BAB II
TINJAUAN PUSTAKA
2.1 Definisi Cedera Otak
Cedera kepala dapat didefinisikan secara luas yang meliputi setiap hal berikut ini: 1. Bukti riwayat pukulan terhadap kepala
2. Bukti trauma terhadap kulit kepala dalam bentuk bengkak, lecet ataupun memar 3. Bukti patah pada tulang kepala dengan foto schedel atau CT Scan kepala atau
bukti cedera otak dengan CT Scan yang dibuat segera setelah trauma. 4. Bukti klinis patah tulang dasar tengkorak
5. Bukti klinis cedera otak (hilang atau terganggunya kesadaran, lupa ingatan, defisit neurologis, kejang) (Selladurai et al, 2007).
Definisi cedera otak adalah proses patologis pada jaringan otak yang bukan bersifat degeneratif ataupun kongenital, melainkan akibat kekuatan mekanis dari luar yang menyebabkan gangguan fisik, fungsi kognitif dan psikososial yang sifatnya menetap atau sementara dan disertai dengan hilangnya atau berubahnya tingkat kesadaran (Narayan et al, 1996)
2.2 Klasifikasi cedera otak
Berat tidaknya cedera otak paling umum digunakan modalitas dari GCS (Glasgow Coma Scale) post resusitasi, yaitu ringan (GCS 13-15), Sedang (GCS 9-12) dan Berat (GCS ≤8). Bila berdasarkan mekanismenya cedera otak dibagi atas tumpul dan tembus/tajam ( penetrating head injury) (Narayan et al, 1996).
2.3 Patofisiologi cedera otak
Perubahan patofisiologi setelah cedera kepala adalah kompleks. Trauma bisa disebabkan oleh mekanisme yang berbeda, dan sering berkombinasi. Perubahan-perubahan setelah trauma adalah terjadi pada tingkat molekuler, biokimia, seluler, dan pada tingkat makroskopis (Selladurai et al, 2007).
2.3.1 Cedera Otak Primer dan Kontusio Serebri
Cedera otak primer disebabkan oleh kerusakan mekanik pada jaringan otak dan pembuluh darah pada saat terjadinya trauma. Pada tingkat makroskopis bisa terlihat terputusnya jaringan otak; pada tingkat mikroskopik bisa terlihat kerusakan parenkhim sel (sel neuron, axon, dan glia) dan mikrosirkulasi (arteriol, capiler, dan venula) (Selladurai,et al,2007).
Kontusio serebri adalah tipe kerusakan otak fokal terutama disebabkan oleh kontak antara permukaan otak dan tonjolan permukaan tulang dasar tengkorak, menuerut ICD-9 kontusio cerebri adalah luka memar pada otak akibat tubrukan / impact terhadap kepala atau suatu trauma acceleration/deceleration (Narayan et al,1996).
Diantara banyak peristiwa molekouler paskacedera otak hal yang paling penting adalah Sur-1 yang memberi kontribusi berkembangnya kontusio serebri. Secara umum area kontusio serebri dibagi tiga yaitu; Epicenter, Pericotusional penumbra, dan Parapenumbra area. Pada epicenter terputusnya pembuluh darah terjadi segera. Pada penumbra dan parapenumbra area pukulan energi tidak merobek jaringan, tetapi mengawali peristiwa molekuler sensitif-mekanik yang mengiduksi overexpresi dari Sur-1. Sur-1 adalah regulator subunit dari non-selektif kation channel (NCCa-ATP) yang ditemukan oleh Simard group dan berimplikasi pada patophisiologi edema serebri dan bertransformasi dari kontusio menjadi hemoragic. Induksi overekspresi Sur-1 meningkatkan pembengkakan sel dan kematian onkotik sel astrocyte, neuron, dan sel endothelial. Pecahnya endotelial sel mengakibatkan microhemoragic yang berakibat terbentuknya perdarahan baru dan konsekuensi perdarahan menjadi progresif pada traumatik kontusio serebri ( Kurland.D., 2012).
Gambaran CT scan pada kontusio serebri lokasi biasanya tanpak pada permukaan korteks dan terlibat gray matter, pada sentral area terlihat hiperdense dan bercampur dengan area hipodense yang merupakan bagian dari hemoragic necrosis atau bagian jaringan otak yang rusak dan bagian otak yang edema (pericontusional edema) ( Selladurai.B., 2007).
Tidak ada aliran darah pada area sentral kontusio serebri dan pengurangan aliran darah pada daerah perikontusional edema, dimana autoregulasi terganggu (vasoparalysis). Oleh karena itu pada daerah perilesional ada kerusakan parsial sel yang rentan terhadap setiap pengurangan perfusi oleh pengurangan MAP (mean arterial pressure), peningkatan tekanan intrakranial atau vasokonstriksi setelah hipocapnia akibat dari hiperventilasi ( Selladurai.B., 2007).
Perkembangan dari lesi kontusio serebri adalah (1) Komponen perdarahan berkembang; penyatuan fokus –fokus perdarahan kecil dapat terjadi; komponen perdarahan dari kontusio serebri dapat mencapai maximal dalam waktu 12 jam pascatrauma pada 84% pasien; koangolopati dan alkoholik dapat memperbesar risiko bertambahnya komponen perdarahan pada kontusio serebri, (2) Meningkatnya pembengkakan zona sentral kontusio dan zona perikontusional; kerusakan parsial sel parenkim pada sentral kontusio juga pada zona perikontusional bisa menyebabkan bengkak (cytotoxic edema). Pada area nekrotik dari kontusio makromolekuler yang didegradasi menjadi molekul yang lebih kecil dapat meningkatkan osmolaritas jaringan dan bisa menyebabkan perpindahan cairan dari intravasculer ke area necrosis kontusio (osmolar edema). Pembengkakan area sentral kontusio menyebabkan penekanan zona perikontusional dan menyebabkan iskhemik lebih lanjut dan edema. Perikontusional edema dapat mencapai maximal 48-72 jam setelah cedera ( Selladurai.B., 2007).
2.3.2 Cedera Otak Sekunder
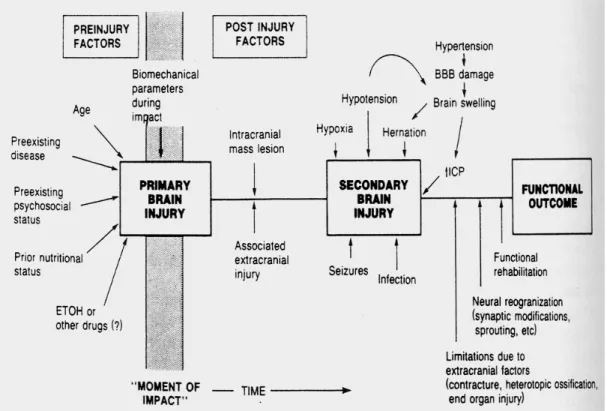
Cedera otak sekunder merujuk kepada efek setelah peristiwa cedera primer, secara klinis efek diaplikasikan setelah postraumatik hematom intrakranial, edema otak dan peningkatan tekanan intrakranial dan pada fase lebih lambat hidrocephalus dan infeki. Cedera otak sekunder adalah peristiwa sistemik yang terjadi setelah trauma yang potensial cedera ini dapat menambah kerusakan neuron, axon, dan pembuluh darah otak. Cedera otak sekunder yang terpenting adalah hipoxia ,hipotensi, hipercarbia, hiperexia, dan gangguan elektrolit (Selladurai et al, 2007).
Gambar 1 : Faktor-Faktor yang memengaruhi Prognosis Setelah Cedera Otak (Vollmer.D.G., 1993).
2.4 Penilaian Tingkat Kesadaran pada Cedera Otak
Teasde dan Jannet telah mengevaluasi secara hati-hati pasien-pasien dengan cedera kepala dan gangguan kesadaran. Hasil yang mereka kembangkan telah dikenal sebagai Glasgow Coma Scale pada 1974. Pada skala ini dilnilai tingkat numerik respon buka mata, motoric, dan verbal dengan rentang nilai 3-15 (Becker et al, 1989).
a. Respon buka mata Nilai
Spontan 4
Atas perintah / suara 3
Rangsangan nyeri 2
Tidak ada 1
b. Respon Motorik Nilai
Menurut perintah 6
Melokalisir nyeri 5
Fleksi normal (menarik anggota yang dirangsang) 4
Fleksi abnormal (dekortikasi) 3
Ekstensi abnormal (deserebrasi) 2
Tidak ada (flasid) 1
c. Respon bicara Nilai
Berorientasi baik 5
Berbicara mengacau / bingung 4
Kata-kata tidak teratur 3
Suara tidak jelas 2
Tidak ada 1
Tabel.1. Diambil dari: American College of Surgeons 1997, Advance Trauma Life Support Program Student Manual, Komisi Trauma ”IKABI” (Ikatan Ahli Bedah Indonesia), 6th ed, Komisi Trauma ”IKABI”, Jakarta.
Skala lain yang bisa dipakai untuk mengukur keparahan cedera kepala adalah Glasgow Liege Scale, Glasgow Pittsburg Coma Scoring system, Head Injury Watch Sheet, Maryland Coma Scale, Leeds Coma Scale dan Glasgow Coma Scale. Kelebihan GCS adalah cukup konsisten dan objektif ketika dilakukan oleh penilai yang berbeda, sederhana dan berguna sebagai pedoman terapi dan memberi informasi tentang prognosis (Stein, 1996). Kendala GCS antara lain adalah jika penderita mengalami edema palpebra atau terintubasi, ada variabel yang tidak bisa dinilai (Feldman, 1996).
2.5 Terapi Standar pada Cedera Kepala Sedang 1. Pemberian antibiotika bila ada luka,
2. Pemberian analgetik NSAID,
3. Pemberian sedatif/transquilizer bila diperlukan untuk memperbaiki kenaikan TIK dan penenang,
4. Pemberian manitol untuk menurunkan TIK secara bolus 0,25-1 gram/kgBB, serum osmolaritas harus diperiksa bawah 320 mmol/l untuk mencegah gagal ginjal,
5. Pemberian nutrisi dini secara bertahap yang harus tercapai untuk kebutuhan total dalam waktu 7 hari setelah trauma, adalah 140% dari kebutuhan basal pada pasien yang tidak dilumpuhkan dan yang diberikan secara parenteral dan enteral, sedikitnya 15% dari asupan energi harus mengandung protein,
6. Pemberian Gastric Mucosal Protector dan Acid Supressor Agent dengan H2 Blocker dan pemberian PPI (proton Pump Inhibitrt) yang dapat menurunkan insiden perdarahan gastrointestinal dan stress related mucosal damage (SRMD).
Bagan.1. Penatalaksanaan Cedera Kepala Sedang Nonoperatif
2.6 Skala Prognosis Glasgow(Glasgow Outcome Scale=GOS)
Glasgow outcome scale (GOS) paling luas digunakan untuk menilai hasil akhir secara umum pada cedera otak GOS dikelompokkan dalam lima katagori: mati, persistent vegetative state, ketidakmampuan yang berat, ketidakmampuan sedang, dan kesembuhan yang baik. Penilaian secara tepat diperoleh pada 3, 6, dan 12 bulan setelah cedera otak. Validitas GOS sebagai suatu penilai hasil akhir cedera otak didukung oleh kuatnya hubungan dengan lamanya koma, beratnya kondisi pada awal trauma (diukur dengan GCS), dan tipe lesi intrakranial. GOS katagori juga berkorelasi dengan lamanya postraumatik amnesia. Kritikan terhadap GOS relatif tidak sensitif terhadap kondisi
pasien yang membaik secara signifikan dan secara klinis terutama 6 bulan setelah cedera otak (Narayan et al, 1995).
Skala pengukuran GOS ini pertama kali ditemukan oleh Jennet dan Bond pada tahun 1975. Prognosis pascacedera otak yang didasarkan kapabilitas sosial pasien pascacedera otak dikombinasikan dengan efek mental spesifik dan defisit neurologis. Derajat skala ini mencerminkan suatu kerusakan otak secara umum, dimana juga mampu menilai prognosis pascakoma traumatik ataupun nontraumatik (Bullock, 2004; Narayan, Michel,2002; Jennet, 2005).
Telaah pada penderita adalah sebanyak 150 orang yang bertahan hidup setelah cedera otak di Glasgow oleh spesialis saraf dan bedah saraf . Keduanya memutuskan bahwa penilaian ini sangat tepat pada 3 bulan, 6 bulan, dan 12 bulan pascatrauma (Jennet, 2005).
Skala penilaian prognosis Glasglow terdiri atas lima kategori (Jennet ,2005)
(1) Pemulihan baik (good recovery= GR) diberi nilai 5.Pasien dapat berpartisipasi pada kehidupan sosial, kembali bekerja seperti biasa. Pemeriksaa ini dapat disertai komplikasi neurologis ringan, seperti defisit minor saraf kranial dan kelemahan ekstremitas atau sedikit gangguan pada uji kognitif atau perubahan personal. (2) Ketidakmampuan sedang (Moderate disability=MD, independent but disabled)
diberi nilai 4. Kondisi pasien jelas berbeda sebelum cedera dan mampu menggunakan transportasi umum, tetapi tidak dapat bekerja seperti biasa. Pasien defisit memori/perubahan personal, hemiparesis, disfasia, ataksia, epilepsi paska traumatika, atau defisit mayor saraf kranial. Derajat ketergantungan pasien pada orang lain lebih baik dibandingkan dengan lansia dan kemampuan kebutuhan personal sehari-hari dapat dikerjakan tetapi, mobilitas dan kapasitas berinteraksi tidak dapat dilakukan tanpa asisten.
(3) Ketidakmampuan berat (Severe disability=SD, conscious but dependent) diberi nilai 3. Pasien mutlak bergantung pada orang lain setiap saat (memakai baju, makan, dll), paralisis spastik, disfasia, disatria, defisit fisik dan mental yang mutlak memerlukan supervisi perawat/keluarga.
(4) Vegetative State=PVS diberi nilai 4. Pasien hanya mampu menuruti perintah ringan saja atau bicara sesaat. Pada perawatan sering ditemukan grasping reflek,
withdrawal sebagai pencerminan menuruti perintah, mengerang, menangis, kadang mampu mengatakan tidak sebagai bukti proses kembali berbicara.
(5) Meninggal dunia (dead) diberi nilai 1. Pada tahun 1981 Jennet menelaah dan memodifikasi ulang skala GOS karena masalah sensitivitas statistik dan penggunaan yang lebih praktis pada uji klinis obat neuroproteksi, yaitu distribusi bimodal (dikotomisasi) antara hidup (GR, MD, SD) dan mati (PVS, Dead) dan penilaian ekstensi (GOS Extended), yaitu
GOS asli Nilai GOS – E Nilai
Meninggal dunia 1 Meninggal dunia 8
Status vegetative 2 Status vegetative 7
Ketidakmampuan Berat 3 Ketidakmampuan berat
Ekstremitas atas
Ekstremitas bawah
6
5
Ketidakmampuan Sedang 4 Ketidakmampuan sedang
Ekstremitas atas
Ekstremitas bawah
4
3
Pemulihan Baik 5 Pemulihan baik
Ekstremitas atas
Ekstremitas bawah
2
1
Tabel. 2. Nilai GOS Asli dan Extended 2.7 Skala Fungsional Barthel’s Index
Skala Barthel atau Index ADL (Activities of Daily Living) Barthel merupakan suatu skala untuk mengukur kemampuan seseorang dalam melakukan aktivitas dasar dan mobilisasi. Semakin tinggi nilai yang diperoleh dalam pemeriksaan, semakin tinggi pula kecenderungan atau kemampuan seseorang untuk hidup mandiri setelah pulang dari rumah sakit. Skala pengukuran ini diperkenalkan pada tahun 1965 oleh Mahoney dan
Barthel dengan manampilkan rentang penilaian dari 0-20. Meskipun versi aslinya telah dipergunakan secara luas, skala ini telah mengalami modifikasi oleh Granger dkk pada tahun 1979 menjadi 0-10 point untuk tiap variabelnya dan perbaikan selanjutnya diperkenalkan pada tahun 1989. Skala ini dikenal cukup reliable (Mahoney, Barthel ,1965). Barthel index diukur pada saat awal terapi dan secara berkala selama terapi sampai diperoleh keuntungan yang maksimum (Mahoney and Barthel, 1965).
2.7.1 Penilaian Kondisi Mental Sederhana
The mini mental state examination(MMSE), digunakan untuk screening gangguan cognitive. Setiap nilai lebih atau sama dengan 25 adalah efektif normal, gangguan cognitive berat (≤ 9),sedang (10-20) dan ringan (21-24). MMSE merupakan suatu skala terstruktur yang terdiri atas tiga puluh poin yang dikelompokan menjadi tujuh kategori: orientasi terhadap tempat (negara, provinsi, kota, gedung dan lantai), orientasi terhadap waktu (tahun, musim, bulan, hari, dan tanggal), registrasi (mengulang dengan cepat tiga kata), perhatian dan konsentrasi (secara berurutan mengurangi tujuh, dimulai dari angka seratus, atau mengeja kata WAHYU secara terbalik), mengingat kembali (mengingat kembali tiga kata yang telah diulang sebelumnya), bahasa (memberi nama dua benda, mengulang kalimat, membaca dengan keras dan memahami suatu kalimat, menulis kalimat dan mengikuti perintah tiga langkah), dan kontruksi visual (menyalin gambar) (Lezak, 2004; Tombaugh, 1992).
2.8 Mediator-Mediator dan Mekanismenya pada Cedera Otak Sekunder 2.8.1 Nekrosis/Apoptosis
Nekrosis sel terjadi sebagai respon terhadap toxic atau cedera fisik dan iskemik. Nekrosis dikarakteristikkan dengan pembengkakan sel dan kerusakan membran yang berkaitan dengan lisis nuclear kromatin. Ketika kelompok-kelompok sel terlibat secara simultan, isi sel yang banyak tumpah dalam jaringan yang cedera dapat membangkitkan respon inflamasi dalam area lokal. Apoptosis adalah kematian sel yang terprogram yang terjadi dengan respon terhadap aktifasi dari sinyal sel dan juga terlihat memberi konstribusi terhadap kematian sel SSP setelah cedera otak. Kematian dengan mekanisme apoptosis secara normal digunakan dalam perkembangan dan mempertahankan populasi sel. Berbeda dengan kematian karena nekrosis sel membengkak dan pecah. Ketika sel mengalami apoptosis, sel menjadi menciut dan integritas membran dipertahankan sampai akhir setelah kematian sel. Bangkai sel apoptosis mengandung sisa sitoplasma, organella, dan nucleus cromatin dihilangkan dan difagosit. Kematian sel dengan proses apoptosis yang memerlukan energi, sedangkan kematian sel karena nekrosis karena tidak adekuatnya persediaan energi (Hatton J, 2001).
Kebutuhan energi yang sangat banyak meningkat cepat setelah cedera otak dan protokol resusitasi setelah cedera otak meningkatkan kemungkinan bahwa terdapat lebih dari satu mekanisme kematian sel. Sel dapat merespon bermacam-macam ransangan stres dan kekacauan metabolisme yang dapat memicu program apoptosis. Zat yang merusak DNA dan zat kimia tertentu yang dapat mengaktifkan mekanisme memerintahkan sel untuk apoptosis. Mekanisme ini termasuk up-regulasi protein apoptosis. Misalnya, aktivasi dari Caspase. Sekali caspase cascade dimulai, proses kematian sel tidak dapat dibalikkan lagi. Walaupun data yang ada menunjukkan bahwa sekali caspase telah diaktifkan, cedera sel otak tidak dapat distop lebih jauh dengan intervensi farmakologi. Akan tetapi caspase antagonis telah memperlihatkan efek neuroproteksi pada model yang berbeda dari iskemik otak (Alzheimer, 2002)
Gambar. 2. Beberapa Gambaran Cedera Apoptosis pada Neuron(Alzheimer, 2002: 23)
2.8.2 Reperfusi Injuri/Cytokines
Sintesis proinflamasi cytokines, aktivasi leukosit, vasogenik edema, dan kerusakan sawar darah otak adalah salah satu yang bisa memberi konstribusi edema paskacedera otak. Perubahan sinyal untuk repair, regenerasi dan proteksi telah dilaporkan dengan reperfusi yang berkaitan dengan respon inflamsi. Proinflamasi cytokine bisa memperberat iskemik pada cedera CNS melalui efek lansung pada neuron, astrosit dan sel mikroglial, atau melalui induksi molekul proinflamsi lain seperti TNFα dan interleukin-1β. Mereka terlihat langsung memodulasi apoptosis sel CNS, dan differensiasi dan, proliferasi dan memengaruhi infiltrasi leukosit. Cytokine juga terlibat dalam produksi protein untuk apoptosis. Aktivasi NFκB menyebabkan up-regulasi cyclo-oxygenase-2(COX-2), intercellular adhesion molecule 1 (ICAM-1)dan, IL-1 β, IL-6, dan juga dapat menginduksi sintesa Nitric Oxide (NOS), TNFα dan Fas Ligand (Hatton.J., 2001).
Interaksi di antara mediator-mediator ini menyebabkan siklus terus berlansung cedera sekunder, necrosis , dan apoptosis. Infiltrasi sel mononuclear dapat dijumpai dalam 6-12 jam pascaiskemik fokal SSP. Cytokines produksi terjadi 12 jam sekunder terhadap infiltrasi monosite. TNFα mRNA dihasilkan dalam 1 jam iskemik dan mencapai
puncak dalam 6-12 jam pascaiskemik dan menurun kembali dalam 1-2 hari. Bukti yang dihasilkan sejauh ini mengesankan bahwa obat yang menekan produksi TNFα akan mengurangi infiltrasi leukosit dalam area iskemik otak dan mengurangi kehilangan jaringan. Pada hewan percobaan cedera otak tertutup, inhibisi TNFα memberikan suatu neuroproteksi. Pentoxifilline telah digunakan untuk mengurangi produksi TNFα dan berhasil menurunkan TNFα otak 80%. Setelah iskemik CNS peningkatan produksi IL-6 terlihat menonjol pada daerah yang kehilangan sel-sel neuron (Hatton J, 2001).
Kerja IL-6 telah dilaporkan sebagai neuroprotektif dan juga sebagai neurotoxic. IL-6 mempromosikan ketahan hidup sel neuron dan menghambat NMDA yang terinduksi toxin in vitro. Konsentrasi yang tinggi dari IL-6 bisa berperan sebagai prediktor pemulihan fungsional pasien dan berkorelasi dengan ukuran infark. Pada reperfusi IL-6 memberi konstribusi terhadap produksi ICAM-1. IL-1 β , IL-6, dan TNFα dapat meningkatkan ekspresi ICAM-1 pada sel endotelial dan astrocyte, memfasilitasi infiltrasi leukosit, dan meningkatkan aktivasi leukosit. Eselectine dan ICAM-1 ter up-regulasi pada endotelial cerebrovascular pasca kontusio fokal otak pada tikus. ICAM-1 antagonis telah memberi keuntungan melawan apoptosis neuron pada fokal iskemik otak (Hatton J,2001).
Gambar. 3. Mekanisme dan Mediator-Mediator Sekunder pada Cedera Saraf LKT = leukotrienes;NMDA = N-methyl-D-aspartate; PG = prostaglandins (Hatton , 2001).
2.8.3 Excitotoxicity/Glutamate
Walaupun excitotoxic neurotrasmitter yang lain ada, glutamate adalah penyebab paling dasar terhadap profile toxicity yang berkaitan dengan cedera otak. Ketika kultur
sel neuron dipapar sementara dengan glutamte, aktifasi NFκB terjadi dan ekspresi gen proapoptosis yang ter-upregulasi menyebakan kematian sel. Glutamat yang berlebihan dengan cepat merusak neuron postsynaptik karena influx kalsium berlebihan. Glutamat dapat mengaktifkan NMDA ,α- amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA), dan reseptor kainate. Pada aktivasi AMPA atau reseptor kainate, ion channels terbuka dan memungkinkan sodium, potassium, dan hidrogen masuk kedalam sel. Pembengkakan sel terjadi karena pergeseran osmotik cairan dan masuknya sodium dengan cepat dapat mendepolarisasi membran sel. Blok pada tipe reseptor AMPA dan kainate telah memperlihatkan keuntungan pada iskemik fokal dan global hewan percobaan. Aktivasi glutamat pada NMDA reseptor yang membuka kunci ion channel dapat menyebabkan peningkatan kalsium dan sodium (Hatton J, 2001).
Dalam keadaan normal, aktivasi reseptor NMDA kompleks yang melibatkan ikatan glutamat dan glysin diperlukan depolarisasi sel yang cukup untuk melawan penghambatan dari magnesium. Ketika teraktivasi oleh glutamat, magnesium bergeser dari channel dan memungkinkan secara elektris diisi ion kalsium. Masuknya ion kalsium yang banyak mengubah aktivitas elektris neuron dan stimulasi sinyal konduksi mengaktifkan neuron-neuron yang berdekatan. Ketika transportasi kalsium terjadi secara berlebihan, seperti pada cedera akut dan level glutamat yang cepat meningkat, neuron bengkak dan pecah. Glutamat melebur kembali pada proses ini dan siklus excitotoxicity terus berlansung. Glysine telah diistilahkan sebagai suatu coagonis karena ia berperan dalam memfasilitasi aktivasi glutamat yang teriduksi. Pada model cedera otak diffuse tanpa sequele sekunder, glutamat hanya meningkat untuk sementara. Dampak pada model cedera dengan sequele sekunder memperlihatkan peningkatan cepat glutamat ke dalam cairan ekstraseluler. Meskipun pada pasien dengan cedera otak levelnya 10-50 kali lipat lebih tinggi daripada nontrauma, Peningkatan ini telah diobservasi selama 96 jam pascacedera otak. Sejumlah target obat pada tempat ikatan NMDA glisine dan yang memengaruhi pelepasan glutamat presinaptik memperlihatkan neuroproteksi pada hewan percobaan dengan iskemik dan cedera axonal diffuse. Reseptor opoid-κ agonis terlihat menekan pelepasan glutamat presinaptik dan telah diperiksa secara klinis. Modulator presinaptik lainnya adalah termasuk endoline, sodium channel antagonis, dan agonis reseptor adenosine. Glisine antagonis meperlihatkan beberapa potensi dan memunyai
toleransi yang baik dibandingakan dengan glutamat antagonis. Glutamat terus menerus menjadi target strategi terapi yang diteliti dan sering digunakan sebagai marker rujukan respon obat (Hatton.J., 2001).
Bagan. 2. Induksi Glutamate pada Cedera Saraf Akut (Alzheimer, 2002).
2.8.4 Kalsium
Setiap terjadi iskemik otak, N-type voltage-sensitif calsium channel terbuka. Hal ini memungkinkan kalsium dan sodium masuk ke dalam terminal neuron yang cedera. Peningkatan kalsium intraseluler memberi konstribusi terhadap depolarisasi membran dan kerusakan saraf terminal. Dalam kondisi normal, kalsium di dalam sitosol sel ditranspor keluar sel dengan pompa membrane yang bergantung pada energi atau dibuang oleh mitokhondria dan retikulum endoplasma. Metabolisme seluler diturunkan sehubungan dengan ikatan kalsium dengan membrane mitokhondria dan berlawanan dengan transpor elektron dan produksi ATP. Hilangnya sumber daya energi sel dapat, mengeluarkan sinyal kematian sel dan melapaskan toksin sehingga siklus cedera otak berlansung terus. Phospholipase mengaktifkan sinyal untuk merusak phospholipid dan menghasilkan kerusakan dinding membran sel yang mengandung phospholipide dan juga pada organella interna. Perubahan struktur yang terjadi pada phosphatide, phosphat
anorganik, dan asam amino merupakan petanda sudah terjadi pelepasan. Hal ini kemudian memicu mekanisme internal selular untuk siklus kematian sel (Hatton.J., 2001).
Sinyal dari kerusakan axon secara lambat dapat terlihat dalam 2-3 jam setelah cedera otak dan menetap selama 24 jam atau lebih. Peningkatan kalsium intraseluler dan bisa mengaktifkan protease netral spesifik kalsium. Kerja protease dapat menyebabkan peningkatan jangka lama pada calpain-induced spectrin degradation breakdown products, kerusakan sel dan perubahan tingkah laku. Degradasi yang termediasi kalsium dari cytoskleton dianggap menjadi penting pada cedera axonal secara lambat yang mana kerusakan menjadi lebih buruk pada 24 jam pertama pascacedera otak. Actine adalah suatu protein struktur besar polymerase dari bentuk microfilamen yang dapat membatasi masuknya kalsium dari voltage-gated channel atau NMDA. Gelsoline suatu enzyme intraseluler yang menghambat masuknya kalsium lebih banyak lagi. Gelsoline memotong microfilamen dan menggangu fungsi sel normal. Gelsoline juga bisa menjadi mediator apoptosis karena dia secara spesifik diaktifkan oleh caspase-3 sebagai enzyme kunci pada cascade apoptosis (Hatton.J., 2001).
Fungsi mitokhondria adalah untuk mempertahankan kalsium intraseluler dan perlindungan sel. Kalsium masuk ke dalam sel dengan low capasitor antiporter atau electronic uniporter. Pompa mitokhondria mengeluarkan kalsium ketika kadar kalsium dalam sitosol tinggi. Masuknya kalsium yang berlebihan setelah aktivasi glutamat pada NMDA reseptor menyebabkan cedera sekunder pada neuron. Pada kondisi normal mitokhondria terlindungi dari cedera cytotoxic oleh akumulasi kalsium ketika terpapar oleh glutamate. Cyclosporin telah diteliti potensialnya terhadap neuroproteksi karena efeknya pada mekanisme pengangkutan kalsium di mitochondria (Hatton.J., 2001).
2.8.5 Radikal Bebas
Kerusakan membran phopholipid merupakan kerusakan sekunder terhadap aktivasi phospholipase-C yang dapat menghasilkan spesies radikal bebas yang sangat reaktif. Radikal bebas adalah produk sampingan yang tidak bisa dielakkan dari proses redok transfer-elektron melalui reaksi enzymatik dan nonenzimatik. Enzyme redok paling
banyak menghasilkan hidrogen peroksida dalam otak. Pengubahan hidrogen peroksida ke hydroxiyl radikal dikatalisa oleh metal transisi (contoh: besi dan tembaga). Radikal reaktif OH, Lipid peroxyl radikal, thio atau thio-ferro radikal yang merusak sel dengan menyebakan:
1. Peroksidasi lemak
2. Oksidasi protein atau proteolysis 3. Mengurangi adenosine triphosphate 4. Memecahkan DNA
Enzyme antioxidant endogen meliputi superoxida dismustase(SOD), Catalase, gluthation peroxidase dan reduktase, thiol-spesifik antioxidan enzyme, thioredoxin dan protease inhibitor. Antioxidatif seluler ini adalah sistem perlindungan yang secara aktif melindungi sel otak dan neuron dari cedera oxidant. Otak mengandung sejumlah besar asam lemak polyunsaturasi, target untuk peroxidase, dan pembentukan species radikal bebas reaktif. Selama iskemik, katekolamin oksidasi, extravasasi oksidasi haemoglobin, neutrophil infiltrasi, dan nitric oxide memberi konstribusi dalam mempercepat produksi radikal bebas. Ketersediaan nitric oxide selama iskemik juga meningkatkan potensial toxisitas dari superoxida radikal. Antioxidan endogen normal, superoxida dismustase, catalase dan gluthathion peroxidase tidak dapat sepenuhnya menetralisasi reaksi ini dalam keadaan iskemik. Seluruh produksi radikal ini, peroxidasi lemak secara lansung merusak membrane sel dan juga mengubah kontraktilitas vaskuler dan mengurangi aliran darah. Setiap radikal cascade diakhiri, anyaman menyilang di dalam membrane atau dengan komponen sel lain dapat terjadi. Hal ini bisa menyebabkan inflamasi sel, edema, dan merubah sistem enzyme. Perubahan ini memengaruhi permiabelitas seluler dan merubah kemotaxis sehingga menyebabkan kerusakan tidak langsung. Radikal bebas mengawali peroxidasi membrane sel, melibatkan myelin, dikatalisa oleh ion besi bebas yang dilepas oleh hemoglobin, transferin, dan ferritin oleh setiap penurunan PH atau oksigen jaringan (Chieueh.C., 1999).
Peningkatan induksi kalsium dapat melepaskan radikal bebas dari mitokhondria dan memicu kalsium teraktivasi protease dan lipase. Hal ini menyebabkan degradasi asam arachidonat menjadi phospholipase A2 aktif, lipoxigenase dan COX, dan menyebabkan
produksi thromboxan A2, Prostaglandin, dan leukotrine. Hal ini tidak begitu jelas apakah peristiwa ini menyebabkan kerusakan yang sama pada pembuluh darah dan neuron. Nitic oxide dihasilkan in vivo pada sel endotelial, astroglia dan sedikit pada neuron dengan tiga bentuk berbeda dari NOS. Nitric oxide bisa berkerja melalui second messengers, contohnya, cyclic guadinosine monophosphate (GMP), yang menyebabkan sinyal neurogenik vasodilatasi. NFκB dan IL-1 dapat mengiduksi sinyal NOS (iNOS) dapat menghasilkan Nitric Oxide. Ada bukti menunjukkan bahwa aktivasi reseptor NMDA bisa meransang pembentukan Nitric Oxide. Nitric oxide telah berakibat pada pelepasan ikatan phosphorilasi oxidatif pada mitokhondria, memicu apoptosis, dan pengurangan produksi energi melalui aktivasi polyadenosin diphosphate (ADP)-ribose sintetase (PARS). Kadar nitrit dan nitrat (produk stabil nitric oxide) dalam CSF meningkat antara 30 dan 42 jam setelah cedera otak manusia. Lubeluzol, yaitu suatu obat yang diteliti dapat menghambat induksi glutamat pada cedera otak dan diusulkan sebagai mekanisme yang terlibat dalam patway nitric oxide intraseluler. Nitric oxide adalah sumber untuk produksi radikal bebas dan NO. Radikal ini dihasilkan selama iskemik dan secara umum telah dikenal sebagai neurotoxic (Chieueh.C., 1999).
2.8.6 Ekspresi Gen, Sintesa Protein dan Growth Factors
SSP memunyai mRNA hampir 30.000 gen dan diperkirakan ada 20.000 protein berbeda di otak. Semua sintesis protein berkurang setelah cedera otak, tetapi urutan mRNA baru diekspresikan dan protein tertentu secara khusus disintesis. Dalam hitungan menit setelah cedera otak, stimulasi glutamat dari NMDA reseptor meningkatkan kalsium intraseluler atau degradasi membran lemak yang memicu up-regulasi kelompok stres protein dan yang melewati area cedera akut. Stress protein (heat-shock protein ) berkerja memobilisasi pertahanan sel dan stabilisasi cytoskleton. Gen tertentu dianggap terlibat dalam dalam respon yang lebih lambat terhadap cedera otak dan efek nya meliputi proteksi dan pencegahan apoptosis. Gen growth factor sudah ada dalam beberapa jam cedera kepala, tetapi produksi protein memerlukan waktu beberapa hari. Banyak neurotrophic growth factor telah diidentifikasi. Selain itu, dikenal dengan baik nerve growth factor(NGF). Yang lainnya termasuk Brain- Derivate Neurotrophic Factor(BDNF), Insuline Like Growth Factor-1(IGF-1), Glial Derived Neurotrophic
Factor(GDNF), dan Neurotrophic Factor-3), neurotrophic factror bisa meng up-regulasi sintesis protein baru untuk berkerja menguragi efek kerusakan akibat masuknya ion kalsium dalam jumlah banyak yang menyebabkan sekunder injury. Ada bukti dari percobaan in vitro bahwa growth factor dapat memproteksi neuron untuk melawan cedera dari energi yang hilang atau kalsium yang berlebihan. Transmisi growth factor bisa dipengaruhi oleh kerusakan axon setelah cedera otak. Pada manusia dengan cedera otak, kadar sistemik IGF-1 endogen menurun setelah cedera otak dan tetap rendah sampai 14 hari. IGF-1 yang ada sekarang hanya growth factor yang diperiksa secara klinis untuk potensial neuroprotektif nya pada pasien dengan cedera otak (Hatton.J., 2001).
2.9 Strategi Farmakologi Pemilihan Neuroproteksi
Mekanisme kematian sel hal penting untuk meningkatkan mamfaat ketika dipertimbangkan intervensi target terapi. Mediator inflamasi, cytokine, growth factor, glutamate, dan neurotrasmitter lainnya telah dieksplorasi sebagai tempat yang potensial untuk memutuskan siklus kematian sel setelah cedera otak akut. Obat yang menghambat reseptor untuk mediator ini berusaha memutuskan siklus kerusakan sel yang terus berlangsung dengan cara menghambat sinyal eksternal inflamasi sel. Banyak sekali susunan mekanisme dan mediator-mediator berperan terhadap komplikasi berkembang trauma otak sekunder yang menjadi latar belakang strategi efektif untuk pemilihan obat neuroproteksi. Mekanisme terapi yang berkerja tunggal untuk usaha beberapa multipel proteksi mungkin tidak adequate. Kemungkinan target untuk intervensi adalah termasuk antagonis kalsium channels sensitif-voltage, reseptor antagonis NMDA, glysine, atau reseptor antagonis polyamine, antagonis radikal bebas atau scavengers, modulator leukosit, dan growth faktor atau gen terapi. Tantangan terapi meliputi klarifikasi target sinyal yang paling sesuai, waktu pemberian, metode pemberian, dosis optimal yang akan mencapai konsentrasi sistemik atau sentral yang mampu membangkitkan suatu respon yang diinginkan di susunan saraf pusat (Hatton.J., 2001).
2.9.1 Apoptosis Inhibibisi
Target obat pada program sel bunuh diri dalam kondisi iskemik meliputi inhibisi caspase, inhibisi protein modulator neuronal apoptosis(NAIP, dan inhibisi poly (ADP-ribose) polymerase (PARP) .Aktifasi Caspase-3 telah diobservasi pada penderita stroke, trauma medula spinalis dan cedera otak. NAIP diekspresikan hampir secara khusus dalam sel saraf dan menghambat aktifitas enzyme caspase-3. Pada stroke hewan percobaan , modulasi NAIP expresi dapat mencegah kematian sel saraf (Hatton.J., 2001).
.
2.9.2 Agonis α-Adrenoceptor
Agonis α2-Adrenoceptor menginduksi vasokonstriksi pembuluh darah otak dan mengurangi ICP pada cedera kepala hewan percobaan. Dexmedotomidine pada tikus percobaan menurunkan volume iskemik 40% walaupun hipotensi dan hiperglikemia telah diobservasi pada beberapa hewan percobaan. Arginin juga telah diberikan pada hewan percobaan ini dan efektivitasnya mengurangi volume kontusio tanpa mengubah ICP. Penelitian ini mengesankan bahwa ada terapi alternatif yang bisa diharapkan mengurangi aliran darah otak tanpa memengaruhi hipotensi sistemik untuk mempertahankan tekan perfusi otak (Hatton.J., 2001).
2.9.3 Agonis cholinergic
Kadar acetilcholin meningkat pada jaringan otak dan cairan otak setelah cedera otak. Pada penderita yang bertahan hidup, gangguan kognitif mungkin berkaitan dengan penurunan aktivitas choline acethyl transferase karena autopsi specimen dari pasien dengan cedera otak memperlihatkan perlindungan reseptor muscarinik pada tempat ikatannya di temporal kortek. Juga, pengurangan ikatan pada reseptor cholinergic di hipocampus dan batang otak menetap sampai dua minggu setelah cedera otak. Pusat inhibisi selektif acetylcholine estaras dan rivastigmin, dan mempercepat penyembuhan fungsi motorik, perbaikan Morris Water Maze Performance dan dapat mengurangi edema serebri dengan cedera otak. Blok selektif reseptor muscarinik M2 postsynap menggunakan BIBN 99 dalam 24 jam cedera otak dan diteruskan selama 11-15 hari untuk mengurangi defisit kognitif dari pada yang dijumpai pada kontrol. Obsevasi ini
menunjukkan pengurangan aktivitas kholinergik yang berperan terhadap sequele nerologis setelah cedera otak (Hatton.J., 2001).
2.9.4 Antagonis kinin
Bradikinin menyebabkan dilatasi vaskuler otak dan dengan nyata meningkatkan permiabelitas vaskuler otak. Efek dimediasi oleh reseptor bradikinin yang terletak pada endotelium vasculer. Aktivasi reseptor bradikinin-2 memediasi edema otak. LF 160687 adalah reseptor antagonis bradikinin-2 yang jelas mengurangi edema otak pada tempat cedera. Satu penelitian yang menggunakan LF 160687 pada dosis 100μ gr/kg/mnt pada tikus dengan edema otak fokal memperlihatkan penyembuhan fungsional yang secara signifikan membaik dalam 6-7 hari. Percobaan klinis dari antagonis kinin Deltibant(CP 0127) telah sempurna. Dua puluh pasien dengan cedera otak ringan sampai sedang (GCS 9-14) diterapi selama 7 hari dengan Deltibant 3 μgr/kg/mnt dalam 24-96 jam cedera otak. Kontrol ICP lebih baik dan sedikit menurun pada score GCS dapat terlihat pada kelompok yang menerima dengan plasebo ( Pruneau.D., 1999).
2.9.5 Inhibisi Cyclo-Oxygenase-2(COX-2)
Kadar COX-2 meningkat dalam neuron dan astrocyte setelah cedera otak pada tikus. Inhibisi COX-2 telah memperlihatkan hasil yang beragam. Celecoxib terlihat memperburuk penampilan motorik, tetapi tidak pada fungsi kognitif tes. Nimesulide, COX-2 inhibitor lain menurunkan pembentukan prostaglandin E2 pada hypotalamus dan jaringan kortek tetapi relatif tidak memunyai efek pada edema otak atau aktivitas fungsional setelah cedera otak pada tikus. Proses inflamasi yang dipicu oleh cedera otak tidak hanya mengaktifasi produksi asam arachidonat, tetapi juga mengaktifasi platelet-factor(PAF). Yang terakhir adalah aktifator transkripsional yang dapat diinduksi gen COX-2. Antagonis PAF telah digunakan untuk mengurangi expresi COX-2 setelah cedera otak iskemik reperfusi fokal pada hewan percobaan. Pada hewan percobaan prostacycline infus mengurangi volume lesi cortex 45% dibandingkan dengan kontrol. Neuroproteksi ini mungkin berkaitan dengan perubahan microsirkulasi berhubungan dengan vasodilatasi dan efek antiagregasi dari prostaglandin (Pruneau.D., 1999).
2.9.6 Antagonis Adhesion Molekul Intraseluler
Pada Citicoline secara alamiah ditemukan senyawa endogen yang dilaporkan dapat memberi efek neuroprotektif setelah iskemik otak. Obat ini terlihat tergantung dosis pada hewan percobaan. Pada dosis lebih tinggi citicoline dapat mengurangi edema dan kerusakan sawar darah otak. Efek ini diobservasi pada kasus cedera dan noncedera. Walaupun citicoline tidak memperlihatkan efek yang signifikan pada percobaan klinis stroke (phase II/III) harapan ke depan pada cedera otak akibat trauma masih ada (Hatton.J., 2001).
2.9.7 Antagonis reseptor NMDA (N-Methyl –D-Aspartate)
Fokus besar penelitian neuroprotektif ditargetkan pada reseptor NMDA. Pada keadaan normal kompleks reseptor NMDA terlibat dalam proses belajar dan memori. Dengan menghambat proses transduksi sinyal dalam neuron, antagonis NMDA bisa menginduksi halusinasi, phisikosis, dan efek samping CNS lainya pada pasien sadar. Kompetitif antagonis NMDA memunyai affinitas yang tinggi terhadap reseptor glutamate. Secara teoritis obat ini jika diberikan dalam konsentrasi yang cukup, akan menghambat glutamat dari ikatan pada reseptornya dan mencegah pembukaan NMDA channel. Saat ini target baru bentuk modulasi efek glutamat pada reseptor NMDA telah diidentifikasi. Penelitian sekarang menghubungkan usaha untuk mengurangi akumulasi glutamat selama iskemik dengan menghambat kunci enzyme intraseluler, N-acetylated alpha-linked acidic dipeptidase (NAALADase). Pendekatan ini potensial menguntungkan secara selektif pada tempat di mana glutamat diproduksi berlebihan dari pada keseluruhan otak. Ini bisa diusulkan perbaikan yang substansial dalam profile yang aman modulator glutamat dan tergantung pada bukti lanjut keefektifan setiap penelitian kedepan yang sempurna (Hatton.J., 2001).
2.9.8 Antagonis Receptor α-Amino-3-Hydroxy-5-Methyl-4- Isoxazolepropionate
(AMPA)
Perkembangan dari antagonis reseptor AMPA baru dimulai. Talampenel (LY 300164) adalah antagonis selektif nonkompetitif reseptor AMPA dengan spektrum luas dan beraktifitas sebagai antikejang. Obat ini memperlihatkan efek anti kejang yang
sangat baik pada hewan percobaan dan berpotensial sebagi neuroprotektif setelah cedera otak. Obat ini diabsorbsi dengan pemberian oral dan berinteraksi dengan beberapa obat lain yang dimetabolisme oleh Cytocrome P450 (CYP) 3 A isoenzyme. Percobaan klinis obat ini pada cedera otak belum dimulai (Hatton.J., 2001).
2.9.9 Magnesium Sulfate
Magnesium mengatur masuknya kalsium ke dalam sel melalui reseptor NMDA. Kadar magnesium intraseluler baik yang bebas maupun total lebih rendah pasca cedera otak dan berkorelasi dengan beratnya cedera axon diffuse. Penurunan magnesium bebas dalam sel menetap sampai 4 hari dan akhirnya menurun ke nilai dasar hari ke 6. Pemberian magnesium chlorida 125 μ mol pada tikus dalam 60 menit dari cedera otak, mampu memperbaiki kadar magnesium terionisasi darah sampai ke nilai dasar dalam waktu 24 jam pasca cedera otak , berkorelasi pada 1-2 minggu dengan perbaikan motorik tetapi tidak mempengaruhi hal yang berkaitan dengan belajar. Waktu pemberian setelah cedera otak penting, ketika magnesium diberikan antara 8-12 jam setelah cedera otak tikus terus memperlihatkan perbaikan motorik. Konsentrasi magnesium terionisasi dalam darah bisa menjadi indikator prognostik motorik outcome. Konsentrasi serum 1,49 mmol/L dan telah memperlihatkan efek neuroprotektif pada penelitian preklinis. Skema dosis pemberian magnesium chlorida 0,5 mmol/kg intravena sebagai loading dose diikuti dengan 0,12 mmol/kg/jam untuk mempertahankan konsentrasi magnesium serum 2 mmol/l (Hatton.J., 2001).
2.9.10 Dexanabinol
Dexanabinol (HU 211) adalah nonpsychotropic sintetis cannabinol dengan sifat farmakologi yang sama dengan non kompetitif antagonis reseptor NMDA; walaupun ia stereoselective inhibisi reseptor. Mekanisme lain dari neuroprotektif telah ditemukan dengan obat ini yang meliputi scavenging dari peroxide, hydroxi radikal dan dapat menghambat produksi TNFα pada tikus cedera otak tertutup. Injeksi tunggal yang diberikan pascacedera otak menghasilkan perbaikan fungsional jangka panjang dan meningkatkan ketahanan hidup neuronal pada hewan percobaan dengan kerusakan otak karena iskemik. Pada tahun 2000, 101 pasien penelitian fase II telah sempurna.
Dexanabinol pada psien memperlihatkan efektif yang terbatas pada hipertensi intrakranial dalam episode 4 jam pascacedera otak. Obat diberikan dalam 6 jam cedera otak dengan dosis 48 mg, 150 mg dan 200 mg (Hatton.J., 2001).
2.9.11 Stratrienes
Estradiol secara lokal dibentuk di jaringan saraf dan ekspresi dipengaruhi dalam astrocyte setelah cedera otak. Estrogen telah dilaporkan memberikan beberapa tingkat neuroprotektif melawan induksi toxisitas glutamat dan juga melawan neurotoxisitas akibat induksi amyloid peptida. Walaupun mekanisme pasti tidak diketahui, downn-regulasi pembentukan jaringan gliosis telah diobservasi. Efek ini menyebabkan akumulasi astrocyte pada daerah cedera otak lebih rendah. Estratrienes adalah salah satu kelas baru neurosteroid yang telah dikembangkan untuk dimanfaatkan sebagai neuroprotektif potensial (Hatton J, 2001).
2.9.12.Antagonis Kalsium
Peran terpadu kalsium dalam memicu sejumlah besar urutan yang berperan utama terhadap cedera otak sekunder secara alamiah menyebabkan penelitian berbagai target terapi mediator ini. Penelitain awal diperiksa antagonis spesifik channel-kalsium, yaitu Nimodipine suatu antagonis kalsium channel type-L. Obat ini dijumpai lebih berproteksi pada pasien dengan perdarahan subarachnoid berkaitan dengan cedera otak, walaupun hasilnya masih diperdebatkan. Fase I penelitain aman telah lengkap dengan DP b 99, prodrug kalsium chelator BEPTA. Ziconotide (SNX 111/CI 1009) bekerja pada presinaptik kalsium channel type –N untuk menghambat ransangan pelepasan neurotransmitter pasca cederaotak, pada tikus percobaan obat ini efektif menurunkan akumulasi kalsium (Hatton.J., 2001).
2.9.13 LOE 908
LOE 098 adalah senyawa baru yang memunyai spektrum luas dan yang menghambat voltage- and store-operated cation channels controlling intracellular calcium levels. Penelitian sebelumnya telah memperlihatkan perbaikan fungsi motorik dengan obat ini, tetapi tidak ada perbaikan pada test kognitif (Hatton.J., 2001).
2.9.14 MS 153
MS 153 yaitu suatu obat yang diteliti baru-baru ini dijelaskan memungkinkan target pada sodium voltage-gated atau channel kalsium teraktifasi setelah iskemik otak. Mekanisme kerjanya kurang dapat dipahami, mekanisme kerja obat ini juga menurunkan kadar glutamate ekstraseluler. Kerjanya pada protein kinase-C menunjukkan sebagai hal yang memberikan kontribusi pada kerja dari MS 153 (Hatton.J., 2001).
2.9.15 Cyclosporine
Cyclosporin dapat mencegah kematian sekunder sel saraf dengan menghambat pembukaan pori dan pencegahan keluarnya kalsium. Cyclosporin secara luas digunakan sebagai obat immunosupressi yang menghambat aktivasi lymphosit-T dan memunyai peran multiple penting dalam regulasi sel saraf. Bukti penelitian pada hewan percobaan menunjukkan efek protektif pada cedera saraf. Cyclosporin diberikan pada kelinci dan tikus setelah kontussio kortek berat, yaitu penurunan cedera saraf sampai 50%. Sifat psychochemical cyclosporin penetrasi ke CNS yang terbatas adalah dalam keadaan psychological normal. Meskipun demikian, sawar darah otak terganggu setelah cedera otak. Hasil dari penelitian telah terindikasi bahwa rentang waktu intervensi terapi adalah paling cepat dan bahkan selambat-lambatnya 24 jam. Pemberian pascacedera otak cyclosporin menghasilkan pengurangan signifikan(40%) volume lesi (Hatton.J., 2001).
2.9.16 Antioxidants
Antusiasme proteksi antioksidan pascacedera kepala menurun secara signifikan setelah ada hasil penelitian klinis fase dari pergorgetein dan tirilazad. Walaupun demikian beberapa strategi baru untuk scavenging radikal bebas pascacedera otak ditemukan dalam penelitian. OPC 14117 adalah scavenger yang telah dievaluasi pada kontusio kortex serebri binatang percobaan. Pada kondisi ini terapi pasca cedera mengurangi progresifitas edema, ukuran kontusio, dan perluasan necrosis dan juga membatasi defisit perilaku. Paling sedikit satu antioxidan baru termasuk edaravone (MC 186) yang telah dimulai fase I penelitian. Penelitian perkembangan dari peroxynitrite scavengers sedang dilakukan. Radikal bebas ini dapat mengoxidatif lemak, protein dan
asam nukleat sel yang cedera. Obat yang melindungi mikrovaskuler bentuk ini dari spesies oksigen reaktif pasca cederaotak dapat memberikan keuntungan ( Chieueh.C., 1999).
2.9.17 Inhibisi Nitric Oxide
Derivat Nitric oxide dan modulatornya dianggap sebagai calon neuroprotektif setelah cedera otak. Antiosidan dan anti radikal bebas dianggap sebagai potensial menguntungkan. Radikal nitroxide stabil yang dievaluasi pada tikus memperlihatkan beberapa efek perbaikan klinis, termasuk pengurangan edema dan kerusakan sawar darah otak. Mekanisme neuroprotektif dapat melibatka nitroxide dalam transisi logam, radikal bebas, atau oksigen. NXY 059 adalah nitrone yang bisa menembus sawar darah otak dan memperlihatkan pengurangan volume infark dan nekrosis ketika diberikan setelah atau sebelum cedera otak pada tikus percobaan. Lubeluzole adalah modulator dari sirkuit sintesis nitric oxide. Volume kontusio, bengkak hemispher dan kandungan air tidak berubah dengan dosis pascacedera kontusio kortek pada tikus. Corticotropin-releasing factor adalah neuroprotektif peptida endogen, yaitu bukti baru yang menunjukkan bahwa ia bisa melawan kematian sel akibat oxidatif di samping fungsi fisiologis normalnya. Pada tingkat molekuler, kortikotropin menurunkan aktivitas ikatan DNA dan aktivitas transkripsi dari NFκB. Mekanisme ini dapat bertanggungjawab untuk efek protektif corticotrophine melawan stress oksidatif yang diinduksi oleh NFκB. Corticotrophine realising factor(CRF) dapat merangsang pelepasan kortikotropin. CRP adalah neuropeptida hipotalamus yang dikenal untuk menghambat kebocoran cairan dari plasma yang melewati membrane. Hal ini mengurangi edema pada jaringan cedera. Corticorelin yang berfungsi sebagai releasing factor dapat meningkatkan kortikotropin, termasuk penelitian fase II pada pasien dengan edema otak peritumoral. Untuk yang akan datang dalam pengobatan edema otak sampai saat ini masih diteliti (Kroppenstedt.S.N.,1999).
2.9.18 Growth Factors
Glial-secreted peptide trophic factors secara potensial memiliki neuroprotektif termasuk BDNF, CNTF, IGF-1, fibrin growth factor (FGF), bone morphogenetic protein (BMP)-4, BMP-7, amphiregulin, cerebellum derived growth factor (neuregulin- 2), and
GDNF. Mengirimkan konsentrasi adekuat ke CNS telah menjadikan tabir dari obat ini untuk dibuat penelitain yang lebih besar. Keterbatasan yang lain adalah kurangnya dokumentasi yang spesifik dari defisiensi growth factor pasca cederaotak dan juga apakah suplemen eksogen growth factor akan memberikan keuntungan. Dosis, waktu pemberian awal, dan lamanya terapi masih dalam penelitian pada banyak hewan percobaan untuk berbagai endogen dihasilkan growth factor. Basic fibroblast growth factor (BFGF) diberikan setelah fluid percussion injury pada tikus, secara signifikan mengurangi jumlah kerusakan neuron kortikal dan ukuran kontusio dengan hanya diinfus 3 jam. IGF-1 diberikan dalam 2 minggu dengan menggunakan pompa subcutan untuk memperbaiki fungsi motorik, kemampuan belajar dan retensi memori pada tikus dengan cedera otak. Penelitian yang terakhir menunjukkan pengobatan jangka panjang setelah cedera otak mungkin manjur. Manusia dengan cedera otak secara signifikan menurunkan IGF-1 serum (Hatton et al, 2001). Penelitian randomise terhadap 33 pasien dengan cederera otak sedang sampai berat (GCS 4-10) tidak diterapi dengan kortikosteroid terhadap dukungan nutrisi saja atau kombinasi dengan kontinyu infus IGF-1(0,01 mg/kg/jam). Terapi dimulai 72 jam pasca cedera otak dan terus sampai 14 hari. Obat yang diberikan mendapat toleransi baik pada semua pasien dan perbaikan metabolik dilaporkan dalam 3 hari cedera otak pada pasien yang menerima obat ini. Ini laporan pertama dari efektifitas anabolik dengan dukungan nutrisi pada populasi ini (Alzheimer, 2002).
Bagan. 3. Ringkasan Jalur Sinyal Neuroprotektif yang Dipengaruhi oleh Fibrine Growth Factor(FGF) (Alzheimer, 2002).
Ditemukan bahwa walaupun IGF-1 yang diberikan kontinyu secara sistematis, konsentrasi serum cepat menurun. Growth hormon meningkat dan konsentrasi ikatan protein utama diperifer (IGFBP3) turun. IGF-1 eksogen yang terlihat mulai negatif biofeedback pada fungsi pituitary dapat menyebabkan perubahan pada pembersihan. Strategi dosis ini disetujui karena konsentrasi farmakologi (> 450 μg/L) tidak dapat dipertahankan. Keseimbangan negatif nitrogen dan perbaikan hasil akhir neurologis pada 6 bulan terlihat berhubungan dengan pencapaian konsentrasi serum lebih tinggi daripada nilai normal endogen (150-450 μg/L). Tes lebih lanjut dengan potensial IGF-1 untuk perbaikan parameter metabolik dan neurologis pada cedera otak berat, secara prospektif, randomisasi dan, double blind dukungan nutrisi saja atau kombinasi IGF-1 dengan terapi growth hormon dan nutrisi telah dimulai. 98 pasien telah dirandomise ke dalam penelitian ini dengan metodelogi identik terhadap penelitian IGF-1 sebelumnya (Alzheimer, 2002).
Kombinasi IGF-1 dan growth hormon cepat mencapai serum farmakologis dan parameter metabolik yang secara signifikan lebih baik pada kelompok terapi. Sistemik IGF-1 /growth hormon terapi terlihat dapat meningkatkan konsentrasi IGF-1 dalam CNS. Hasil akhir klinis neurologis, tidak ada perbedaan signifikan dari yang dicapai dengan dukungan nutrisi saja. Penambahan growth hormon pada regimen dalam usaha mengoptimalkan konsentrasi sistemik telah dapat memengaruhi hasil akhir. Penelitian telah ditempatkan pada penanganan klinis karena laporan growth hormon dapat meningkatkan kematian pasien pada penyakit kritis. Potensial penuh dari terapi IGF-1 dalam hal neuroprotektif masih memerlukan penelitian lebih lanjut (Hatton.J., 2001).
2.10 Statin sebagai Neuroprotektor
Statin yang juga dikenal sebagai 3-hydroxy-3-methylglutaryl co-enzyme A (HMG-CoA) reductase inhibitors digunakan sebagai obat penurun kholesterol dan memperlihatkan penurunan insiden penyakit jantung koroner pada percobaan klinis. Statin berkerja menghambat produksi enzyme penting L-Mevalonat, yaitu suatu hasil sampingan metabolisme kolesterol. Walaupun bukti tertentu menunjukkan pengurangan kadar kolesterol, tidak seluruhnya efek dari statin. Indikasi ini menunujukkan bahwa
statin memunyai mekanisme kerja lain yang mungkin melalui jalur produksi mevalonat yang berperan dalam pemberi isyarat seluler (cellular signalling). Selama beberapa dekade yang lalu memunyai bukti yang jelas bahwa statin juga memunyai efek neuroprotektif. Telah dilaporkan bahwa pemberian statin berhubungan dengan insiden penyakit Alzaimer. Hal ini tidak dipersoalkan lagi, tetapi beberapa penelitian memperlihatkan bukti bahwa statin mengurangi produksi Amyloid-β peptida in vitro. Penelitian terbaru juga memunyai harapan efek statin pada penyakit parkinson. Lebih lanjut pada hewan percobaan tampak bahwa statin mungkin menguntungkan pada terapi multiple sclerosis dan stroke akut. Statin dikenal efektif dan mempunyai sedikit efek samping. Efek samping adalah yang paling sering adalah gejala gatrointestinal dan merasa mules, Hepatotoxic bisa ditandai dengan peningkatan serum SGOT, terjadi kurang 1% pasien dengan pemberian dosis tinggi (Most et al, 2009).
Jenis-jenis statin adalah lovastatin, pravastatin dan simvastatin yang berasal dari jamur; atorvastatin, rosuvastatin, fluvastatin, pravastatin adalah sintetis (Schachter, 2005). Atorvastatin dan simvastatin meskipun farmakokinetiknya berbeda, tidak banyak menunjukkan perbedaan dalam penanganan cedera otak. Simvastatin yang diberikan secara oral akan diserap oleh usus antara 30% hingga 85%. Simvastatin diserap dalam bentuk laktone inaktif sehingga perlu ditransformasi di hati menjadi bentuk aktif, yaitu ß-hydroxy acid dan asam simvastatin. Hampir seluruh Simvastatin yang diserap akan mengalami first pass metabolism di hati. Mekanisme simvastatin masuk ke dalam hati melalui difusi sederhana karena sifat simvastatin yang lipofilik, Akibat first pass metabolism, bioavabilitas sistemik simvastatin dan metabolitnya bervariasi antara 5% hingga 30% dari dosis yang diberikan. Farmakokinetik statin di dalam plasma, lebih dari 95% simvastatin dan metabolitnya akan berikatan dengan protein. Setelah pemberian oral, konsentrasi simvastatin dalam plasma akan mencapai puncak dalam waktu 1 hingga 4 jam. Begitu juga dengan waktu paruh dari simvastatin, yaitu 1 hingga 4 jam. Simvastatin dan metabolitnya diekskresikan melalui feses sebanyak 70% dan sisanya melaui urin (Suzy. Rr, 2012; Brunton, 2006).
Simvastatin dan atorvastatin memiliki sifat relative lipohilic yang setara sehingga memiliki daya penetrasi yang baik ke sawar darah otak (bood brain barrier) , sementara rosuvastatin dan pravastatin memiliki sifat hydrophilic sehingga minimal penetrasi ke
sawar darah otak (bood brain barrier). (Suzy. Rr, 2012; Wible.E.F., et al, 2010). Walaupun simvastatin dan atorvastatin memiliki keunggulan yang sama sebagai neuroprotektor, pada penelitian ini digunakan simvastatin karena harga murah dan sudah tersedia dalam bentuk generik di indonesia.
Bagan 4. Jalur metabolisme mevalonat (Most et al, 2009).
2.10.1 Neuroprotective Signalling Pathways yang Diaktifkan oleh Statin
Statin dapat mengaktifkan beberapa jalur pemberi sinyal neuroprotektif. Zacco dkk, menunjukkan bahwa beberapa statin menjadikan neuron kortek primer tikus yang resisten terhadap excitotoxicity pada jalur cholesterol-dependent. Potensi neuroprotektif kurang lebih berkorelasi dengan efektivitas block HMG-CoA telah dilaporkan oleh McTaggal dkk (2001) dan efek neuroprotektif dapat dibalikkan dengan penambahan mevalonat dan cholesterol. Meskipun demikian percobaan in vitro oleh kelompok Chopp
menunjukkan terapi statin setelah cedera otak meningkatkan neurogenesis dan synaptogenesis tanpa perubahan serum kolesterol. Penemuan mereka juga menunjukkan induksi neuroproteksi oleh statin dengan mempromosikan pelepasan neurotrophic factor. Sesungguhnya Simvastatin telah memperlihatkan induksi ekspresi dari BDNF (Brain Derived Neurotrophic Factor) setelah cedera otak dan injeksi LPS ke dalam substansia nigra. Beberapa penelitian in vitro dan in vivo memberikan bukti bahwa statin mengaktifkan neuroprotektif protein kinase-B (PKB/Akt). Aktifasi dari PKB/Akt oleh statin terbukti pada saat sekarang dan lampau pada sel endotel dan prognitor dan sekarang telah dilakukan pada kultur neuron dan pada CNS binatang yang diterapi dengan statin (Most et al , 2009).
Bagan 5. Mekanisme Statin pada Metabolisme Lemak dan Selular Signalling Neuroprotektif (Most et al, 2009).
2.10.2.Efek Neuroinflamasi Inhibitor HMG CoA Reduktase (Statin)
2.10.2.1.Menurunkan Inflamasi Sitokin
Statin mengurangi ekspresi induksi-sitokain dari ko-stimulasi molekul pada sel imun (termasuk mikroglia) dan endotelium. Statin juga mengurangi induksi molekul ekspresi dari MHC kelas II. Efek yang terakhir bergantung pada GGPP, tetapi beberapa penelitian mengindikasikan lemak bisa terlibat dan bisa juga tidak. Statin bisa juga efek menghambat ekspresi MHC kelas-I, tetapi buktinya terbatas untuk asumsi ini dan masih kontroversi (Most et al, 2009).
Tidak mengherankan, inhibisi presentasi antigen berakibat pada pengurangan proliferasi sel-T, tetapi statin juga bisa memengaruhi sel-T phenotype, walaupun ini kontroversi. Statin mempromosikan differensiasi sel-T menjadi Th2 phenotype. Statin mengurangi sekresi pro-inflamasi cytokine Th1 dan meningkatkan sekresi anti inflamsi cytokine Th2. Ini juga paling mungkin akibat kekurangan isoprenoid, walaupun tidak semua bukti setuju. Penelitian tunggal pada binatang dilaporkan bahwa statin pelindung melawan multiple sclerosis dan proteksi dapat ditransfer ke binatang lainnya dengan mentransfer Th2 defferensiasi sel. Pada hewan percobaan multiple sclerosis juga memperlihatkan bahwa statin mengurangi migrasi leukosit dan infiltrasi pada jaringan inflamasi. LFA-1 memainkan peran penting pada infiltrasi jaringan, jadi ini mungkin disebabkan oleh ikatan LFA-1 dan mencegah interaksi nya dengan ICAM-1 (Most et al, 2009).
2.10.2.2 Meningkatkan Integritas Sawar Darah Otak
Penyebab utama hilangnya integritas sawar darah otak pascatrauma adalah terjadinya upregulasi mediator-mediator inflamsi seperti tumor nekrosis factor (TNF-α), interleukin-6 (IL-6) interleukin-1β (IL-1β) dan interseluler adhesion molekul-1 (ICAM-1). Pada percobaan preklinis pemberian inhibitor HMG CoA reduktase (statin) memperlihatkan penurunan kadar TNFα, IL-6, IL-1β, dan ICAM-1 fase akut dan subakut setelah cedera otak (Wible,E.F., et al, 2010).
2.10.2.3 Mengurangi Edema Serebri
Respon neuroinflamasi setelah cedera otak menyebabkan kematian sel sekunder subakut melalui proses eksitotoksik, peroksidasi lemak, kerusakan sawar darah otak, dan akhirnya menyebabkan edema serebri. Pada percobaan preklinis inhibitor HMG CoA reduktase (simvastatin dan atorvastatin) memperlihatkan penurunan translokasi sukrosa dan albumin. Dengan demikian, mengurangi edema serebri pascacedera otak (Wible, E.F., et al, 2010).
2.10.2.3 Mengurangi Oksidatif Stres
Statin dapat melindungi sel dari kerusakan akibat osidatif. Statin telah meperlihatkan mempunyai efek antiokidatif pada jaringan dan in vivo binatang percobaan juga pada percobaan kinis. Pada neuron statin dilaporkan mengurangi peroksidasi lemak setelah kehilagan oksigen, gula, dan reperfusi. Statin dapat mengurangi produksi species oksigen reaktif dengan menghambat pembentukan dan aktivasi NADPH commplex. Selain itu statin dapat mengurangi kerusakan akibat oksidasi dengan mengontrol produksi nitric oxide dan memungkinkan pengurangan respon inflamasi (Most et al, 2009).
Oksigen radikal nitric oxide (NO) berfungsi sebagai molekul pemberi sinyal pada sistem vaskuler. NO secara lokal memperbaiki aliran darah dengan mengiduksi respon vasodilator poten. Karena gambaran hipoperfusi pada penyakit Alzaimer dan dikaitkan dengan penurunan fungsi kognitif, hal ini menunjukkan vasodilatasi dapat mengurangi kematian sel. NO diproduksi oleh tiga enzyme : Endotelial Nitric Oxide synthase(eNOS), Neuronal Nitric Oxide Synthase(nNOS) dan Inducible/Inflammatory Nitric Oxide Synthase(iNOS) yang membentuk ekspresi makrophag. Segera setelah cedera iskemik eNOS diaktifkan dan usaha keras untuk efek melindungi sangat berkurang karena efek vasodilatasinya. Meskipun demikian, cedera iskemik mengaktifkan ekspresi nNOS, induksi, ekspresi dan aktivasi iNOS secara menetap. Pada akhir overproduksi NO menyebabkan kerusakan oxidatif. Statin juga mengurangi produksi superoxida radikal dan peroxynitrite (Most et al, 2009).
2.10.3 Respon Neuroinflamasi pada Kontusio Serebri
Ringkasan efek neuroprotektif statin Aktifitas tombosist
↓Agregasi trombosit
↓Deposit trombosit pada dinding pembuluh darah yang rusak Pembentukan trombin
↓Penumpukan peptida dari pemecahan trombin
↓Penumpukan trombus (menekan penumpukan abnormal tromboxan A2) Pembentukan nitric oxide(NO)
↑Upregulasi sintesa endotelial NO dengan ↑ bioavailabilitas NO (Peran fisiologis protektif) ↑Aliran darah otak(CBF)
↓Produksi toxic dari NO melalui iNOS Efek anti inflamsi
↓Menurunkan potensial kerusakan cytokine dari macrophage selama iskemik otak ↓Pembentukan pro-inflamsi isoprenoid
↓Ekspresi adhesion molekul ↓Interaksi endotelial leukosit Efek anti oksidan
↓Oksidasi lipoprotein
↓Kerusakan akibat radikal bebas
Tabel 5. Efek Neuroprotektive Statin (Cucchiara et al , 2001; Indharty.S, 2012)
Bagan-6; Efek statin pada cedera otak (Wible et al, 2010; Indharty.S, 2012 ), Bax/Bcl-2= Bcl-2
associated X protein; BBB= blood brain barrier; BDNF= brain-derived neurotrophic factor; eNOS= endothelial isoform of nitric oxide synthase; PKB= protein kinase B; PI3K =phosphoinositide-3-kinase; VEGF= vascular endothelial growth factor; VEGFR2 vascular endothelial growth factor receptor 2; vWF =von Willebrand Factor
2.11 Interleukin
Interleukin adalah suatu kelompok cytokine yang pertama sekali diekspresikan oleh leukosit, istilah interleukin berasal dari inter- yang artinya komunikasi dan –leukin yang artinya turunan protein yang dihasilkan oleh leukosit. Fungsi sistem imun sebagian tergantung pada interleukin. Mayoritas interleukin disintesis oleh helper CD4 T limphosit juga oleh monosit, makrophag, dan sel endotelial. Interleukin reseptor pada astrosit dihipocampus juga diketahui terlibat dalam perkembangan memori pengenalan ruang / lingkungan pada tikus (Wikipedia, 2013).
Banyak interleukin yang sudah ditemukan pada manusia; interleukin-1 dihasilkan oleh makrphage, B sel, monosit dan dendritic sel. Interleukin-2 dihasilkan oleh sel T-helper (th-1). Interleukin-3 dihasilkan oleh T-sel T-helper yang teraktivasi, sel mast, sel NK, sel endotelium dan sel eosinophil. Interleukin-4 dihasilkan oleh sel th-2, sel CD4+, sel mast dan makrophag. Interleukin-5 dihasilkan oleh sel th-2, sel mast dan eosinophil. Interleukin-7 dihasilkan dari stromal sel sumsum tulang dan sel thymus. Interleukin-8 dihasilkan oleh makrophag, limphosit, sel epitelial CXCL8 dan sel endotelial. Inteleukin-9 dihasilkan oleh sel th-2 dan th-2 CD4+. Interleukin-10 dihasilkan oleh monosit, th-2, T sel CD8+, sel mast, makrophag dan sel B. Interleukin -11 dihasilkan oleh sel stroma sumsum tulang, Interleukin-12 dihasilkan oleh dentritik sel, B sel, T sel dan makrophag. Interleukin-13 diahasilkan oleh T helper sel aktif dan NK sel. Interleukin-14 dihasilkan oleh T sel dan B sel malignan tertentu, Interleukin-15 dihasilkan oleh mononuklear phagosit. Interleukin-16 dihasilkan oleh limphosit, sel epitel, eosinoiphil dan T sel CD8+. Interleukin-18 dihasilkan oleh makrophag. Interlaukin-20 dihasilkan oleh keratinosit dan monosit teraktifasi. Interleukin-21 dihasilkan oleh sel T helper teraktifasi dan sel NK. Interleukin-36 dihasilkan oleh T sel. Sejumlah interleukin belum diketahui sel yang memproduksi seperti interleukin-19 (19), 22, 22, 23, 24, 25, 26, IL-27, IL-28, IL-29, IL-30, IL-32, IL-33 dan IL-36 (Wikipedia, 2013).
2.11.1 Gambaran Umum Interleukin-6
Interleukin-6 (IL-6) adalah glikoprotein dengan berat molekul 20-30 kDa, dan tergantung pada sumber sel. Interleukin-6 juga adalah suatu cytokine dengan daya pleiotropic dan memainkan peran utama dalam mempertahankan host. Tergantung pada target sel, Interleukin-6 (IL-6) dapat menginduksi pertumbuhan dapat menghambat pertumbuhan, dan juga efek menginduksi differensiasi. Interleukin-6 (IL-6) dihasilkan oleh banyak tipe sel meliputi monosit, makrophage, fibroblas, keratinosit, sel endotelial, sel mesangial, khondrosit, osteoblas, sel otot polos, T sel, B sel, granulosit, mast sel, dan sel tumor tertentu. Sejumlah sel kultur telah dijumpai melepaskan interleukin-6 (IL-6) dalam jumlah kecil, dan menjadi sangat tinggi setelah stimulasi (Suzuki, 2009).
Sitokin dihasilkan juga oleh susunan saraf pusat (SSP) dan terlibat dalam patogenesis banyak penyakit SSP, seperti peradangan, autoimun dan penyakit degeneratif, infeksi, neoplasma, dan stroke. Pada hewan percobaan dijumpai peningkatan akibat iskemik stroke. Walaupun Suzuki dkk; menjumpai peningkatan tidak hanya oleh inflamasi tetapi juga efek neutrophil pada iskemik otak, peran sebenarnya dari Interleukin-6 (IL-6) pada patofisiologi iskemik otak belum sepenuhnya dapat dijelaskan. Dengan kata lain apakah Interleukin-6 (IL-6) berkerja sebagai mediator peradangan cedera atau zat neuroprotektif pada iskemik otak belum dapat ditentukan. Interleukin-6 (IL-6) adalah satu dari cytokine inflamsi SSP dan telah berakibat pada respon seluler. Sitokin ini atau chestrate adalah suatu respon inflamsi antara sel darah, endotelium vaskuler, dan sel penghuni parenkhim otak dan dapat menginduksi beberapa chemokine dan sel adhesion molekul, bersama dengan kebocoran sawar darah otak dapat menyebabkan infiltrasi leukosit (Suzuki, 2009).
Baik penelitian klinis dan exsperimental bahwa iskemik otak meningkatkan exspresi Interleukin-6 6) pada otak dan serum. Secara keseluruhan Interleukin-6 (IL-6) memainkan peran ambivalen, tergantung pada fase iskemik otak. Selama fase akut Interleukin-6 (IL-6) bekerja sebagai cytokine inflamasi dan memberi kontribusi terhadap cedera iskemik. Walaupun Interleukin-6 (IL-6) mungkin mempunyai efek penting pada perbaikan dan regenerasi hanya berhasil sangat kecil jika peran sel inflamsi mengambil
porsi yang lebih besar. Selanjutnya, selama fase subakut Interleukin-6 (IL-6) bekerja sebagai mediator neuroprotektif bersama dengan leukemia inhibitory factor (LIF) dan ciliaryneurotrophic factor (CNTF) (Suzuki, 2009).
Peningkatan serum interleukin-6(IL-6) dimulai dalam 24 jam dan mencapai puncak 2-4 hari setelah serangan stroke. Kadar tinggi menetap sampai 90 hari stroke. Biomarker inflamasi meningkat paralel dengan C-reaktif protein, fibrinogen, reseptor antagonis IL-1 dan tumor nekrosis faktor(TNF-α). Beberapa percobaan independent dilaporkan bahwa tingginya serum interleukin-6(IL-6) berhubungan dengan volume infark, perburukan neurologis dini, suhu tubuh dan hasil akhir yang buruk dalam jangka lama. Interleukin-6(IL-6) meningkat dalam CSF pada pasien stroke akut. Secara sistemik produksi interleukin-6(IL-6) dapat potensial secara pasif masuk ke CSF melalui sawar darah otak yang rusak. Akan tetapi penelitian lain melaporkan interleukin-6(IL-6) dalam serum secara signifikan lebih rendah dari pada CSF selama minggu pertama setelah serangan stroke. Penemuan ini sangat kuat sebagai bukti bahwa bukan produksi primer IL-6 disistemik yang masuk ke CSF setelah iskemik otak (Suzuki, 2009).
2.11.2 Interleukin-6(IL-6) pada Cedera Otak Akibat Trauma
Pro-inflamasi cytokine termasuk interleukin-6(IL-6), adalah mediator penting dari neuroinflamasi dan dihasilkan pada trauma otak akut oleh astrocyte, sel makrofage/mikroglia, neuron dan endotelium SSP. Puncak peningkatan Interleukin-6 (IL-6) mRNA dan protein telah dijumpai pada 6-8 jam pasca cedera kepalatertutup, akhirnya penelitian telah mendokumentasikan peningkatan Interleukin-6 (IL-6) ,soluble Interleukin-6 (IL-6) reseptor dan TNF-α dalam CSF, plasma atau parenkhim dari pasien cedera kepala sampai 7 hari setelah trauma. Ekspresi kronis berlebihan dari TNF-α dan IL-6 menyebabkan neurodegeneratif inflamasi encephalopathy dan IL-6 sendiri mempromosikan demyelinasi, trombosis, infiltrasi leukosit , rusaknya sawar darah otak dan mengganggu neurogenesis pada dewasa (Marklunda, 2005).
2.11.3.Kondisi Lain yang Dapat Meningkatkan Kadar Interleukin-6
Selama latihan fisik IL-6 disintesis dan dilepaskan oleh otot, kontraksi otot memproduksi IL-6 dan dilepaskan ke dalam aliran darah dan efeknya sampai ke organ lain. Nilai normal dibuktikan 1 pg/ml, juga meningkat pada orang gemuk, aktivitas fisik, wanita menstruasi, hiperglikemia akut, selama dan sesudah pembedahan dan, juga pada pasien arteriosklerosis (Fisman, 2010).
2.11.4.Peran Inhibitor HMG CoA Reduktase Dalam Menurunkan Kadar IL-6
Respon neuroinflamsi setelah cedera kepala menyebabkan kematian sel neuron sekunder subakut melalui excitotoxic injury, lipid perosidasi, kerusakan sawar darah otak dan edema cerebri. Pada percobaan preklinis cedera kepala menunjukkan upregulasi dari mediator inflamasi. Terutama tumor necrosis faktor (TNF-α), interleukinn-6 (IL-6) dan IL-1 β meningkat dan berhubungan dengan hilangnya integritas sawar darah otak yang memberi kontribusi terhadap edema otak. Baik terapi preinjury dan postinjury pada hewan percobaan bahwa statin menurunkan kadar IL-1 β, TNF α , IL- 6 and ICAM-1 pada akut dan subakut setelah cedera otak traumatik. Mikroglia marker (mediator inflamasi) meningkat setelah percobaan trauma otak, puncak pada 24 jam pasca trauma dan menetap untuk 7 hari (Wible, 2010).
Inhibitor HMG CoA reduktase dapat menurunkan expresi IL-6, IL-8 dan monocyte chemoattractant protein-1(MCP-1) mRNA dalam darah perifer sel mononuclear. IL-6 adalah pleotropic cytokine dan mediator sentral dari respon fase akut, dengan rentang yang luas dari diversi sel imun simvastatin, atrovastatin, atau cerivastatin dalam 12-24 jam signifikan yang menurunkan ekspresi dan sekresi IL-6, IL-8 dan (MCP-1)mRNA. Kenyataanya semua statin yang diberikan mengurangi kadar IL-6, IL-8 dan (MCP-1)mRNA dan tidak ada perbedaan signifikan pemberian dosis 20mg atau 40 mg dalam hal produksi cytokine (Majd, 2002).
2.12 Kerangka teori
Berdasarkan teori-teori pendukung yang telah dikemukakan sebelumnya, maka dapatlah disusun suatu kerangka teori yang akan mendasari dilakukan penelitian ini.