCl,
50 mM Tris, 10 mM EDTA), then incubated
for about 30 minutes at 65 C in high salt
buffer (1 M NaCl, 50 mM Tris, 10 mM EDTA)
to elute the DNA.
Progress in binding DNA to the membrane
and eluting it can be monitored with UV light
to detect the ethidium bromide bound to
DNA. After elution, DNA is precipitated with
ethanol.
This procedure is simple and provides very
clean DNA. However, fragments larger than
about 5 kb do not elute well from the
Strand-separating Gels
Strand-separating Gels
For some purposes, eg. sequencing by
Maxam-Gilbert procedure. It is
necessary to obtain separated stands of fragment of DNA. Often this can be
achieved by electrophoresis of denatured DNA through neutral agarose.
The strands of DNA fragment less than
1kb in length are separated on polyacrilamide gel.
Polyacrylamide gel necessary to obtain
separated the each nucleotide of DNA sequence
Polyacrylamide Gels
Polyacrylamide Gels
A commonly-used means of recovering DNA
from polyacrylamide gels is by the so-called "crush and soak" method. The slice of
polyacrylamide containing DNA is crushed in a microcentrifuge using a plastic pipet tip, and
incubated with constant shaking in elution buffer (high salt) at 37ºC for several hours. The
polyacrylamide pieces are then eliminated by
centrifugation or by passing the mixture through a plug of siliconized glass wool. Finally, DNA is recovered by ethanol precipitation.
DNA can also be recovered from polyacrylamide
by use of certain types of silica gel particles, as described above for recovery from agarose.
However, small (< 100 bp) fragments of DNA are very difficult to elute from standard glass particles.
Pulse-field gel Electrophoresis
Pulse-field gel Electrophoresis
(PFGE)
(PFGE)
Ideally, the DNA should separate in straight lanes to
simplify lane-to-lane comparisons.
The original pulsed-field systems used inhomogeneous
electric fields that did not produce straight lanes, making interpretation of gels difficult (Schwartz and Cantor, 1984).
Again, the simplest approach to straight lanes is FIGE,
which uses parallel electrodes to assure a homogeneous electric field.
Although extremely useful for separating relatively small
DNA, 4- 1,000 kb (fig. 2),
FIGE's reorientation angle of 180ø results in a separation
range most useful under 2,000 kb. Furthermore, like other PFGE techniques, FIGE has mobility inversions in which
larger DNA can move ahead of smaller DNA during electrophoresis.
11/23/18 fatchiyah, JB-UB 28
29
pulsed-field gel
pulsed-field gel
electrophoresis
electrophoresis
Switching between two orientations: the
larger the DNA is, the longer it takes to
reorient
PFGE System: Electrode
PFGE System: Electrode
Configuration
Configuration
11/23/18 fatchiyah, JB-UB 30
PFGE Result
PFGE Result
Figure 2. Increased
separation of the 20-50 kb range with field
inversion gel
electrophoresis (FIGE). Run conditions: 230 V, 7.9 V/cm, 16 hrs., 50 msec. pulse,
forward:reverse pulse ratio = 2.5:1, 1% GTG agarose, 0.5X TBE, 10 C.a) 1 kb ladder, 0.5-12 kb; b) Lambda/Hind III, 0.5-23 kb; and c) High molecular weight
markers, 8.3-48.5 kb.