SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Ilmu Farmasi
Oleh : Jayanti Micell NIM : 068114094
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
ii SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Ilmu Farmasi
Oleh : Jayanti Micell NIM : 068114094
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
iii
MENGGUNAKAN METODE KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK
oleh :
Jayanti Micell NIM : 068114094
v
Ia
memberikan
kekekalan
dalam
hati
mereka.
Tetapi
manusia
tidak
dapat
menyelami
pekerjaan
yang
dilakukan
Allah
dari
awal
sampai
akhir.
Pengkotbah 3 : 11
vi
Nama : Jayanti Micell
Nomor Mahasiswa : 068114094
Demi pengembangan ilmu pengetahuan, saya memberikan kepada Perpustakaan Universitas Sanata Dharma karya ilmiah saya yang berjudul :
“VALIDASI METODE PENETAPAN KADAR CAMPURAN PARASETAMOL DAN IBUPROFEN DENGAN PERBANDINGAN 7:4
MENGGUNAKAN METODE KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK”
beserta perangkat yang diperlukan (bila ada), dengan demikian saya memnerikan kepada Perpustakaan Universitas Sanata Dharma hak untuk menyimpan, mengalihkan dalam bentuk media lain, mengelolanya dalam bentuk pangkalan data, mendistribusikan secara terbatas, dan mempublikasikannya di Internet atau media lain untuk kepentingan akademis tanpa perlu meminta ijin dari saya meupun memberikan royalti kepada saya selama tetap mencantumkan nama saya sebagai penulis.
vii
berkat, kasih, kekuatan, dan penyertaan-Nya maka skripsi yang berjudul “Validasi
Metode Penetapan Kadar Campuran Parasetamol dan Ibuprofen Dengan Perbandingan 7:4 Menggunakan Metode Kromatografi Cair Kinerja Tinggi Fase Terbalik” dapat terselesaikan dengan baik oleh penulis. Skripsi ini disusun untuk
memenuhi salah satu syarat memperoleh gelar Sarjana Farmasi (S.Farm) di Fakultas Farmasi Universitas Sanata Dharma Yogyakarta.
Selama penyusunan skripsi ini, telah banyak pihak yang membantu penulis, oleh karena itu penulis ingin mengucapkan terima kasih yang sebesar-besarnya kepada :
1. Rita Suhadi, M.Si.,Apt. selaku Dekan Fakultas Farmasi Universitas Sanata Dharma Yogyakarta.
2. Jeffry Julianus, M.Si., selaku pembimbing yang telah membantu dalam penyelesaian skripsi ini. Terima kasih atas waktu, kritik dan saran yang telah diberikan selama penyusunan skripsi.
3. Christine Patramurti, M.Si.,Apt. selaku dosen penguji yang telah banyak membantu dalam penyelesaian skripsi ini, baik kritik maupun sarannya.
4. Dra. M. M. Yetty Tjandrawati, M.Si., selaku dosen penguji atas kritik dan sarannya.
5. Yohanes Dwiatmaka, S.Si., M.Si. yang telah memberikan ijin melakukan
viii
Kunto, dan Mas Otok yang telah membantu penulis selama penelitian.
7. Teman seperjuangan, Angel dan Pungki atas kebersamaan selama penelitian di laboratorium, atas saran dan kritik untuk penulis sehingga dapat terselesaikan dengan baik.
8. Boim, Tony, Yoki dan Aang atas dukungan dan kebersamaannya selama penelitian di laboratorium.
9. Marleen dan Riri, atas bantuan, kebersamaan serta doanya.
10. Meli, Dian, Dani, Hani, Rudy, Nico terima kasih atas bantuan dan doanya. Sahabat dan teman-teman lainnya, terima kasih atas dukungannya.
11. Mas Dwi, Pak Mukmin, Mas Narto atas bantuannya.
12. Anak-anak Kost Gracia serta Ibu dan Bapak kost yang telah banyak
membantu.
13. Teman-teman FST 2006 atas kebersamaannya, saat-saat yang membahagiakan dan tak terlupakan.
14. Semua orang yang mungkin tidak dapat disebutkan satu per satu oleh penulis, terima kasih atas bantuan dan kebersamaannya selama ini sehingga menjadi
pribadi seperti sekarang ini.
ix
x
tidak memuat karya atau bagian karya orang lain, kecuali yang telah disebutkan
dalam kutipan dan daftar pustaka, sebagaimana layaknya karya ilmiah.
Yogyakarta, 12 Maret 2010
Penulis
xi INTISARI
Penetapan kadar campuran parasetamol dan ibuprofen dapat dilakukan dengan metode kromatografi cair kinerja tinggi (KCKT) fase terbalik. Oleh karena itu perlu dilakukan validasi metode terlebih dahulu untuk mengetahui metode yang digunakan dapat memberikan hasil yang terpercaya.
Penelitian ini merupakan penelitian noneksperimental. Parasetamol dan ibuprofen dianalisis secara kuantitatif dengan metode KCKT fase terbalik menggunakan fase diam oktadesilsilan (C18), fase gerak metanol : aquabidest (90:10) dan ditambah asam asetat glasial hingga pH 4,0, kecepatan alir 1,5 ml/menit, detektor UV pada panjang gelombang 230 nm.
Parameter validasi yang diukur adalah akurasi, presisi, spesifisitas dan linearitas. Hasil penelitian menunjukkan metode memiliki linearitas yang baik yaitu 0,998 pada konsentrasi 70-175 ppm untuk parasetamol dan 0,999 pada konsentrasi 40-100 ppm untuk ibuprofen. Nilai recovery dan CV berturut-turut untuk parasetamol 70 ppm; 122,5 ppm; 175 ppm adalah 101,03-111,55% dan 1,78%; 91,46-98,55% dan 5,04%; 85,44-95,97% dan 3,09% sedangkan ibuprofen 40 ppm; 70 ppm; 100 ppm adalah 85,84-100,75% dan 7,38%; 90,14-101,35% dan 7,99%; 92,53-107,89% dan 7,99%. Berdasarkan hasil tersebut maka metode ini valid untuk penetapan kadar campuran parasetamol dan ibuprofen dengan perbandingan 7:4.
xii ABSTRACT
Quantitative analysis paracetamol and ibuprofen combine can be made with high performance liquid chromatography (HPLC) reversed phase method. Therefore in order to have a trusted outcome, necessary to do validation of KCKT reversed phase method previously.
This research is a nonexperimental research. Paracetamol and ibuprofen are analyze quantitatively by using HPLC reversed phase method with static phase oktadesilsilan (C18), mobile phase methanol : aquabidest (90:10 v/v) with added asetic acid until pH 4,0, flow rate 1,5 ml/menit, UV detector at wavelength 230 nm.
Validation parameter to be used are accuracy, precision, specificity and linearity. Result of the research for paracetamol and ibuprofen respectively are linearity 0,998 at range 70-175 ppm and 0,999 at range 40-100 ppm; Recovery and CV value respectively are 101,03-111,55% and 1,78%; 91,46-98,55% and 5,04%; 85,44-95,97% and 3,09% for paracetamol 70 ppm; 122,5 ppm; 175 ppm whereas ibuprofen 40 ppm; 70 ppm; 100 ppm are 85,84-100,75% and 7,38%; 90,14-101,35% and 7,99%; 92,53-107,89% and 7,99%. It shows that HPLC reversed phase method can be used for quantitative analysis of paracetamol and ibuprofen combine with 7:4 ratio.
xiii
HALAMAN PERSETUJUAN PEMBIMBING... iii
HALAMAN PENGESAHAN... iv
HALAMAN PERSEMBAHAN... v
PERNYATAAN PERSETUJUAN PUBLIKASI... vi
PRAKARTA... vii
PERNYATAAN KEASLIAN KARYA... x
INTISARI... xi
BAB I. PENGANTAR... 1
A. Latar Belakang... 1
1. Permasalahan... 3
2. Keaslian penelitian... 3
3. Manfaat penelitian... 3
B. Tujuan Penelitian... 4
BAB II. PENELAAHAN PUSTAKA... 5
xiv
E. Kromatografi Cair Kinerja Tinggi... 11
1. Definisi dan instrumentasi... 11
2. Kromatografi partisi... 15
3. Waktu tambat dan resolusi... 17
4. Analisis kualitatif dan kuantitatif... 18
F. Validasi Metode... 19
G. Landasan Teori... 22
H. Hipotesis... 23
BAB III. METODE PENELITIAN... 24
A. Jenis dan Rancangan Penelitian... 24
B. Variabel Penelitian... 24
C. Definisi Operasional... 24
D. Bahan Penelitian... 25
E. Alat Penelitian... 25
F. Tata Cara Penelitian... 26
1. Pembuatan fase gerak... 26
2. Pembuatan larutan baku parasetamol dan ibuprofen... 26
3. Optimasi metode... 26
4. Validasi metode analisis... 28
xv
C. Optimasi metode... 32
D. Analisis kualitatif... 38
E. Validasi metode analisis... 45
BAB V. KESIMPULAN DAN SARAN... 50
A. Kesimpulan... 50
B. Saran... 50
DAFTAR PUSTAKA... 51
LAMPIRAN... 54
xvi
Tabel II Kriteria rentangrecoveryyang dapat diterima..……… 20
Tabel III Kriteria KV yang dapat diterima...……… 20
Tabel IV Parameter analisis yang harus dipenuhi untuk syarat validasi metode...……… 22
Tabel V Pembuatan larutan campuran parasetamol dan ibuprofen... 28
Tabel VI Data kurva baku parasetamol... 36
Tabel VII Data kurva baku ibuprofen... 36
Tabel VIII Data waktu retensi... 39
Tabel IX Data %recoveryparasetamol dan ibuprofen... 46
xvii
Gambar 2. Struktur ibuprofen... 7
Gambar 3. Tingkat energi elektronik molekul...………... 9
Gambar 4. Peralatan KCKT...………. 12
Gambar 5. Reaksi silanisasi... 16
Gambar 6. Reaksi pembuatan kolom oktadesilsilan... 17
Gambar 7. Pemisahan dua senyawa... 18
Gambar 8. Reaksi kolom oktadesilsilan dengan asam klorida... 30
Gambar 9. Gugus kromofor dan auksokrom pada parasetamol... 33
Gambar 10. Gugus kromofor ibuprofen... 34
Gambar 11. Spektra gabungan parasetamol dan ibuprofen... 35
Gambar 12. Kurva baku parasetamol... 37
Gambar 13. Kurva baku ibuprofen... 38
Gambar 14. Kromatogram pemisahan dari campuran parasetamol dan ibuprofen dengan fase gerak metanol : aquabidest (90:10 v/v) pH 4,0 kecepatan alir 1,5 ml/menit... 40
Gambar 15. Bagian non-polar ibuprofen dan parasetamol... 41
Gambar 16. Interaksi parasetamol dengan fase gerak... 42
Gambar 17. Interaksi parasetamol dengan fase gerak... 43
xix
Lampiran 1. Sertifikat analisis parasetamol... 55
Lampiran 2. Sertifikat analisis ibuprofen... 56
Lampiran 3. Data penimbangan bahan... 57
Lampiran 4. Spektra panjang gelombang tumpang tindih parasetamol-ibuprofen dengan perbandingan 7 : 4... 58
Lampiran 5. Kromatogrampeakibuprofen mengekor pada pH 5,0... 59
Lampiran 6. Kromtogrampeakibuprofen pada pada pH 4,0... 60
Lampiran 7. Kromatogram waktu retensi parasetamol dan ibuprofen...……. 61
Lampiran 8. Kromatogram campuran parasetamol dan ibuprofen ( 7 : 4 )…….... 64
Lampiran 9. Data waktu retensi parasetamol dan ibuprofen... 66
Lampiran 10. Contoh perhitungan resolusi pemisahan parasetamol dan ibuprofen (7:4)... 67
Lampiran 11. Kromatogram fase gerak dan metanol... 68
Lampiran 12. Kromatogram baku parasetamol...……….. 69
Lampiran 13. Kromatogram baku ibuprofen...………. 78
Lampiran 14. Kromatogram validasi metode campuran parasetamol dan ibuprofen...……….... 87
Lampiran 15. Contoh perhitungan kadar parasetamol...………... 96
xx
Lampiran 19. Nilai AUC dan contoh perhitungan kadar terukur parasetamol dan
ibuprofen....……….…….. 100
Lampiran 20. Contoh perhitungan recovery parasetamol dan
ibuprofen.……….…. 101
Lampiran 21. Contoh perhitungan CV parasetamol dan
1
A. Latar Belakang Penelitian
Dewasa ini, telah banyak obat analgesik yang beredar di pasaran, dengan bentuk sediaan obat yang beranekaragam antara lain berupa tablet, kaplet, kapsul, dan lain-lain. Beberapa obat analgesik memiliki zat aktif yang merupakan
campuran dua senyawa, antara lain parasetamol yang dikombinasikan dengan ibuprofen. Salah satu produk obat yang beredar di pasaran adalah tablet merk “X”
dengan kandungan parasetamol (350 mg) dan ibuprofen (200 mg). Menurut Farmakope Indonesia Edisi IV (1995), tablet parasetamol atau ibuprofen mengandung parasetamol atau ibuprofen tidak kurang dari 90,0% dan tidak lebih
dari 110,0% dari jumlah yang tertera dari etiket. Jika kadar parasetamol dan ibuprofen kurang dari yang tertera pada etiket maka efek yang dihasilkan kurang
maksimal sedangkan jika kadar parasetamol dan ibuprofen lebih dari yang tertera pada etiket maka dapat mengakibatkan kerusakan hati (Beers et al., 2003). Oleh karena itu perlu dilakukan analisis kuantitatif untuk menjamin kebenaran kadar
obat tersebut.
Penetapan kadar campuran parasetamol dan ibuprofen merupakan cara
yang digunakan untuk menjamin kesesuaian kadar yang tertera pada etiket. Metode Kromatografi Cair Kinerja Tinggi (KCKT) fase terbalik dapat digunakan untuk menetapkan kadar campuran parasetamol dan ibuprofen karena metode ini
dan dapat dihubungkan dengan detektor yang sesuai (Jhonson and Stevenson, 1978).
Telah dilakukan penelitian mengenai “Optimasi Pemisahan Campuran Parasetamol dan Ibuprofen Dengan Metode Kromatografi Cair Kinerja Tinggi
Fase Terbalik” oleh Prabowo (2010). Berdasarkan penelitian Prabowo (2010) diperoleh kondisi optimal sistem KCKT fase terbalik untuk validasi metode maupun penetapan kadar tablet merk “X”. Kondisi optimal sistem KCKT fase
terbalik yang diperoleh adalah fase diam oktadesilsilan (C18); fase gerak metanol : aquabidest (90:10 v/v) dengan penambahan asam asetat glasial hingga pH 4.0; dan
kecepatan alir 1,5 ml/menit.
Jaminan bahwa metode KCKT fase terbalik yang akan digunakan untuk penetapan kadar campuran parasetamol dan ibuprofen dapat diketahui melalui
validasi metode. Parasetamol dan ibuprofen merupakan zat aktif yang biasa digunakan sebagai analgesik. Menurut United State of Pharmacopeia (2007),
prosedur analisis kuantitatif untuk menetapkan kadar komponen utama bahan obat atau zat aktif dalam sediaan farmasi termasuk kategori 1, dan parameter yang harus dianalisis adalah akurasi, presisi, spesifisitas, dan linearitas. Oleh karena itu,
1. Permasalahan
Berdasarkan latar belakang tersebut maka permasalahan yang muncul
adalah apakah metode KCKT fase terbalik dengan fase gerak metanol : aquabidest (90:10 v/v) dengan penambahan asam asetat glasial hingga pH 4.0 pada penetapan
kadar campuran parasetamol dan ibuprofen dengan perbandingan 7:4 memenuhi parameter validitas yaitu akurasi, presisi, spesifisitas dan linearitas yang baik.
2. Keaslian Penelitian
Sejauh pengamatan penulis, telah dilakukan penelitian mengenai
parasetamol dan ibuprofen dalam suatu tablet menggunakan KCKT fase terbalik dengan fase gerak acetonitrile : phosphate buffer (60:40, v/v, pH 7.0), pada kecepatan alir 0,8 ml/menit dengan detektor Ultraviolet pada 260 nm. Namun
penelitian mengenai parasetamol dan ibuprofen dalam suatu tablet menggunakan KCKT fase terbalik dengan fase gerak metanol : aquabidest (90:10 v/v), dengan
asam asetat glasial pH 4.0 belum pernah dilakukan sebelumnya.
3. Manfaat Penelitian
a. Manfaat teoritis. Penelitian ini diharapkan dapat memberikan informasi mengenai parasetamol dan ibuprofen dengan metode KCKT fase
terbalik.
parameter-parameter validitas parasetamol dan ibuprofen menggunakan metode KCKT fase terbalik.
B. Tujuan Penelitian
Berdasarkan latar belakang dan permasalahan yang muncul maka penelitian ini bertujuan untuk mengetahui parameter validitas yaitu akurasi, presisi, spesifisitas dan linearitas pada metode KCKT fase terbalik dengan fase
gerak metanol : aquabidest (90:10 v/v) dengan penambahan asam asetat glasial hingga pH 4.0 pada penetapan kadar campuran parasetamol dan ibuprofen dengan
5
A. Parasetamol
Parasetamol secara umum dikenalkan pada tahun 1955 untuk mengobati rasa sakit dan demam pada anak-anak, namun menjadi obat bebas pada tahun 1960. Parasetamol atau yang juga disebut asetaminofen, bukan merupakan suatu
Non-Steroidal Anti Inflammantory Drug(NSAID), namun biasanya dibandingkan dengan aspirin dalam mengobati rasa sakit dan demam. Walaupun parasetamol
hampir tidak mempunyai efek samping di bagian pencernaan, tetapi mengkonsumsi parasetamol dalam waktu yang lama mempunyai beberapa resiko termasuk kerusakan hati (Beerset al.,2003).
Parasetamol mengandung tidak kurang dari 98,0% dan tidak lebih dari 101,0% C8H9NO2, dihitung terhadap zat anhidrat. Pemeriannya berupa serbuk
hablur, putih; tidak berbau; rasa sedikit pahit. Kelarutannya yaitu larut dalam air mendidih, dalam natrium hidroksida 1 N, dan mudah larut dalam etanol (Anonim, 1995).
Parasetamol memiliki pKa 9,5. Serapan maksimum parasetamol pada
daerah ultraviolet di larutan asam adalah 244 nm ( = 668) dan dalam larutan
basa adalah 257 nm ( = 715) (Clarke, 1986). atau serapan jenis adalah serapan dari larutan 1% zat terlarut dalam sel dengan ketebalan 1 cm (Anonim,
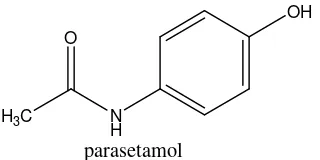
O
Gambar 1. Struktur Parasetamol (Anonim, 1995)
Parasetamol atau N-(4-hidroksifenil) asetamida merupakan agen analgesik maupun antipiretik. Parasetamol efektif dalam mengobati sakit kepala,
neuralgia dan sakit pada otot dan persendian (Battu and Reddy, 2009).
Penggunaan normal atau dosis yang dianjurkan adalah 650 mg/dosis
tunggal dan di atas 3250 mg untuk dewasa. Kelebihan dosis parasetamol dapat menyebabkan kerusakan hati bahkan kematian. Kombinasi parasetamol dan alkohol bisa meracuni liver, maka orang yang mengonsumsi lebih dari tiga gelas
minuman beralkohol disarankan untuk mengurangi asupan parasetamol dari dosis biasa (Anonim, 2009b).
B. Ibuprofen
Ibuprofen mengandung tidak kurang dari 97,0% dan tidak lebih dari
103,0% C13H18O2, dihitung terhadap zat anhidrat. Pemeriannya berupa serbuk hablur, putih hingga hampir putih; berbau khas lemah. Kelarutannya yaitu praktis
CH3
Gambar 2. Struktur Ibuprofen (Anonim, 1995)
Ibuprofen mempunyai berat molekul 206 g/mol. Ibuprofen termasuk NSAID, biasa digunakan untuk gejala arthritis, primary dysmenorrheal, demam
dan sebagai analgesik (Battu and Reddy, 2009).
Kelebihan dosis ibuprofen dapat menyebabkan kerusakan pada bagian perut maupun usus halus. Dosis maksimum ibuprofen untuk dewasa adalah 800
mg/dosis atau 3200 mg/hari (4 kali dosis maksimum). Dianjurkan untuk menggunakan dosis terendah untuk menghilangkan rasa sakit, kembung atau
demam. Ibuprofen tidak dikonsumsi jika sedang mengkonsumsi aspirin untuk mencegah stroke atau gangguan hati. Ibuprofen dapat membuat efektivitas dari aspirin berkurang dalam melindungi hati dan pembuluh darah. Ketika
mengkonsumsi ibuprofen tidak meminum alkohol karena dapat meningkatkan resiko pendarahan pada bagian perut (Anonim, 2009a).
C. Penelitian Terdahulu
Penelitian mengenai parasetamol dan ibuprofen dalam tablet pernah
dilakukan di India. Battu dan Reddy mengemukakan penelitian parasetamol dan ibuprofen dalam tablet menggunakan KCKT fase terbalik dengan fase gerak
dengan detektor UV pada panjang gelombang 260 nm memperoleh hasil yang baik. Hasil validasi yang ditunjukkan untuk parasetamol dan ibuprofen
berturut-turut yaitu persen recovery 98,90 ± 0,815% dan 96,01 ± 0,580%; koefisien korelasi 0,999 dan 0,998; nilai resolusi 1,30 dan 1,30; nilai Limit of Detection
(LOD) 6 ng/ml dan 10 ng/ml; dan nilaiLimit of Quantitation(LOQ) 15 ng/ml dan 25 ng/ml (Battu and Reddy, 2009).
Penelitian mengenai “Optimasi Pemisahan Campuran Parasetamol dan
Ibuprofen Dengan Metode Kromatografi Cair Kinerja Tinggi Fase Terbalik” juga telah dilakukan oleh Prabowo di Yogyakarta. Kondisi optimal sistem
Kromatografi Cair Kinerja Tinggi (KCKT) fase terbalik yang didapat adalah fase gerak metanol : aquabidest dengan perbandingan 90:10 dengan penambahan asam asetat glasial hingga pH 4 pada kecepatan alir 1,5 ml/menit, dengan nilai resolusi
sebesar ± 6,40 menggunakan detektor UV pada panjang gelombang 230 nm (Prabowo, 2010).
D. Spektrofotometri UV
Spektroskopi adalah salah satu teknik analisis fisika-kimia yang
mengamati tentang interaksi atom atau molekul dengan radiasi elektromagnetik (REM). Spektrofotometri UV merupakan suatu teknik analisis spektroskopi yang
banyaknya radiasi elektromagnetik yang diserap dengan panjang gelombangnya, yang disebut dengan spektrum absorpsi (Mulja dan Suharman, 1995).
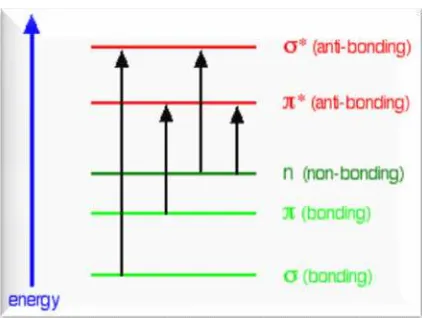
Suatu molekul jika dikenai radiasi elektromagnetik maka akan terjadi eksitasi ke tingkat energi yang lebih tinggi yang dikenal sebagai orbital elektron
antibonding. Terdapat 4 tipe transisi elektronik yang mungkin terjadi yaitu σ
σ*
, n σ*, n π*, π π*. Eksitasi elektron (σ σ*) memberikan energi terbesar dan terjadi pada daerah ultraviolet jauh yang diberikan oleh ikatan
tunggal, misalnya alkana. Eksitasi elektron (n σ*) terjadi pada gugus karbonil (dimetil keton dan asetaldehid) yang terjadi pada daerah ultraviolet jauh. Eksitasi
elektron (π π*) diberikan oleh ikatan rangkap dua dan tiga, yang juga terjadi pada daerah ultraviolet jauh (Mulja dan Suharman, 1995).
Gambar 3. Tingkat energi elektronik molekul (Skooget al.,1998)
Senyawa yang dapat diukur secara spektrofotometri ultraviolet harus
memiliki gugus kromofor dan auksokrom (Mulja dan Suharman, 1995). Molekul suatu senyawa dapat memberikan serapan radiasi elektromagnetik jika memiliki
auksokrom, yaitu gugus yang tidak menyerap radiasi namun terikat bersama dengan kromofor dan dapat meningkatkan penyerapan oleh kromofor atau
mengubah panjang gelombang serapan maksimum (Christian, 2004).
Dalam praktek spektrofotometri ultraviolet digunakan terbatas pada
sistem terkonjugasi. Meskipun demikian terdapat keuntungan yang selektif dari serapan ultraviolet, yaitu gugus-gugus karakteristik dapat dikenal dalam molekul yang sangat kompleks (Sastrohamidjojo, 2002).
Spektrofotometri ultraviolet melibatkan energi elektronik yang cukup besar pada molekul yang dianalisis, sehingga spektrofotometer ultraviolet lebih
banyak untuk analisis kuantitatif daripada kualitatif. Analisis kuantitatif selalu melibatkan pembacaan absorban radiasi elektromagnetik oleh molekul, atau radiasi elektromagnetik yang diteruskan, yang disebut absorben (A) tanpa satuan
dan transmitan dengan satuan persen (%T). Bouger, Lambert, dan Beer membuat formula secara matematik hubungan antara transmitan atau absorben terhadap
intensitas radiasi atau konsentrasi zat yang dianalisis dan tebal larutan yang mengabsorpsi sebagai berikut (Mulja dan Suharman, 1995):
(1)
Dimana T = persen transmitan
I0 = intensitas radiasi yang datang It = intensitas radiasi yang diteruskan
= daya serap molar (Lt.mol-1.cm-1) c = konsentrasi (mol. Lt-1)
b = tebal larutan (cm) A = serapan/absorbansi
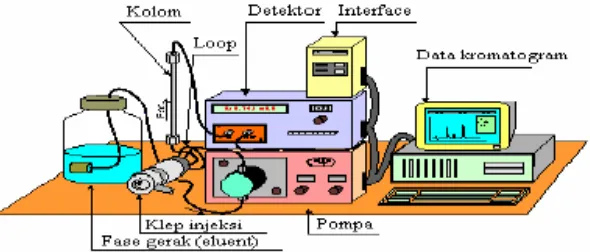
E. Kromatografi Cair Kinerja Tinggi 1. Definisi dan Instrumentasi
Kromatografi adalah teknik pemisahan fisik suatu campuran zat-zat kimia yang berdasar pada perbedaan migrasi dari masing-masing komponen
campuran yang terpisah pada fase diam di bawah pengaruh pergerakan fase yang bergerak. Kromatografi bertujuan untuk memisahkan komponen dari matriks
sampel dan tetap dibiarkan dalam fase diam kemudian ditentukan untuk analisis (Mulja dan Suharman, 1995).
Kemajuan dalam teknologi kolom, sistem pompa tekanan tinggi, dan
detektor yang sensitif telah menyebabkan perubahan kromatografi kolom cair menjadi suatu sistem pemisahan dengan kecepatan dan efisiensi yang tinggi.
Metode ini dikenal sebagai kromatografi cair kinerja tinggi (KCKT) (Anonim, 1995).
Kromatografi cair kinerja tinggi merupakan salah satu metode
kromatografi cair yang fase geraknya dialirkan secara cepat dengan bantuan tekanan dan hasilnya dideteksi dengan instrumen. Tidak seperti kromatografi gas,
lebih banyak interaksi spesifik untuk terjadinya pemisahan senyawa (Willard et al., 1988).
Metode KCKT banyak digunakan karena mempunyai banyak keuntungan, antara lain: mampu memisahkan molekul-molekul dari suatu
campuran, mudah melaksanakannya, kecepatan analisis dan kepekaan tinggi, dapat menghindari terjadinya dekomposisi atau kerusakan bahan yang dianalisis, resolusi yang baik, dapat digunakan untuk bermacam-macam detektor, kolom
dapat digunakan kembali, mudah melakukan “sample recovery” (Snyder and Kirkland, 1979). Selain itu, zat – zat yang tidak menguap atau tidak tahan panas
dapat dikromatografi tanpa peruraian atau tanpa perlunya membuat derivat yang dapat menguap (Anonim, 1995).
Keterbatasan metode KCKT adalah untuk mengidentifikasi senyawa
kecuali jika KCKT dihubungkan dengan spektrometer massa (MS). Keterbatasan lainnya adalah jika analit yang akan digunakan sangat kompleks maka resolusi
yang baik sulit diperoleh (Rohman dan Gandjar, 2007). Peralatan KCKT dapat dilihat pada gambar 4.
Tiga variabel utama yang harus diperhatikan untuk proses pemisahan dan analisis menggunakan KCKT adalah fase gerak, fase diam dan detektor.
a. Fase gerak. Kemampuan KCKT untuk memisahkan banyak senyawa terutama tergantung pada keanekaragaman fase gerak. Fase gerak pada KCKT
sangat berpengaruh pada tambatan dan pemisahan senyawa (Munson, 1984). Fase gerak untuk analisis secara KCKT harus murni untuk mencegah adanya peak pengganggu yang dapat tumpang tindih dengan peak analit, tidak bereaksi atau
mempengaruhi kolom, dapat melarutkan analit, memiliki titik didih 20-500C di atas temperatur kolom, viskositasnya rendah (tidak lebih dari 50 cP) dan
memungkinkan untuk memperoleh kembali analit dengan mudah (jika diperlukan), tidak mudah terbakar dan toksisitasnya rendah, memiliki harga yang wajar (Skoog et al., 1985). Fase gerak KCKT juga harus bebas dari gas yang
terlarut karena dapat mempengaruhi respon detektor sehingga memunculkan sinyal palsu dan akan mempengaruhi kolom (Gritteret al., 1985). Oleh karena itu,
peralatan degassing diperlukan untuk menghilangkan gas yang terlarut di dalam fase gerak (Dean, 1995).
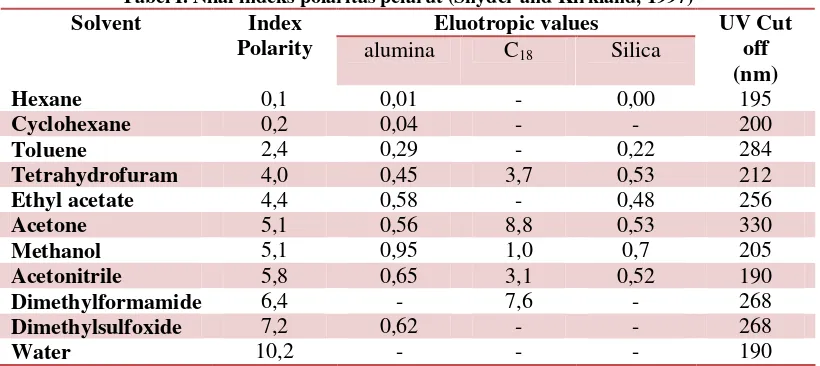
Berikut ini merupakan tabel beberapa nilai indeksi polaritas dari
Tabel I. Nilai indeks polaritas pelarut (Snyder and Kirkland, 1997)
Eluotropic values Solvent Index
Polarity alumina C18 Silica
UV Cut off (nm)
Hexane 0,1 0,01 - 0,00 195
Cyclohexane 0,2 0,04 - - 200
Toluene 2,4 0,29 - 0,22 284
Tetrahydrofuram 4,0 0,45 3,7 0,53 212
Ethyl acetate 4,4 0,58 - 0,48 256
Acetone 5,1 0,56 8,8 0,53 330
Methanol 5,1 0,95 1,0 0,7 205
Acetonitrile 5,8 0,65 3,1 0,52 190
Dimethylformamide 6,4 - 7,6 - 268
Dimethylsulfoxide 7,2 0,62 - - 268
Water 10,2 - - - 190
Tabel di atas menunjukkan bahwa semakin besar eluotropic values dari pelarut
menunjukkan semakin mudah untuk mengelusi sampel. Semakin besar indeks polaritas yang dimiliki oleh pelarut maka semakin bersifat polar pelarut yang digunakan (Snyder and Kirkland, 1997).
b. Fase diam. Kolom merupakan bagian yang sangat penting dalam pemisahan komponen-komponen sampel. Keberhasilan pemisahan komponen
sampel bergantung pada keadaan kolom (Mulja dan Suharman, 1995). Kolom pada KCKT dapat berupa gelas atau baja tidak berkarat. Kolom gelas dapat menahan tekanan sampai 50 atm. Panjang kolom bervariasi antara 15-150 cm.
Pengisi kolom biasanya adalah silika gel, alumina dan elit (Khopkar, 1990). Analit yang polar, terutama yang bersifat basa atau memiliki gugus amin
akan memberikan puncak yang mengekor (tailing peak) pada penggunaan fase diam silika fase terikat. Hal ini disebabkan oleh adanya interaksi adsorbsi antara gugus amin pada analit dengan residual silanol dan pengotor logam yang terdapat
residual silanol dengan gugus trimetilsilil dan menggunakan silika dengan kemurnian tinggi (kandungan logam < 1 ppm) (Rohman dan Gandjar, 2007).
c. Detektor. Detektor UV umumnya digunakan untuk analisis bahan organik bergugus fungsi (Khopkar, 1990). Detektor ini didasarkan pada adanya
penyerapan radiasi ultraviolet oleh spesies analit yang mempunyai struktur atau gugus kromoforik. Detektor dengan panjang gelombang yang bervariasi lebih berguna karena seorang analisis dapat memilih panjang gelombang dengan
sensitifitas yang paling tinggi (Rohman dan Gandjar, 2007).
Persyaratan detektor KCKT adalah sensitifitasnya harus sangat tinggi
(10-8-10-15 g analit/detik); kestabilan dan reprodusibilitas yang baik; memberikan respon yang linier terhadap konsentrasi analit; dapat bekerja pada temperatur kamar sampai 4000C; tidak terpengaruh oleh perubahan temperatur dan kecepatan
fase gerak; mudah didapat dan mudah dioperasikan; selektif terhadap berbagai macam analit di dalam fase gerak; tidak merusak analit; dapat menghilangkan
“zone broadening”dengan adanya pengaruh minimal internal volume (Mulja dan Suharman, 1995).
2. Kromatografi partisi
Prinsip kromatografi partisi didasarkan pada partisi solut di antara dua
fase yang tidak saling campur, karena adanya perbedaan koefisien distribusi dari masing-masing senyawa. Jika solut ditambahkan ke dalam sistem yang terdiri dari dua pelarut tidak saling campur dan keseluruhan sistem dibiarkan setimbang,
(3)
K adalah koefisien distribusi, Cs adalah konsentrasi solut dalam fase diam, dan Cm adalah konsentrasi solut dalam fase gerak (Jhonson and Stevenson, 1978).
Pada kromatografi partisi, fase diam dapat polar atau nonpolar. Bila fase
diam polar dan fase gerak nonpolar disebut kromatografi partisi fase normal, sedangkan bila fase diam nonpolar dan fase gerak polar dinamakan kromatografi
fase terbalik (Munson, 1984).
Penggunaan temperatur kolom hanya beberapa derajat di bawah temperatur kamar akan meningkatkan reprodusibilitas waktu retensi dan
meningkatkan presisi analisis kuantitatif. Permukaan silika pada kolom memilki gugus silanol (Si-OH) sampai 8 μmol per meter persegi. Gugus silanol akan
mengalami disosiasi menjadi bermuatan negatif Si-O- pada pH di atas 3. Gugus Si-O- akan mengikat gugus amin terprotonasi secara kuat dan menyebabkan
tailing.Kromatografi partisi menggunakan fase diam silika yang ditempeli gugus secara kovalen pada permukaannya (Harris, 1999). Gugus yang ditempelkan pada silanol tersebut pada umumnya adalah hidrokarbon rantai panjang sehingga fase
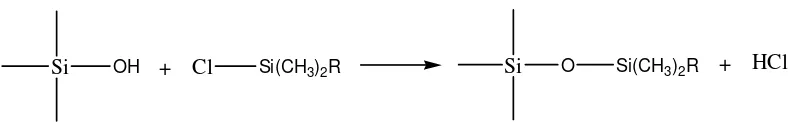
gerak umumnya lebih polar dari fase diam (Skoog et al., 1988). Reaksi yang terjadi adalah sebagai berikut :
Si OH + Cl Si(CH3)2R Si O Si(CH3)2R + HCl
Reaksi tersebut digunakan untuk membuat isian kolom oktadesilsilan (ODS) gugus silanol dan oktadesilklorosilan sebagai berikut :
Si OH + Cl Si (CH2)17CH3 Si O Si (CH2)17CH3 + HCl
Gambar 6. Reaksi pembuatan kolom oktadesilsilan
Pada kromatografi partisi fase terbalik dengan kemasan fase terikat, R
pada siloksan biasanya berupa gugus C18 atau C8. Panjang pendeknya rantai karbon mempengaruhi tertambatnya senyawa pada fase diam (Skooget al.,1998).
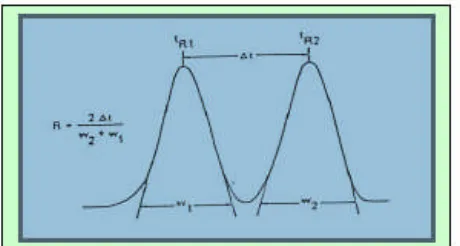
3. Waktu tambat (tR) dan resolusi
Waktu tambat atau retensi (tR) merupakan selang waktu yang diperlukan
oleh analit mulai saat injeksi hingga keluar dari kolom dan sinyalnya ditangkap detektor (Mulja dan Suharman, 1995). Faktor resolusi adalah ukuran pemisahan
dari 2 puncak. Daya pisah, R, diukur dengan persamaan berikut :
(4)
Harga tR2 dan tR1 adalah waktu retensi senyawa, diukur pada titik maksimum
Gambar 7. Pemisahan dua senyawa (Jhonson and Stevenson, 1978)
Harga R > 1,5 disebut baseline resolution, yaitu pemisahan sempurna
dari dua puncak dengan ukuran yang sama. Namun dalam prakteknya, pemisahan dengan harga R = 1,0 (kedua puncak berhimpit lebih kurang 2%) dianggap
memadai (Pescoket al.,1976).
4. Analisis kualitatif dan kuantitatif
Kromatografi Cair Kinerja Tinggi digunakan untuk analisis kualitatif dan kuantitatif dari suatu sampel atau cuplikan selain untuk memisahkan senyawa
dalam sampel. Hasil dari pemisahan adalah kromatogram. Dari kromatogram diperoleh informasi mengenai waktu retensi suatu senyawa (Noegrohati, 1994).
Waktu retensi yang menunjukkan identitas suatu senyawa merupakan
selang waktu yang diperlukan senyawa mulai pada saat injeksi sampai keluar dari kolom dan sinyalnya ditangkap oleh detektor (Gritteret al.,1985). Setiap senyawa
membandingkan waktu retensi senyawa murni dengan waktu retensi senyawa yang dimaksud dalam sampel (Gritteret al.,1985).
Analisis kuantitatif dilakukan berdasarkan perbandingan tinggi atau luas puncak kromatogram senyawa sampel terhadap senyawa standar. Bila variasi
keadaan kolom tidak menyebabkan pelebaran puncak, maka analisis berdasarkan tinggi puncak dapat memberikan ketelitian tinggi. Analisis berdasarkan luas puncak tidak dipengaruhi oleh pelebaran puncak. Oleh karena itu cara ini lebih
disukai dalam perhitungan kuantitatif (Noegrohati, 1994).
F. Validasi Metode
Validasi menurut USP (United States Pharmacopeia) dilakukan untuk menjamin bahwa metode analisis bersifat akurat, spesifik, reprodusibel, dan tahan
pada kisaran analit yang akan dianalisis. Secara singkat, validasi merupakan aksi konfirmasi bahwa metode analisis yang akan digunakan sesuai dengan tujuan
yang diinginkan (Rohman, 2009). Untuk itu diperlukan suatu pedoman mengenai kesahihan metode analisis yang didukung oleh parameter – parameter validitas yaitu akurasi, presisi, linearitas, spesifisitas, LOD (Limit of Detection) dan LOQ
(Limit of Quantitation). 1. Akurasi
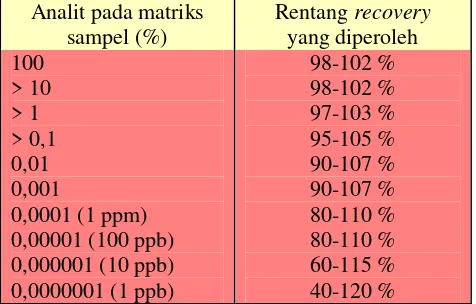
2004). Persen perolehan kembali yang dapat diterima bergantung pada matriks analit, prosedur pengolahan analit dan konsentrasi analit (Anonim, 2004).
Tabel II. Kriteria rentangrecoveryyang dapat diterima (Anonim, 2007)
Analit pada matriks
Presisi atau keseksamaan adalah ukuran yang menunjukkan derajat
kesesuaian antara hasil uji individual, diukur melalui penyebaran hasil individual dari rata-rata jika prosedur diterapkan secara berulang pada sampel-sampel yang
diambil dari campuran yang homogen. Presisi biasanya dinyatakan dalam koefisien variasi (KV) atau persen Relative Standard Deviation (RSD) (Harmita, 2004).
Tabel III. Kriteria KV yang dapat diterima (Anonim, 2007)
Kadar Analit KV (%) ≥1 %
Linearitas merupakan kemampuan suatu metode (pada rentang tertentu)
konsentrasi (jumlah) analit di dalam sampel (Anonim, 2007). Linearitas suatu metode merupakan ukuran seberapa baik kurva kalibrasi yang menghubungkan
antara respon (y) dengan konsentrasi (x) (Rohman, 2009). Persyaratan data linearitas yang bisa diterima jika memenuhi nilai koefisien korelasi (r) > 0,99 atau
r2≥0,997 (Chanet al., 2004).
4. Spesifisitas
Spesifisitas suatu metode adalah kemampuannya yang hanya mengukur zat tertentu saja secara cermat dan seksama dengan adanya komponen lain yang
mungkin ada dalam matriks sampel. Selektivitas metode ditentukan dengan membandingkan hasil analisis sampel yang mengandung cemaran, hasil urai, senyawa sejenis, senyawa asing lainnya atau pembawa plasebo dengan hasil
analisis sampel tanpa penambahan bahan-bahan tadi (Harmita, 2004).
5. LOD (Limit of Detection) dan LOQ (Limit of Quantitation)
Limit of Detectionadalah jumlah terkecil analit dalam sampel yang dapat dideteksi dan masih memberikan respon signifikan dibandingkan dengan blanko.
Limit of Quantitation merupakan kuantitas terkecil analit dalam sampel yang masih dapat memenuhi kriteria akurasi dan presisi. LOD dan LOQ dapat dihitung
a. Kategori I. Mencakup prosedur analisis kuantitatif, untuk menetapkan kadar komponen utama bahan obat atau zat aktif dalam sediaan farmasi.
b. Kategori II. Mencakup prosedur analisis kualitatif dan kuantitatif yang digunakan untuk menganalisis impurities ataupun degradation compounds dalam
sediaan farmasi.
c. Kategori III. Mencakup prosedur analisis yang digunakan untuk menentukan karakteristik penampilan suatu sediaan farmasi, misalnya disolusi dan
pelepasan obat.
d. Kategori IV (tes identifikasi)
Tabel IV. Parameter analisis yang harus dipenuhi untuk syarat validasi metode (Anonim, 2007)
* = Mungkin diperlukan (tergantung sifat spesifik tes)
G. Landasan Teori
Beberapa obat analgesik yang beredar di pasaran adalah obat tunggal maupun campuran, antara lain campuran parasetamol dan ibuprofen. Parasetamol dan ibuprofen merupakan senyawa non-volatile dan tidak tahan pada suhu tinggi
suatu senyawa dengan kadar yang sangat kecil dan dan selektif terhadap senyawa yang akan dianalisis.
Salah satu sistem KCKT yang dapat untuk memisahkan parasetamol dan ibuprofen adalah metode KCKT fase terbalik, yaitu fase diam (C18) lebih
non-polar dibandingkan dengan fase geraknya (metanol dan aquabidest). Kondisi optimal sistem KCKT fase terbalik adalah fase diam oktadesilsilan (C18); fase gerak metanol : aquabidest (90:10 v/v) dengan penambahan asam asetat glasial
hingga pH 4.0; dan kecepatan alir 1,5 ml/menit. Parameter validitas yang akan diukur adalah akurasi, presisi, spesifisitas dan linearitas.
H. Hipotesis
Metode KCKT fase terbalik dengan fase gerak metanol : aquabidest (90 :
10 v/v) dengan penambahan asam asetat glasial hingga pH 4.0 pada penetapan kadar campuran parasetamol dan ibuprofen dengan perbandingan 7:4 memenuhi
24
A. Jenis dan Rancangan Penelitian
Penelitian yang dilakukan bersifat noneksperimental deskriptif karena tidak terdapat manipulasi dan perlakuan terhadap subjek uji.
B. Variabel Penelitian
1. Variabel bebas adalah sistem KCKT yang meliputi komposisi fase gerak dan
kecepatan alir.
2. Variabel tergantung adalah parameter validasi yaitu akurasi, presisi, spesifisitas dan linearitas.
3. Variabel pengacau terkendali adalah pelarut dan asal parasetamol dan ibuprofen.
C. Definisi Operasional
1. Sistem KCKT fase terbalik yang digunakan dalam penelitian adalah fase diam
berupa kolom oktadesilsilan (C18) dan fase gerak berupa campuran metanol p.a. dan aquabidest dengan perbandingan 90:10 dengan penambahan asam
asetat glasial hingga pH 4,0.
3. Parameter validasi yang digunakan adalah akurasi, presisi, spesifisitas dan linearitas.
D. Bahan Penelitian
Baku parasetamol dan ibuprofen (Working standard, PT. Konimex), metanol (p.a., E. Merck), aquabidestilata (Pharmaceutical laboratories, PT. Ikapharmindo Putranas), dan asam asetat (glasial, E. Merck), asam sulfat pekat
(p.a.,E. Merck), dan kalium bikromat (p.a.,E. Merck).
E. Alat Penelitian
Spektrofotometer uv/vis (Optima SP 3000 F), sistem KCKT (model LC-2010C HT, CAT No. 228-46703-38, SERIAL No. C21254706757 LP, Shimadzu
Coorporation), kolom oktadesilsilan (C18) merkKNAUER, seperangkat komputer (merk Dell B6RDZ1SConnexant SystemRD01-D850 A03-0382 JP France S.A.S,
UPS Prolink Model PR0650P, printer HP Deskjet D2566 HP-024-000 625 730), ultrasonikator (Retsch tipe T460 no V935922013 Ey), vaccum (gaast model DOA-P104-BN), organic solvent membrane filter (Whatman) ukuran pori
(0,45μm ; diameter 47mm), indikator pH, penyaring Millipore, mikropipet Socorex,neraca kasar, neraca analitik (Scaltecmax 60/210 g; d = 0,01/0,1 mg; e =
F. Tata Cara Penelitian 1. Pembuatan fase gerak
Fase gerak yang digunakan dalam penelitian menggunakan campuran metanol : aquabidest dengan perbandingan 90:10 ditambah dengan asam asetat
hingga pH 4. Fase gerak dibuat dalam labu 500,0 ml kemudian digojog dan disaring dengan penyaring Whatman organik dengan bantuan pompa vakum dan didegassingselama 15 menit.
2. Pembuatan larutan baku parasetamol dan ibuprofen
a. Larutan stok parasetamol. Parasetamol lebih kurang 10,0 mg ditimbang seksama dan dilarutkan dengan metanol dalam labu takar 10,0 ml hingga tanda.
b. Larutan stok ibuprofen. Ibuprofen baku lebih kurang 10,0 mg ditimbang seksama dan dilarutkan dengan metanol dalam labu takar 10,0 ml
hingga tanda.
3. Optimasi metode
a. Penetapan panjang gelombang pengamatan. Larutan stok parasetamol diambil 0,105 ml dan diencerkan dengan metanol dalam labu takar 10,0 ml hingga
tanda, sehingga didapatkan konsentrasi sebesar 10,5 ppm. Larutan stok ibuprofen diambil 0,060 ml dan diencerkan dengan metanol dalam labu takar 10,0 ml hingga tanda, sehingga didapatkan konsentrasi sebesar 6 ppm. Dari kadar parasetamol
gelombang 220-280 nm, sehingga dapat diketahui absorbansi masing-masing larutan kemudian dibuat spektra serapan antara panjang gelombang versus
absorbansi. Selanjutnya dari kurva parasetamol dan ibuprofen tersebut, spektra ditumpangtindihkan untuk mengetahui panjang gelombang pengamatan pada
deteksi dengan KCKT fase terbalik.
b. Penetapan kurva baku. Larutan stok parasetamol diambil 0,350; 0,525; 0,615; 0,700; 0,790 dan 0,875 ml dan diencerkan dengan metanol dalam labu
takar 5,0 ml hingga tanda, sehingga didapatkan konsentrasi sebesar 70; 105; 122,5; 140; 157,5; dan 175 ppm. Saring dengan milipore dan didegassing selama
15 menit. Masing-masing seri larutan baku parasetamol sebanyak 50,0 μl disuntikkan ke dalam sistem KCKT dengan kolom ODS (5mm×30cm) menggunakan fase gerak metanol : aquabidest dengan perbandingan 90:10
ditambah dengan asam asetat hingga pH 4 dan kecepatan alir 1,5 ml/menit. Replikasi dilakukan sebanyak 3 kali dan dipilih persamaan kurva baku
parasetamol yang paling baik.
Larutan stok ibuprofen diambil 0,2; 0,3; 0,35; 0,4; 0,45; dan 0,5 ml dan diencerkan dengan metanol dalam labu takar 5,0 ml hingga tanda, sehingga
didapatkan konsentrasi sebesar 40; 60; 70; 80; 90; dan 100 ppm. Saring dengan milipore dan didegassing selama 15 menit. Masing-masing seri larutan baku
1,5 ml/menit. Replikasi dilakukan sebanyak 3 kali dan dipilih persamaan kurva baku ibuprofen yang paling baik.
4. Validasi Metode Analisis
a. Pembuatan campuran parasetamol dan ibuprofen. Larutan campuran parasetamol dan ibuprofen dibuat dalam 3 konsentrasi yang berbeda yaitu campuran parasetamol 70 ppm dan ibuprofen 40 ppm; campuran parasetamol
122,5 ppm dan ibuprofen 70 ppm; dan campuran parasetamol 175 ppm dan ibuprofen 100 ppm.
Tabel V. Pembuatan larutan campuran parasetamol dan ibuprofen
Konsentrasi Ambil dari larutan stok Parasetamol 70 ppm 0,350 ml Rendah Ibuprofen 40 ppm 0,200 ml
Parasetamol 122,5 ppm 0,615 ml Tengah Ibuprofen 70 ppm 0,350 ml
Parasetamol 175 ppm 0,875 ml Tinggi Ibuprofen 100 ppm 0,500 ml
Ad dengan metanol hingga
tanda dalam labu takar 5,0
ml
Masing-masing campuran tersebut disaring dengan milipore dan didegassing
selama 15 menit. Replikasi dilakukan sebanyak 6 kali.
b. Penetapan recovery dan CV (Coefficient of Varriation). Masing-masing seri larutan campuran parasetamol dan ibuprofen sebanyak 50,0 μl disuntikkan ke
dalam sistem KCKT dengan kolom ODS (5mm×30cm) menggunakan fase gerak metanol : aquabidest dengan perbandingan 90:10 ditambah dengan asam asetat
hingga pH 4 dan kecepatan alir 1,5 ml/menit. Recovery dan CV (Coefficient of Varriation) parasetamol dan ibuprofen yang ada dalam campuran dihitung dengan
G. Analisis Hasil
Kesahihan dari metode yang digunakan dalam penetapan kadar
parasetamol dan ibuprofen dalam campuran secara KCKT fase terbalik dapat ditentukan berdasarkan parameter berikut :
1. Akurasi
Akurasi dinyatakan sebagai persen perolehan kembali (recovery) analit yang ditambahkan. Recovery dihitung dari kadar yang terukur pada kurva baku
dibandingkan dengan kadar yang diketahui dikalikan 100%.
2. Presisi
Presisi diukur sebagai simpangan baku relatif (koefisien variansi).
3. Linearitas
Linearitas dinyatakan dengan koefisien korelasi (r) pada analisis regresi linear.
30
A. Pembuatan Fase Gerak
Pemilihan fase gerak pada penelitian ini mengacu pada penelitian Prabowo (2010) yaitu menggunakan fase gerak metanol : aquabidest (90:10 v/v) dengan penambahan asam asetat glasial hingga pH 4,0. Fase gerak pada penelitian
bersifat polar sedangkan fase diam C18 bersifat non-polar sehingga sistem kromatografi yang digunakan adalah kromatografi partisi fase terbalik.
Fase gerak diatur pada pH 4,0 sebab bila fase gerak terlalu asam (pH≤2) dapat menyebabkan kolom kromatografi menjadi rusak. Oktadesilsilan akan melepaskan kembali gugus oktadesilnya sehingga akan mengakibatkan kolom
terdegradasi. Asam akan bereaksi dengan oktadesilsilan sehingga kembali ke bentuk silanol. Berikut ini gambaran reaksinya:
Si O Si (CH2)17CH3 Si OH + Cl Si (CH2)17CH3 H2O/HCl
+ H2O
Gambar 8. Reaksi kolom oktadesilsilan dengan asam klorida
Ibuprofen dengan fase gerak (metanol : aquabidest dengan perbandingan 90:10 v/v) tanpa pengaturan pH yaitu pH 5,0 menghasilkan peak yang mengekor (tailing) yang dapat dilihat pada lampiran 5. Oleh karena itu, dilakukan
pengaturan pH untuk ibuprofen menjadi pH 4,0 sebab pada pH 4,0 peak yang dihasilkan lebih bagus, yaitupeak runcing dan tidak mengekor yang dapat dilihat
Pengaturan pH dilakukan karena dari hasil studi pustaka diketahui bahwa dalam pemisahan ibuprofen digunakan fase gerak dengan pH 4 atau larutan buffer
pH 7. Pemilihan pH 4 dilakukan karena lebih efisien daripada penggunaan buffer dikarenakan buffer dapat tertambat kuat pada fase diam (kolom C18)
mengakibatkan waktu elusi menjadi lebih lama, tekanan pompa lebih tinggi, dan perlu waktu lebih lama saat mencuci kolom dari hasil sisa fase gerak dengan buffer.
Proporsi metanol pada komposisi fase gerak pada penelitian ini memiliki jumlah paling besar karena metanol memiliki viskositas yang rendah yaitu 0,54cP
sehingga dapat mengurangi tekanan pada kolom sehingga kolom tidak rusak, pompa menjadi lebih awet serta dapat memisahkan parasetamol dan ibuprofen dengan baik.
B. Pembuatan Larutan Baku
Larutan baku parasetamol dan ibuprofen dibuat dalam konsentrasi tertentu dengan menggunakan pelarut metanol p.a.. Pelarut yang digunakan memenuhi syarat pelarut yang dapat digunakan dalam sistem KCKT yaitu
memiliki kemurnian yang tinggi, dapat bercampur dengan fase gerak dan mudah terelusi.
Pembuatan kurva baku parasetamol dan ibuprofen disesuaikan dengan kadar yang tertera pada kemasan obat merk “X” yaitu 350 mg parasetamol dan 200 mg ibuprofen tiap tablet, sehingga perbandingan parasetamol dan ibuprofen
konsentrasi yang berbeda. Kadar parasetamol adalah 70 ppm; 105 ppm; 122,5 ppm; 140 ppm; 157,5 ppm; dan 175 ppm sedangkan kadar ibuprofen adalah 40
ppm; 60 ppm; 70 ppm; 80 ppm; 90 ppm; dan 100 ppm. Pemilihan seri konsentrasi kurva baku ini bertujuan agar kadar yang terdapat dalam sampel dapat masuk ke
dalam seri kurva baku sehingga persamaan kurva baku yang diperoleh dapat digunakan untuk penetapan kadar dalam sampel.
C. Optimasi metode 1. Penetapan Panjang Gelombang Pengamatan
Penetapan panjang gelombang tumpang tindih bertujuan untuk mengetahui panjang gelombang dimana parasetamol dan ibuprofen memiliki serapan yang optimal menggunakan spektrofotometer UV. Pengukuran panjang
gelombang tumpang tindih didasarkan pada perbandingan 7 : 4 untuk parasetamol dan ibuprofen. Konsentrasi yang digunakan untuk parasetamol dan ibuprofen
berturut-turut adalah 10,5 ppm dan 6 ppm.
Pelarut yang digunakan untuk mengukur panjang gelombang maksimum pada spektrofotometer UV dan untuk analisis pada sistem KCKT sama yaitu
metanol, dan diharapkan penggunaan pelarut yang sama dapat meningkatkan ketelitian pada pengukuran panjang gelombang tumpang tindih yang selanjutnya
Senyawa yang dapat ditetapkan kadarnya secara spektrofotometri UV harus memiliki gugus kromofor agar dapat menyerap radiasi sinar pada daerah
UV. Parasetamol dan ibuprofen memiliki gugus kromofor yang dapat menyerap radiasi sinar pada daerah UV sehingga dapat diukur secara spektrofotometri UV.
Pada gugus kromofor terdapat ikatan rangkap yang mengandung elektron π yang jika terkena radiasi elektromagnetik mudah bereksitasi ke tingkat energi yang lebih tinggi yaitu orbital π*. Selain memiliki gugus kromofor, parasetamol juga
mempunyai gugus auksokrom yang terikat langsung pada gugus kromofor. Gugus auksokrom adalah gugus yang tidak menyerap radiasi namun terikat bersama
dengan kromofor. Oleh karena itu, dengan adanya gugus auksokrom maka dapat mengubah panjang gelombang atau meningkatkan intensitas maksimum dari senyawa tersebut. Berikut adalah gambar gugus kromofor dan auksokrom dari
parasetamol.
Gambar 9. Gugus kromofor dan auksokrom pada parasetamol
--- = kromofor = auksokrom
Pada gambar 9 terlihat gugus kromofor dan auksokrom dari parasetamol. Parasetamol memiliki gugus kromofor yang merupakan ikatan rangkap yang
memiliki pasangan elektron bebas pada elektron n yang dapat berinteraksi dengan elektron π pada kromofor. Oleh karena itu, gugus auksokrom dapat mengubah
panjang gelombang maksimum dan intensitas serapan maksimum dari parasetamol.
Gambar 10. Gugus kromofor ibuprofen
--- = kromofor
Pada gambar 10 terlihat ibuprofen hanya mempunyai gugus kromofor dan tidak ada gugus auksokrom yang terikat langsung dengan gugus kromofor sehingga panjang gelombang ibuprofen lebih pendek dibandingkan dengan
parasetamol.
Spektra yang dihasilkan dari tumpangtindih parasetamol 10,5 ppm dengan ibuprofen 6 ppm (perbandingan 7:4) ditunjukkan pada gambar 11. Gambar
11 menunjukkan absorbansi antara parasetamol dan ibuprofen saling tumpang tindih antara 220 nm sampai 240 dan berpotongan pada panjang gelombang 230
nm. Analisis dengan sistem KCKT yang dilakukan menggunakan panjang gelombang 230 nm sebagai panjang gelombang pengamatan karena parasetamol dan ibuprofen memberikan hasil yang optimal. Campuran parasetamol dengan
ibuprofen (7:4) memiliki perbandingan yang lebih kecil untuk ibuprofen sehingga pemilihan penjang gelombang pengamatan lebih diutamakan untuk memperoleh
pada panjang gelombang 223 nm berdekatan dengan panjang gelombang serapan dari metanol sampai dengan 205 nm, sehingga spektra kemungkinan berasal dari
metanol bukan ibuprofen, hal tersebut mengakibatkan hasil penelitian menjadi kurang maksimal. Bila panjang gelombang pengamatan dilakukan pada panjang
gelombang maksimum parasetamol maka kemungkinan serapan ibuprofen terlalu kecil atau bahkan tidak memberikan serapan.
Gambar 11. Spektra serapan gabungan parasetamol (A) konsentrasi 10,5 ppm dan ibuprofen (B) konsentrasi 6 ppm
2. Penetapan kurva baku
Pembuatan kurva baku parasetamol dan ibuprofen untuk penetapan kadar dilakukan replikasi 3 kali. Persamaan kurva baku yang diperoleh menyatakan
hubungan yang linier antara konsentrasi analit dengan AUC (Area Under Curve) yang dihasilkan. Parameter linearitas yang digunakan adalah koefisien korelasi (r),
yang menunjukkan korelasi hubungan antara konsentrasi dengan AUC. Dari ketiga replikasi kurva baku yang diperoleh, dipilih salah satu kurva baku yang kemudian akan digunakan untuk analisis kuantitatif. Pemilihan kurva baku
persamaan kurva baku yang diperoleh dari parasetamol dan ibuprofen dapat dilihat pada tabel berikut.
Tabel VI. Data kurva baku parasetamol
Baku Parasetamol
Replikasi 1 Replikasi 2 Replikasi 3
Seri baku
67,900 2437397 69,300 2307560 71,400 2047792 101,850 3927465 103,950 3648424 107,100 3486008 119,310 4606396 121,770 4284769 125,460 3878446 135,800 5323163 138,600 4935837 142,800 4178335 153,260 6049820 156,420 5328015 161,160 4905496 169,750 7062694 173,250 6050790 178,500 5561752
A -617076,429 A -72879,641 A -82861,082
B 44271,589 B 35363,588 B 31223,768
r 0,998 r 0,997 r 0,993
Berdasarkan hasil yang diperoleh pada tabel VI, data kurva baku yang digunakan untuk parasetamol adalah replikasi 1, dikarenakan koefisien korelasi
yang diperoleh memiliki nilai tertinggi daripada replikasi lainnya yaitu 0,998. Kurva baku tersebut memenuhi persyaratan linearitas karena memiliki nilai (r) > 0,99 yaitu hasil uji yang secara langsung proporsional dengan konsentrasi
(jumlah) analit di dalam sampel (Chanet al., 2004).
Tabel VII. Data kurva baku ibuprofen
Baku Ibuprofen
Replikasi 1 Replikasi 2 Replikasi 3 Seri baku
38,000 781714 39,600 723600 40,400 878381 57,000 1029707 59,400 1031771 60,600 1218188 66,500 1153765 69,300 1203848 70,700 1409751 76,000 1319124 79,200 1336962 80,800 1636694 85,500 1507308 89,100 1514881 90,900 1847692 95,000 1646629 99,000 1671321 101,000 1965876
A 165051,100 A 89238,929 A 112782,971
B 15425,695 B 15948,001 B 18631,602
Berdasarkan hasil yang diperoleh pada tabel VII, data kurva baku yang digunakan untuk ibuprofen adalah replikasi 2, dikarenakan koefisien korelasi yang
diperoleh memiliki nilai tertinggi daripada replikasi lainnya yaitu 0,999. Kurva baku tersebut memenuhi persyaratan linearitas karena memiliki nilai (r) > 0,99
yaitu hasil uji yang secara langsung proporsional dengan konsentrasi (jumlah) analit di dalam sampel (Chanet al., 2004).
Persamaan kurva baku yang digunakan untuk analisis kuantitatif
parasetamol adalah y = 44271,589x – 617076,429 dan persamaan kurva baku untuk analisis kuantitatif ibuprofen adalah y = 15948,001x + 89238,929.
Gambar 13. Kurva baku ibuprofen
Berdasarkan gambar kurva baku di atas, dapat dilihat bahwa AUC
parasetamol maupun ibuprofen meningkat seiring dengan meningkatnya kadar sehingga linearitas yang diperoleh baik.
D. Analisis Kualitatif
Campuran parasetamol dan ibuprofen dipisahkan dengan KCKT jenis
kromatografi partisi fase terbalik. Fase diam yang digunakan adalah oktadesilsilan (C18) yang bersifat non-polar sedangkan fase gerak yang digunakan adalah campuran metanol dan aquabidest ditambah asam asetat glasial hingga pH 4,0
yang bersifat polar. Pemisahan pada KCKT dipengaruhi oleh interaksi suatu analit dengan fase diam dan fase geraknya. Oleh karena itu, sampel yang bersifat lebih
polar akan terelusi lebih dahulu sedangkan sampel yang bersifat lebih non-polar akan terelusi belakangan karena tertambat pada fase diam (Willard dkk, 1988). Berdasarkan sifat kepolarannya maka parasetamol akan terelusi lebih dahulu
Pemisahan yang optimal dapat diketahui dari nilai resolusinya (resolusi yang baik adalah lebih dari 1,5 dimana pemisahannya≥99,7%) (Sastrohamidjojo,
2002). Kromatogram pemisahan campuran parasetamol dan ibuprofen dapat dilihat pada gambar 15. Dari kromatogram sampel tersebut dapat dilihat waktu
retensi tiap senyawa dan dibandingkan dengan waktu retensi baku yang ditunjukkan pada tabel VIII.
Tabel VIII. Data waktu retensi (tR) masing-masing senyawa baku dan dalam sampel
Senyawa tRbaku (menit) tRsampel (menit)
Parasetamol 1,679 1,681
Ibuprofen 2,955 2,987
Keterangan: sampel merupakan campuran parasetamol dan ibuprofen dengan perbandingan 7:4
Pada tabel VIII dapat diketahui bahwa tR baku parasetamol adalah 1,679
menit sedangkan tR sampel parasetamol adalah 1,681 menit maka dapat disimpulkan bahwa pada sampel terdapat parasetamol. Sedangkan tR baku ibuprofen adalah 2,955 menit dan tRsampel ibuprofen adalah 2,987 menit maka
dapat disimpulkan bahwa pada sampel terdapat ibuprofen. Berdasarkan data tersebut disimpulkan bahwa dalam sampel mengandung parasetamol dan
A
B
C
Gambar 14. Kromatogram pemisahan dari campuran parasetamol dan ibuprofen dengan fase gerak metanol : aquabidest (90:10 v/v) pH 4,0 kecepatan alir 1,5 ml/menit
A. Baku parasetamol konsentrasi tengah (122,5 ppm) B. Baku ibuprofen konsentrasi tengah (70 ppm)
Perbedaan waktu retensi kedua senyawa dikarenakan adanya perbedaan interaksi senyawa tersebut dengan fase gerak dan fase diamnya. Parasetamol
cenderung bersifat lebih polar dibandingkan dengan ibuprofen sehingga parasetamol akan lebih dulu terelusi daripada ibuprofen sebab fase gerak yang
digunakan bersifat lebih polar dibandingkan fase diamnya. Pemisahan antara campuran parasetamol dan ibuprofen sempurna karena nilai resolusi yang diperoleh memenuhi syarat yang telah ditentukan yaitu 5,624 (lebih dari 1,5) dan
peak kedua senyawa runcing (tidak tailing). Penggunaan kecepatan alir 1,5 ml/menit sangat efisien karena pemisahan hanya dilakukan dalam waktu elusi
selama 5 menit. Berikut ini gambar bagian non-polar dari parasetamol dan ibuprofen.
Gambar 15. Bagian non-polar ibuprofen dan parasetamol Keterangan : = gugus non-polar
Pada campuran, sifat dari parasetamol lebih polar dari ibuprofen, hal ini dapat dilihat dari strukturnya diatas. Keduanya memiliki gugus non-polar namun terlihat bahwa gugus non-polar dari ibuprofen lebih banyak dibandingkan dengan
mempunyai log P sebesar 3,75 (diperoleh dari Chemical properties dalam program ChemOffice 2004) dapat diketahui bahwa parasetamol lebih polar dari
ibuprofen karena semakin kecil nilai log P maka senyawa tersebut semakin polar.
O
CH3
Gambar 17. Interaksi ibuprofen dengan fase gerak metanol : aquabidest
Pada gambar 16 dan gambar 17 di atas dapat diketahui bahwa terjadi interaksi antara parasetamol dan ibuprofen dengan fase gerak yang digunakan
yaitu metanol dan aquabidest. Parasetamol dan ibuprofen memiliki gugus polar dan gugus non-polar. Gugus polar dari parasetamol dan ibuprofen akan berinteraksi dengan fase gerak yaitu campuran metanol dan aquabidest. Metanol
sendiri berinteraksi dengan aquabidest dengan ikatan hidrogen. Kemudian campuran dari metanol dan aquabidest akan berinteraksi dengan parasetamol dan
dengan fase gerak lebih banyak dibandingkan interaksi ibuprofen dengan fase
Gambar 18. Interaksi ibuprofen dengan fase diam
O
Gambar 19. Interaksi parasetamol dengan fase diam
Gugus non-polar dari parasetamol dan ibuprofen akan berinteraksi
lebih kuat pada fase diam dan akan terelusi lebih lama dari parasetamol. Dari keseluruhan interaksi parasetamol-ibuprofen dengan fase diam dan fase gerak
dapat diketahui bahwa interaksi analit dengan fase gerak lebih kuat dibandingkan dengan fase diamnya dikarenakan sifat dari interaksi hidrogen yang lebih kuat dari
interaksi van der Waals. Oleh karena itu, analit dapat terelusi dari kolom KCKT. Pada sistem KCKT fase terbalik ini, parasetamol akan memiliki tR 1,681 menit lebih pendek dibandingkan dengan ibuprofen akan memiliki tR2,987 menit karena
sifat dari parasetamol yang lebih polar. Dengan sifatnya yang semakin polar maka interaksi dengan fase gerak semakin kuat dan semakin cepat terelusi.
E. Validasi Metode Analisis
Validasi metode bertujuan untuk membuktikan bahwa metode yang
digunakan memenuhi persyaratan penggunaannya sehingga data yang diperoleh terpercaya. Validasi dilakukan dengan 3 seri konsentrasi sebanyak 6 replikasi.
Campuran tiga seri konsentrasi ini mewakili konsentrasi rendah, tengah dan tinggi dari konsentrasi seri baku. Seri konsentrasi untuk parasetamol dan ibuprofen berturut-turut adalah 70 ppm; 122,5 ppm; 175 ppm dan 40 ppm; 70 ppm; 100
ppm.
Kesahihan metode KCKT yang digunakan dalam penetapan kadar
1. Akurasi
Akurasi dinyatakan dengan persen perolehan kembali (recovery) dari
kadar yang terukur terhadap kadar sebenarnya. Jika % recovery dari 6 replikasi parasetamol dan ibuprofen pada konsentrasi 100 ppm berada pada rentang
90-107% (Anonim, 2007) maka metode ini memiliki akurasi yang baik.
Tabel IX. Data %recoveryparasetamol dan ibuprofen
Recovery(%) Parasetamol 111,03 105,49 106,21 101,03 111,55 106,09 70 ppm
40 ppm Ibuprofen 91,66 94,08 90,99 100,75 97,87 85,84
Parasetamol 98,55 94,01 105,73 91,46 96,51 96,60 122,5 ppm
70 ppm Ibuprofen 90,54 94,62 100,88 101,35 99,00 90,14
Parasetamol 94,25 91,58 94,62 85,44 94,05 95,97 175 ppm
100 ppm Ibuprofen 92,53 96,41 107,89 102,99 96,19 96,67
Pada tabel IX dapat dilihat bahwa %recoveryparasetamol dan ibuprofen sebanyak 6 replikasi yang masuk pada rentang 90-107% adalah pada konsentrasi
tengah yaitu parasetamol 122,5 ppm dan ibuprofen 70 ppm, dengan rentang 91,46-105,73% untuk parasetamol dan 90,14-101,35% untuk ibuprofen. Pada konsentrasi rendah yaitu parasetamol 70 ppm dan ibuprofen 40 ppm diketahui %
recovery berturut-turut adalah 101,03-111,55% dan 85,84-100,75%, dengan demikian parasetamol dan ibuprofen pada konsentrasi rendah kurang memiliki
akurasi yang baik dikarenakan tidak memenuhi persyaratan rentang yang diperbolehkan sedangkan pada konsentrasi tinggi yaitu parasetamol 175 ppm dan ibuprofen 100 ppm diketahui % recoveryberturut-turut adalah 85,44-95,97% dan
diperbolehkan. Oleh karena itu, dapat disimpulkan bahwa metode ini memiliki akurasi yang baik pada konsentrasi tengah untuk parasetamol dan ibuprofen.
2. Presisi
Presisi suatu metode diukur dengan parameter % koefisien variasi (KV). Menurut Anonim (2007), % KV yang dapat diterima untuk konsentrasi analit 1-1000 ppm adalah 5-16%.
Tabel X. Data % CV parasetamol dan ibuprofen
Konsentrasi
(ppm)
SD CV (%)
Parasetamol 75,4007 1,3397 1,78
70 ppm
40 ppm Ibuprofen 37,3062 2,7519 7,38
Parasetamol 120,3617 6,0671 5,04
122,5 ppm
70 ppm Ibuprofen 67,1107 5,3636 7,99
Parasetamol 163,3892 5,0542 3,09
175 ppm
100 ppm Ibuprofen 98,5288 7,8691 7,99
Pada tabel X, dapat dilihat bahwa semua konsentrasi baik rendah, tengah, dan tinggi untuk parasetamol dan ibuprofen berturut-turut adalah 1,78% dan 7,38%; 5,04% dan 7,99%; 3,09% dan 7,99%. Persyaratan yang telah ditetapkan
yaitu range 5-16% untuk konsentrasi 1-1000 ppm. Berdasarkan hasil yang diperoleh dapat disimpulkan bahwa semua konsentrasi parasetamol dan ibuprofen
masuk dalam range yang ditentukan (memenuhi persyaratan). Oleh karena itu, metode ini memiliki presisi yang baik.
3. Spesifisitas
Spesifisitas merupakan kemampuan suatu metode untuk hanya mengukur
mungkin ada dalam matriks sampel. Pada penelitian ini, senyawa yang akan diukur adalah parasetamol dan ibuprofen. Berikut adalah kromatogram campuran
parasetamol dan ibuprofen.
Gambar 20. Kromatogram campuran parasetamol dan ibuprofen
Pada gambar 20 dapat dilihat terdapat 2peak senyawa yaitu parasetamol dan ibuprofen. Pada tabel VII mengenai waktu retensi diketahui bahwa tR baku
parasetamol adalah 1,679 menit dan tRbaku ibuprofen adalah 2,955 menit. Pada kromatogram campuran parasetamol dan ibuprofen di atas diketahui bahwa tR
parasetamol adalah 1,681 menit dan tRibuprofen adalah 2,987 menit maka dapat disimpulkan bahwa senyawa pada kromatogram di atas adalah parasetamol dan ibuprofen. Oleh karena itu, metode ini mempunyai spesifisitas yang baik.
4. Linearitas
diketahui bahwa nilai r untuk parasetamol dan ibuprofen yaitu 0,998 untuk parasetamol dan 0,999 untuk ibuprofen sehingga dapat disimpulkan bahwa
50
A. Kesimpulan
Metode KCKT yang digunakan dengan fase diam oktadesilsilan (C18), fase gerak metanol : aquabidest (90:10 v/v) dengan penambahan asam asetat glasial hingga pH 4,0 dan kecepatan alir 1,5 ml/menit mempunyai akurasi yang
baik pada konsentrasi 122,5 ppm untuk parasetamol dan 70 ppm untuk ibuprofen; presisi yang baik pada konsentrasi 70-175 ppm untuk parasetamol dan 40-100
ppm untuk ibuprofen; spesifisitas dan linearitas yang baik untuk parasetamol dan ibuprofen. Berdasarkan hal tersebut maka metode KCKT tersebut mempunyai validitas yang baik untuk menetapkan kadar campuran parasetamol dan ibuprofen
dengan perbandingan 7:4.
B. Saran
Perlu dilakukan penelitian lebih lanjut untuk mengaplikasikan metode penetapan kadar campuran parasetamol dan ibuprofen dalam sediaan obat,
Daftar Pustaka
Anonim, 1995, Farmakope Indonesia, edisi IV, 449, 649, 660, 1009, Departemen Kesehatan Republik Indonesia, Jakarta.
Anonim, 2004, Guidlines for Validation of Analytical Methods for Active Constituent, Agricultural and Veterinary Chemical Products, 4-5, http://www.apvma.gov.au/guidlines/downloads/g169analyticalmethoda.pd f., diakses tanggal 22 Desember 2009
Anonim, 2007,The United States Pharmacopeia 30th, United States Pharmacopeal Convention, Inc., New York.
Anonim, 2009a,Ibuprofen,
http://www.drugs.com/search.php?searchterm=ibuprofen&is_main_search =1, diakses tanggal 30 Oktober 2009
Anonim, 2009b, Paracetamol, http://www.dechacare.com/Parasetamol-Tak-Seaman-yang-Dikira-I588.html,diakses tanggal 26 Februari 2009
Anonim, 2010, Motrin (Ibuprofen),
http://www.druglib.com/Motrin%20%28Ibuprofen%29%20-%20Drug%20Info,%20User%20Reviews,%20Side%20Effects,%20Resear ch,%20Clinical%20Trials_files/urchin.js, diakses tanggal 26 Maret 2010
Battu, P. R., and Reddy, M. S., 2009a, RP-HPLC Method for Simultaneous Estimation of Paracetamol and Ibuprofen in Tablets,
http://www.ajrconline.org/AJRC%20V0l2%20(1)%20PDF%20Final/19.pd f, diakses tanggal 29 Agustus 2009
Battu, P. R., and Reddy, M. S., 2009b,Simultaneous Determination of Tizanidine and Ibuprofen in Tablets,
http://sphinxsai.com/CTVOL3/CT=17,PRASANNA%20REDDY%20(499 -501).pdf, diakses tanggal 29 Agustus 2009
Beers, M.H., Fletcher, A. J., Jones, T. V., Porter, R., Berkwits, M., and Kaplan, J. L., 2003,The Merck Manual of Medical Information,2nd ed., 95, Merck & Co., Inc., New York.
Chan, C.C., Lam, H., Lee, Y.C., and Zhang, X., 2004, Analytical Method Validation and Instrument Performance Verification, 16, John Wiley & Sons, Inc., USA.
Clarke, E. G. C., 1986, Isolation and identificationof Drugs, 2nd ed., 849-850, 883-884, The Pharmaceutical Press, London.
Dean, J. A., 1995, Analytical Chemistry, 6th ed., 234, 253, 465, John Willey & Sons, Inc., USA.
Fessenden, R.J., Fessenden, J.S., 1982, Kimia Organik, edisi III, 24-27, Penerbit Erlangga, Jakarta.
Gritter, R. J., Bobbit, J. M., dan Schwarting, A. E., 1985, Introduction to Chromatography,diterjemahkan oleh Kosasih Padwawinata, edisi II, 205-219, Penerbit ITB, Bandung.
Harris, D. C., 1999, Quantitative Chemical Analysis, 2nd ed., 643, 648, 716-717, W. H. Freeman Company, New York.
Harmita, 2004, Petunjuk Pelaksanaan Validasi Metode dan Cara, Majalah Ilmu Kefarmasian,Vol.I, No.3, 117-135
Johnson E. L., and Stevenson, R., 1978, Basic Liquid Chromatography, diterjemahkan oleh Kosasih Padmawinata , 2-11, 17-25, 90-91, 99-103, ITB Press, Bandung.
Kazakevich, Y., and Nair, H. M., 1996, Basic Liquid Chromatography Textbook on KCKT, http://KCKT.chem.shu.edu/NEW/KCKT_Book., diakses tanggal 20 Desember 2009
Khopkar, S. M., 1990, Konsep Dasar Kimia Analitik, 3-5, 167-172, Penerbit Universitas Indonesia, Jakarta.
Mulja, M., dan Suharman, 1995,Analisis Instrumental, 19-32, 139-148, 155, 164, 236-270, Universitas Airlangga Press, Surabaya.
Munson, J. W., 1984, Pharmaceutical Analysis Modern Methods, diterjemahkan oleh Harjana Parwa B, Bagian B, 14-16, Airlangga University Press Surabaya.
Noegrohati, S., 1994, Pengantar Kromatografi, dalam Noegrohati, S. Dan Narsito, (Eds.), Risalah Prinsip dan Aplikasi Beberapa Teknik Analisis Instrumental, 16-17, Laboratorium Analisis Kimia dan Fisika Pusat UGM, Yogyakarta.
Prabowo, Y. P., 2010, Optimasi Pemisahan Campuran Parasetamol dan Ibuprofen Dengan Metode Kromatografi Cair Kinerja Tinggi Fase Terbalik, Skripsi, Universitas Sanata Dharma, Yogyakarta
Rohman, A., 2009, Kromatografi untuk Analisis Obat, edisi pertama, cetakan pertama, 217, 230, Penerbit Graha Ilmu, Yogyakarta.
Rohman, A., dan Gandjar, I. G., 2007, Kimia Farmasi Analisis, cetakan kedua, 323-345, 378-389, Penerbit Pustaka Pelajar, Yogyakarta.
Sastrohamidjojo, H., 2002,Spektroskopi, 11, Penerbit Liberty, Yogyakarta.
Skoog, D. A., Holler, F. J., and Nieman, T. A., 1998, Principles of Instrumental Analysis,5thed., 329-351, 785, Harcourt Bace College, Philadelphia. Snyder, L. R., and Kirkland J.J., 1979, Introduction To Modern Liquid
Chromatography, 2nd ed., 208-209, 236, John Wiley & Sons.Inc., NewYork.