Previous edition:Pharmaceutical Process Validation: Second Edition, Revised and Ex-panded(I. R. Berry, R. A. Nash, eds.), 1993.
Library of Congress Cataloging-in-Publication Data
A catalog record for this book is available from the Library of Congress.
ISBN: 0-8247-0838-5
This book is printed on acid-free paper. Headquarters
Marcel Dekker, Inc.
270 Madison Avenue, New York, NY 10016 tel: 212-696-9000; fax: 212-685-4540
Eastern Hemisphere Distribution Marcel Dekker AG
Hutgasse 4, Postfach 812, CH-4001 Basel, Switzerland tel: 41-61-260-6300; fax: 41-61-260-6333
World Wide Web http://www.dekker.com
The publisher offers discounts on this book when ordered in bulk quantities. For more information, write to Special Sales/Professional Marketing at the headquarters address above.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
Neither this book nor any part may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, microfilming, and recording, or by any information storage and retrieval system, without permission in writing from the publisher.
Current printing (last digit):
10 9 8 7 6 5 4 3 2 1
Dedicated to Theodore E. Byers, formerly of the U.S. Food and Drug Administration, and Heinz Sucker, Professor at the University of Berne,
Switzerland, for their pioneering contributions with respect to the pharmaceutical process validation concept. We also acknowledge the past contributions of Bernard T. Loftus and Ira R. Berry toward the
Preface
The third edition of Pharmaceutical Process Validation represents a new ap-proach to the topic in several important respects.
Many of us in the field had made the assumption that pharmaceutical process validation was an American invention, based on the pioneering work of Theodore E. Byers and Bernard T. Loftus, both formerly with the U.S. Food & Drug Administration. The truth is that many of our fundamental concepts of pharmaceutical process validation came to us from “Validation of Manufactur-ing Processes,” Fourth European Seminar on Quality Control, September 25, 1980, Geneva, Switzerland, and Validation in Practice, edited by H. Sucker, Wissenschaftliche Verlagsegesellschaft, GmbH, Stuttgard, Germany, 1983.
There are new chapters in this edition that will add to the book’s impact. They include “Validation for Medical Devices” by Nishihata, “Validation of Biotechnology Processes” by Sofer, “Transdermal Process Validation” by Neal, “Integrated Packaging Validation” by Frederick, “Statistical Methods for Uni-formity and Dissolution Testing” by Bergum and Utter, “Change Control and SUPAC” by Waterland and Kowtna, “Validation in Contract Manufacturing” by Parikh, and “Harmonization, GMPs, and Validation” by Wachter.
I am pleased to have Dr. Alfred Wachter join me as coeditor of this edi-tion. He was formerly head of Pharmaceutical Product Development for the CIBA Pharmaceutical Company in Basel, Switzerland, and also spent a number of years on assignment in Asia for CIBA. Fred brings a very strong international perspective to the subject matter.
Contents
Preface Contributors Introduction
1. Regulatory Basis for Process Validation John M.Dietrick and Bernard T.Loftus
2. Prospective Process Validation
Allen Y.Chao,F.St.John Forbes,Reginald F.Johnson, and Paul Von Doehren
3. Retrospective Validation Chester J.Trubinski
4. Sterilization Validation
Michael J.Akers and Neil R.Anderson
5. Validation of Solid Dosage Forms Jeffrey S.Rudolph and Robert J.Sepelyak
6. Validation for Medical Devices Toshiaki Nishihata
7. Validation of Biotechnology Processes Gail Sofer
8. Transdermal Process Validation Charlie Neal,Jr.
10. Validation of Inhalation Aerosols
Christopher J.Sciarra and John J.Sciarra
11. Process Validation of Pharmaceutical Ingredients Robert A.Nash
12. Qualification of Water and Air Handling Systems Kunio Kawamura
13. Equipment and Facility Qualification Thomas L.Peither
14. Validation and Verification of Cleaning Processes William E.Hall
15. Validation of Analytical Methods and Processes Ludwig Huber
16. Computer System Validation:
Controlling the Manufacturing Process Tony de Claire
17. Integrated Packaging Validation Mervyn J.Frederick
18. Analysis of Retrospective Production Data Using Quality Control Charts
Peter H.Cheng and John E.Dutt
19. Statistical Methods for Uniformity and Dissolution Testing James S.Bergum and Merlin L.Utter
20. Change Control and SUPAC
Nellie Helen Waterland and Christopher C.Kowtna
21. Process Validation and Quality Assurance Carl B.Rifino
22. Validation in Contract Manufacturing Dilip M.Parikh
23. Terminology of Nonaseptic Process Validation Kenneth G.Chapman
Contributors
Michael J. Akers Baxter Pharmaceutical Solutions, Bloomington, Indiana, U.S.A.
Neil R. Anderson Eli Lilly and Company, Indianapolis, Indiana, U.S.A.
James S. Bergum Bristol-Myers Squibb Company, New Brunswick, New Jer-sey, U.S.A.
Kenneth G. Chapman Drumbeat Dimensions, Inc., Mystic, Connecticut, U.S.A.
Allen Y. Chao Watson Labs, Carona, California, U.S.A.
Peter H. Cheng New York State Research Foundation for Mental Hygiene, New York, New York, U.S.A.
Tony de Claire APDC Consulting, West Sussex, England
John M. Dietrick Center for Drug Evaluation and Research, U.S. Food and Drug Administration, Rockville, Maryland, U.S.A.
John E. Dutt EM Industries, Inc., Hawthorne, New York, U.S.A.
Mervyn J. Frederick NV Organon–Akzo Nobel, Oss, The Netherlands
William E. Hall Hall & Pharmaceutical Associates, Inc., Kure Beach, North Carolina, U.S.A.
F. St. John Forbes Wyeth Labs, Pearl River, New York, U.S.A.
*Reginald F. Johnson Searle & Co., Inc., Skokie, Illinois, U.S.A.
Kunio Kawamura Otsuka Pharmaceutical Co., Ltd., Tokushima, Japan
Christopher C. Kowtna DuPont Pharmaceuticals Co., Wilmington, Dela-ware, U.S.A.
*Bernard T. Loftus Bureau of Drugs, U.S. Food and Drug Administration, Washington, D.C., U.S.A.
Robert A. Nash Stevens Institute of Technology, Hoboken, New Jersey, U.S.A.
Charlie Neal, Jr. Diosynth-RTP, Research Triangle Park, North Carolina, U.S.A.
Toshiaki Nishihata Santen Pharmaceutical Co., Ltd., Osaka, Japan
Dilip M. Parikh APACE PHARMA Inc., Westminster, Maryland, U.S.A.
Thomas L. Peither PECON—Peither Consulting, Schopfheim, Germany
Carl B. Rifino AstraZeneca Pharmaceuticals LP, Newark, Delaware, U.S.A.
Jeffrey S. Rudolph Pharmaceutical Consultant, St. Augustine, Florida, U.S.A.
Christopher J. Sciarra Sciarra Laboratories Inc., Hicksville, New York, U.S.A.
John J. Sciarra Sciarra Laboratories Inc., Hicksville, New York, U.S.A.
Robert J. Sepelyak AstraZeneca Pharmaceuticals LP, Wilmington, Delaware, U.S.A.
Gail Sofer BioReliance, Rockville, Maryland, U.S.A.
Edward H. Trappler Lyophilization Technology, Inc., Warwick, Pennsylva-nia, U.S.A.
Chester J. Trubinski Church & Dwight Co., Inc., Princeton, New Jersey, U.S.A.
Merlin L. Utter Wyeth Pharmaceuticals, Pearl River, New York, U.S.A.
Paul Von Doehren Searle & Co., Inc., Skokie, Illinois, U.S.A.
Alfred H. Wachter Wachter Pharma Projects, Therwil, Switzerland
Introduction
Robert A. Nash
Stevens Institute of Technology, Hoboken, New Jersey, U.S.A.
I. FDA GUIDELINES
The U.S. Food and Drug Administration (FDA) has proposed guidelines with the following definition forprocess validation[1]:
Process validation is establishing documented evidence which provides a high degree of assurance that a specific process (such as the manufacture of pharmaceutical dosage forms) will consistently produce a product meet-ing its predetermined specifications and quality characteristics.
According to the FDA, assurance of product quality is derived from care-ful and systemic attention to a number of important factors, including: selection of quality components and materials, adequate product and process design, and (statistical) control of the process through in-process and end-product testing.
Thus, it is through careful design (qualification) and validation of both the process and its control systems that a high degree of confidence can be estab-lished that all individual manufactured units of a given batch or succession of batches that meet specifications will be acceptable.
According to the FDA’s Current Good Manufacturing Practices (CGMPs) 21CFR 211.110 a:
Control procedures shall be established to monitoroutput and to validate performance of the manufacturing processes that may be responsible for causing variability in the characteristics of in-process material and the drug product. Such control procedures shall include, but are not limited to the following, where appropriate [2]:
3. Adequacy of mixing to assure uniformity and homogeneity 4. Dissolution time and rate
5. Clarity, completeness, or pH of solutions
The first four items listed above are directly related to the manufacture and validation of solid dosage forms. Items 1 and 3 are normally associated with variability in the manufacturing process, while items 2 and 4 are usually influenced by the selection of the ingredients in the product formulation. With respect to content uniformity and unit potency control (item 3), adequacy of mixing to assure uniformity and homogeneity is considered a high-priority con-cern.
Conventional quality control procedures for finished product testing en-compass three basic steps:
1. Establishment of specifications and performance characteristics 2. Selection of appropriate methodology, equipment, and
instrumenta-tion to ensure that testing of the product meets specificainstrumenta-tions 3. Testing of the final product, using validated analytical and testing
methods to ensure that finished product meets specifications.
With the emergence of the pharmaceutical process validation concept, the fol-lowing four additional steps have been added:
4. Qualification of the processing facility and its equipment
5. Qualification and validation of the manufacturing process through ap-propriate means
6. Auditing, monitoring, sampling, or challenging the key steps in the process for conformance to in-process and final product specifications 7. Revalidation when there is a significant change in either the product
or its manufacturing process [3].
II. TOTAL APPROACH TO PHARMACEUTICAL
PROCESS VALIDATION
re-quirement of current regulations, such a comprehensive approach with respect to each subpart of the CGMP document has been adopted by many drug firms. A checklist of qualification and control documentation with respect to CGMPs is provided in Table 1. A number of these topics are discussed sepa-rately in other chapters of this book.
III. WHY ENFORCE PROCESS VALIDATION?
The FDA, under the authority of existing CGMP regulations, guidelines [1], and directives [3], considers process validation necessary because it makes good engineering sense. The basic concept, according to Mead [5], has long been
Table 1 Checklist of Qualification and Control Documentation
Qualification and
Subpart Section of CGMPs control documentation
A General provisions
B Organization and personnel Responsibilities of the quality con-trol unit
C Buildings and facilities Plant and facility installation and qualification
Maintenance and sanitation Microbial and pest control
D Equipment Installation and qualification of
equipment and cleaning methods E Control of components, containers Incoming component testing
proce-and closures dures
F Production and process controls Process control systems, reprocess-ing control of microbial contami-nation
G Packaging and labeling controls Depyrogenation, sterile packaging, filling and closing, expire dating H Holding and distribution Warehousing and distribution
pro-cedures
I Laboratory controls Analytical methods, testing for re-lease component testing and sta-bility testing
J Records and reports Computer systems and information
systems K Return and salvaged drug products Batch reprocessing
applied in other industries, often without formal recognition that such a concept was being used. For example, the termsreliability engineeringand qualification have been used in the past by the automotive and aerospace industries to repre-sent the process validation concept.
The application of process validation should result in fewer product re-calls and troubleshooting assignments in manufacturing operations and more technically and economically sound products and their manufacturing processes. In the old days R & D “gurus” would literally hand down the “go” sometimes overformulated product and accompanying obtuse manufacturing procedure, usually with little or no justification or rationale provided. Today, under FDA’s Preapproval Inspection (PAI) program [4] such actions are no longer accept-able. The watchword is to provide scientifically sound justifications (including qualification and validation documentation) for everything that comes out of the pharmaceutical R & D function.
IV. WHAT IS PROCESS VALIDATION?
Unfortunately, there is still much confusion as to what process validation is and what constitutes process validation documentation. At the beginning of this introduction several different definitions for process validation were provided, which were taken from FDA guidelines and the CGMPs. Chapman calls process validation simply “organized, documented common sense” [6]. Others have said that “it is more than three good manufactured batches” and should represent a lifetime commitment as long as the product is in production, which is pretty much analogous to theretrospective process validationconcept.
The big problem is that we use the term validationgenerically to cover the entire spectrum of CGMP concerns, most of which are essentially people, equipment, component, facility, methods, and proceduralqualification. The spe-cific term process validation should be reserved for the final stage(s) of the product/process development sequence. The essential or key steps or stages of a successfully completed product/process development program are presented inTable 2[7].
Table 2 The Key Stages in the Product/Process Development Sequence
Development stage Pilot scale-up phase
Product design 1×batch size
Product characterization Product selection (“go” formula) Process design
Product optimization 10×batch size
Process characterization Process optimization
Process demonstration 100×batch size Process validation program
Product/process certification
With the exception of solution products, the bulk of the work is nor-mally carried out at 10×batch size, which is usually the first scale-up batches in production-type equipment.
sequence provides only the necessary process validation documentation required by the regulatory authorities—in other words, the “Good Housekeeping Seal of Approval,” which shows that the manufacturing process is in a state of control. Such a strategy is consistent with the U.S. FDA’spreapproval inspection program [4], wherein the applicant firm under either a New Drug Application (NDA) or an Abbreviated New Drug Application (ANDA) submission must show the necessary CGMP information and qualification data (including appro-priate development reports), together with the formal protocol for the forthcom-ing full-scale, formal process validation runs required prior to product launch.
Again, the termvalidationhas both a specific meaning and a general one, depending on whether the word “process” is used. Determine during the course of your reading whether the entire concept is discussed in connection with the topic—i.e., design, characterization, optimization, qualification, validation, and/ or revalidation—or whether the author has concentrated on the specifics of the validation of a given product and/or its manufacturing process. In this way the text will take on greater meaning and clarity.
V. PILOT SCALE-UP AND PROCESS VALIDATION
1. Formulation design, selection, and optimization 2. Preparation of the first pilot-laboratory batch 3. Conduct initial accelerated stability testing
4. If the formulation is deemed stable, preparation of additional pilot-laboratory batches of the drug product for expanded nonclinical and/ or clinical use.
The pilot program is defined as the scale-up operations conducted subse-quent to the product and its process leaving the development laboratory and prior to its acceptance by the full scale manufacturing unit. For the pilot program to be successful, elements of process validation must be included and completed during the developmental or pilot laboratory phase of the work.
Thus, product and process scale-up should proceed in graduated steps with elements of process validation (such as qualifications) incorporated at each stage of the piloting program [9,10].
A. Laboratory Batch
The first step in the scale-up process is the selection of a suitable preliminary formula for more critical study and testing based on certain agreed-upon initial design criteria, requirements, and/or specifications. The work is performed in the development laboratory. The formula selected is designated as the (1×) laboratory batch. The size of the (1×) laboratory batch is usually 3–10 kg of a solid or semisolid, 3–10 liters of a liquid, or 3000 to 10,000 units of a tablet or capsule.
B. Laboratory Pilot Batch
After the (1×) laboratory batch is determined to be both physically and chemi-cally stable based on accelerated, elevated temperature testing (e.g., 1 month at 45°C or 3 months at 40°C or 40°C/80% RH), the next step in the scale-up process is the preparation of the (10×) laboratory pilot batch. The (10×) laboratory pilot batch represents the first replicated scale-up of the designated formula. The size of the laboratory pilot batch is usually 30–100 kg, 30–100 liters, or 30,000 to 100,000 units.
It is usually prepared in small pilot equipment within a designated CGMP-approved area of the development laboratory. The number and actual size of the laboratory pilot batches may vary in response to one or more of the following factors:
1. Equipment availability
3. Cost of raw materials
4. Inventory requirements for clinical and nonclinical studies
Process demonstration or process capability studies are usually started in this important second stage of the pilot program. Such capability studies consist of process ranging, process characterization, and process optimization as a prereq-uisite to the more formal validation program that follows later in the piloting sequence.
C. Pilot Production
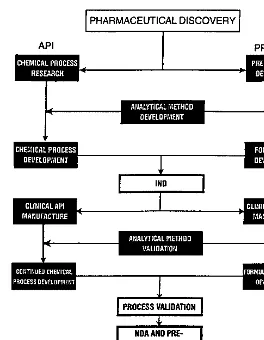
The pilot-production phase may be carried out either as a shared responsibility between the development laboratories and its appropriate manufacturing coun-terpart or as a process demonstration by a separate, designated pilot-plant or process-development function. The two organization piloting options are pre-sented separately in Figure 1. The creation of a separate pilot-plant or process-development unit has been favored in recent years because it is ideally suited to carry out process scale-up and/or validation assignments in a timely manner. On the other hand, the joint pilot-operation option provides direct communication between the development laboratory and pharmaceutical production.
The object of the pilot-production batch is to scale the product and process by another order of magnitude (100×) to, for example, 300–1,000 kg, 300– 1,000 liters, or 300,000–1,000,000 dosage form units (tablets or capsules) in size. For most drug products this represents a full production batch in standard production equipment. If required, pharmaceutical production is capable of scal-ing the product/process to even larger batch sizes should the product require expanded production output. If the batch size changes significantly, additional validation studies would be required. The term product/process is used, since one can’t describe a product with discussing its process of manufacture and, conversely, one can’t talk about a process without describing the product being manufactured.
Usually large production batch scale-up is undertaken only after product introduction. Again, the actual size of the pilot-production (100×) batch may vary due to equipment and raw material availability. The need for additional pilot-production batches ultimately depends on the successful completion of a first pilot batch and its process validation program. Usually three successfully completed pilot-production batches are required for validation purposes.
In summary, process capability studies start in the development labora-tories and/or during product and process development, and continue in well-defined stages until the process is validated in the pilot plant and/or pharmaceu-tical production.
An approximate timetable for new product development and its pilot scale-up program is suggested inTable 3.
VI. PROCESS VALIDATION: ORDER OF PRIORITY
Because of resource limitation, it is not always possible to validate an entire company’s product line at once. With the obvious exception that a company’s most profitable products should be given a higher priority, it is advisable to draw up a list of product categories to be validated.
The following order of importance or priority with respect to validation is suggested:
A. Sterile Products and Their Processes
1. Large-volume parenterals (LVPs) 2. Small-volume parenterals (SVPs)
Table 3 Approximate Timetable for New Product Development and Pilot Scale-Up Trials
Calendar
Event months
Formula selection and development 2–4
Assay methods development and formula optimization 2–4 Stability in standard packaging 3-month readout (1×size) 3–4
Pilot-laboratory batches (10×size) 1–3
Preparation and release of clinical supplies (10×size) and
establishment of process demonstration 1–4
Additional stability testing in approved packaging 3–4 6–8-month readout (1×size)
3-month readout (10×size)
Validation protocols and pilot batch request 1–3
Pilot-production batches (100×size) 1–3
Additional stability testing in approved packaging 3–4 9–12-month readout (1×size)
6–8-month readout (10×size) 3-month readout (100×size)
Interim approved technical product development report with
approximately 12 months stability (1×size) 1–3 Totals 18–36
B. Nonsterile Products and Their Processes
1. Low-dose/high-potency tablets and capsules/transdermal delivery sys-tems (TDDs)
2. Drugs with stability problems 3. Other tablets and capsules
4. Oral liquids, topicals, and diagnostic aids
VII. WHO DOES PROCESS VALIDATION?
Process validation is done by individuals with the necessary training and experi-ence to carry out the assignment.
Table 4 Specific Responsibilities of Each Organizational Structure within the Scope of Process Validation
Engineering Install, qualify, and certify plant, facilities, equipment, and sup-port system.
Development Design and optimize manufacturing process within design limits, specifications, and/or requirements—in other words, the estab-lishment of process capability information.
Manufacturing Operate and maintain plant, facilities, equipment, support sys-tems, and the specific manufacturing process within its design limits, specifications, and/or requirements.
Quality assurance Establish approvable validation protocols and conduct process validation by monitoring, sampling, testing, challenging, and/ or auditing the specific manufacturing process for compliance with design limits, specifications, and/or requirements. Source: Ref. 8.
best approach in carrying out the process validation assignment is to establish a Chemistry, Manufacturing and Control (CMC) Coordination Committee at the specific manufacturing plant site [10]. Representation on such an important lo-gistical committee should come from the following technical operations:
• Formulation development (usually a laboratory function)
• Process development (usually a pilot plant function)
• Pharmaceutical manufacturing (including packaging operations)
• Engineering (including automation and computer system responsibili-ties)
• Quality assurance
• Analytical methods development and/or Quality Control
• API Operations (representation from internal operations or contract manufacturer)
• Regulatory Affairs (technical operations representative)
• IT (information technology) operations
The chairperson or secretary of such an important site CMC Coordination Com-mittee should include the manager of process validation operations. Typical meeting agendas may include the following subjects in the following recom-mended order of priority:
• Specific CGMP issues for discussion and action to be taken
• Technology transfer issues within or between plant sites.
• Pre-approval inspection (PAI) issues of a forthcoming product/process
• Change control and scale-up, post approval changes (SUPAC) with respect to current approved product/process [11].
VIII. PROCESS DESIGN AND CHARACTERIZATION
Process capabilityis defined as the studies used to determine thecriticalprocess parameters or operating variables that influence process output and the range of numerical data for critical process parameters that result in acceptable process output. If the capability of a process is properly delineated, the process should consistently stay within the defined limits of its critical process parameters and product characteristics [12].
Process demonstration formerly called process qualification, represents the actual studies or trials conducted to show that all systems, subsystems, or unit operations of a manufacturing process perform as intended; that all critical process parameters operate within their assigned control limits; and that such studies and trials, which form the basis of process capability design and testing, are verifiable and certifiable through appropriate documentation.
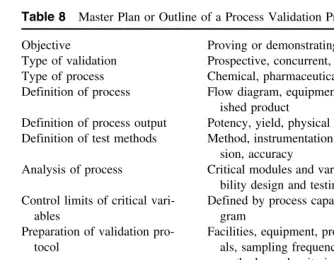
The manufacturing process is briefly defined as the ways and means used to convert raw materials into a finished product. The ways and means also include people, equipment, facilities, and support systems required to operate the process in a planned and effectively managed way. All the latter functions must be qualified individually. The master plan or protocol for process capabil-ity design and testing is presented inTable 5.
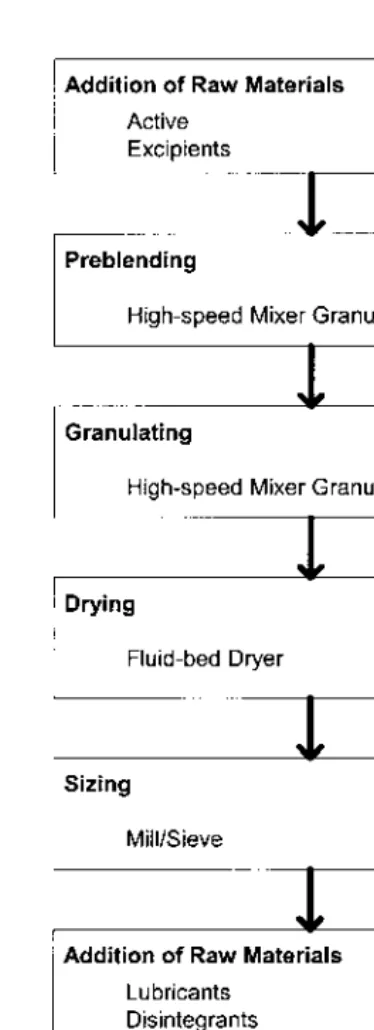
A simple flow chart should be provided to show the logistical sequence of unit operations during product/process manufacture. A typical flow chart used in the manufacture of a tablet dosage form by the wet granulation method is presented inFigure 2.
IX. STREAMLINING VALIDATION OPERATIONS
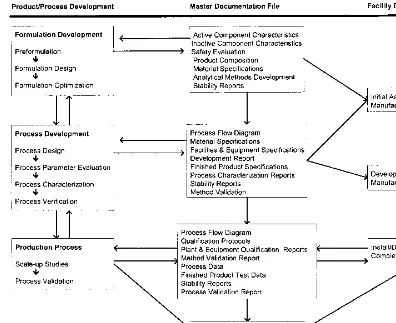
The best approach to avoiding needless and expensive technical delays is to work in parallel. The key elements at this important stage of the overall process are the API, analytical test methods, and the drug product (pharmaceutical dos-age form). An integrated and parallel way of getting these three vitally important functions to work together is depicted inFigure 3.
develop-Table 5 Master Plan or Protocol for Process Capability Design and Testing Objective Process capability design and testing Types of process Batch, intermittent, continuous Typical processes Chemical, pharmaceutical, biochemical Process definition Flow diagram, in-process, finished product Definition of process output Potency, yield, physical parameters Definition of test methods Instrumentation, procedures, precision, and
accuracy
Process analysis Process variables, matrix design, factorial design analysis
Pilot batch trials Define sampling and testing, stable, extended runs Pilot batch replication Different days, different materials, different
equip-ment
Process redefinition Reclassification of process variables
Process capability evaluation Stability and variability of process output, eco-nomic limits
Final report Recommended SOP, specifications, and process limits
Table 6 A List of Useful Pharmaceutical Unit Operations According to Categories Heat transfer processes:Cooking, cooling, evaporating, freezing, heating, irradiating,
sterilizing, freeze-drying
Change in state: Crystallizing, dispersing, dissolving, immersing, freeze-drying, neutral-izing
Change in size: Agglomerating, blending, coating, compacting, crushing, crystallizing, densifying, emulsifying, extruding, flaking, flocculating, grinding, homogenizing, milling, mixing, pelletizing, pressing, pulverizing, precipitating, sieving
Moisture transfer processes: Dehydrating, desiccating, evaporating, fluidizing, humidify-ing, freeze-dryhumidify-ing, washhumidify-ing, wetting
Separation processes: Centrifuging, clarifying, deareating, degassing, deodorizing, dia-lyzing, exhausting, extracting, filtering, ion exchanging, pressing, sieving, sorting, washing
Transfer processes: Conveying, filling, inspecting, pumping, sampling, storing, trans-porting, weighing
Source: Ref. 13.
ment, clinical study, process development, and process validation and into pro-duction. Working individually with separate analytical testing functions and with little or no appropriate communication among these three vital functions is a prescription for expensive delays. It is important to remember that the concept illustrated inFigure 3can still be followed even when the API is sourced from outside the plant site or company. In this particular situation there will probably be two separate analytical methods development functions: one for the API manufacturer and one for the drug product manufacturer [14].
X. STATISTICAL PROCESS CONTROL AND
PROCESS VALIDATION
Figure 3 Working in parallel. (Courtesy of Austin Chemical Co., Inc.)
There are three ways of establishing quality products and their manufac-turing processes:
1. In-process and final product testing, which normally depends on sam-pling size (the larger the better). In some instances, nothing short of excessive sampling can ensure reaching the desired goal, i.e., sterility testing.
stan-dard will often reduce the need for more extensive sampling require-ments.
3. The modern approach, based on Japanese quality engineering [15], is the pursuit of “zero defects” by applying tighter control over process variability (meeting a so-called 6 sigma standard). Most pharmaceuti-cal products and their manufacturing processes in the United States today, with the exception of sterile processes are designed to meet a 4 sigma limit (which would permit as many as eight defects per 1000 units). The new approach is to center the process (in which the grand average is roughly equal to 100% of label potency or the target value of a given specification) and to reduce the process variability or noise around the mean or to achieve minimum variability by holding both to the new standard, batch after batch. In so doing, a 6 sigma limit may be possible (which is equivalent to not more than three to four defects per 1 million units), also called “zero defects.” The goal of 6 sigma, “zero defects” is easier to achieve for liquid than for solid pharmaceutical dosage forms [16].
Process characterization represents the methods used to determine the critical unit operations or processing steps and their process variables, that usu-ally affect the quality and consistency of the product outcomes or product attri-butes. Process ranging represents studies that are used to identify critical process or test parameters and their respective control limits, which normally affect the quality and consistency of the product outcomes of their attributes. The follow-ing process characterization techniques may be used to designate critical unit operations in a given manufacturing process.
A. Constraint Analysis
One procedure that makes subsystem evaluations and performance qualification trials manageable is the application of constraint analysis. Boundary limits of any technology and restrictions as to what constitutes acceptable output from unit operations or process steps should in most situations constrain the number of process variables and product attributes that require analysis. The application of the constraint analysis principle should also limit and restrict the operational range of each process variable and/or specification limit of each product attri-bute. Information about constraining process variables usually comes from the following sources:
• Previous successful experience with related products/processes
• Technical and engineering support functions and outside suppliers
A practical guide to constraint analysis comes to us from the application of the Pareto Principle(named after an Italian sociologist) and is also known as the 80–20 rule, which simply states that about 80% of the process output is governed by about 20% of the input variables and that our primary job is to find those key variables that drive the process.
The FDA in their proposed amendments to the CGMPs [17] have desig-nated that the following unit operations are considered critical and therefore their processing variables must be controlled and not disregarded:
• Cleaning
• Weighing/measuring
• Mixing/blending
• Compression/encapsulation
• Filling/packaging/labeling
B. Fractional Factorial Design
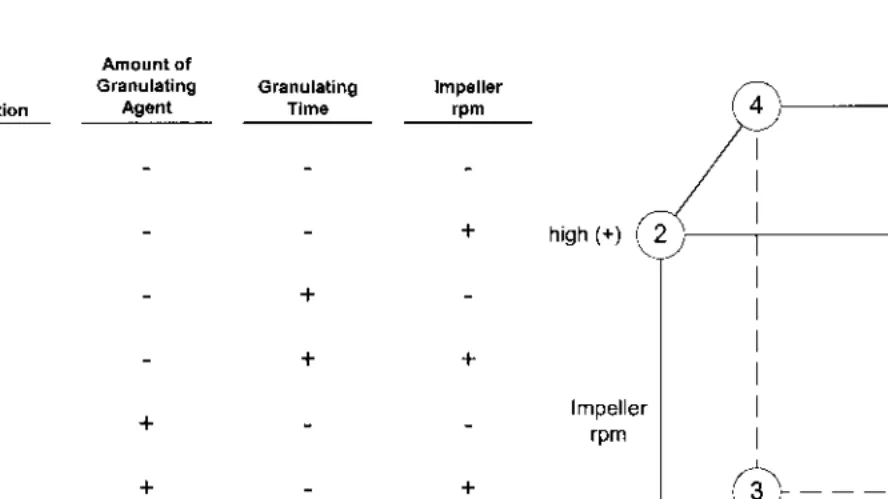
An experimental design is a series of statistically sufficient qualification trials that are planned in a specific arrangement and include all processing variables that can possibly affect the expected outcome of the process under investigation. In the case of afull factorial design,n equals the number of factors or process variables, each at two levels, i.e., the upper (+) and lower (−) control limits. Such a design is known as a 2n factorial. Using a large number of process variables (say, 9) we could, for example, have to run 29
, or 512, qualification trials in order to complete the full factorial design.
The fractional factorial is designed to reduce the number of qualification trials to a more reasonable number, say, 10, while holding the number of ran-domly assigned processing variables to a reasonable number as well, say, 9. The technique was developed as a nonparametric test for process evaluation by Box and Hunter [18] and reviewed by Hendrix [19]. Ten is a reasonable number of trials in terms of resource and time commitments and should be considered an upper limit in a practical testing program. This particular design as presented in Table 7does not include interaction effects.
XI. OPTIMIZATION TECHNIQUES
sta-Table 7 Fractional Factorial Design (9 Variables in 10 Experiments)
Worst-case conditions: Trial 1 (lower control limit). Trial 10 (upper control limit). X variables randomly assigned. Best values to use are RSD of data set for each trial. When adding up the data by columns,+and−are now numerical values and the sum is divided by 5 (number of+s or−s). If the variable is not significant, the sum will approach zero.
ble, least variable, robust process within its proven acceptable range(s) of opera-tion, Chapman’s so-called proven acceptable range (PAR) principle [20].
Optimization techniques may be classified as parametric statistical meth-ods and nonparametric search methmeth-ods.Parametric statistical methods, usually employed for optimization, are full factorial designs, half factorial designs, sim-plex designs, and Lagrangian multiple regression analysis [21]. Parametric methods are best suited for formula optimization in the early stages of product development.Constraint analysis, described previously, is used to simplify the testing protocol and the analysis of experimental results.
The steps involved in the parametric optimization procedure for pharma-ceutical systems have been fully described by Schwartz [22]. Optimization tech-niques consist of the following essential operations:
1. Selection of a suitable experimental design
2. Selection of variables (independent Xs and dependent Ys) to be tested 3. Performance of a set of statistically designed experiments (e.g., 23
or 32
factorials)
4. Measurement of responses (dependent variables)
5. Development of a predictor, polynomial equation based on statistical and regression analysis of the generated experimental data
XII. WHAT ARE THE PROCESS VALIDATION OPTIONS?
The guidelines on general principles of process validation [1] mention three options: (1) prospective process validation (also called premarket validation), (2) retrospective process validation, and (3) revalidation. In actuality there are four possible options.
A. Prospective Process Validation
In prospective process validation, an experimental plan called the validation protocolis executed (following completion of the qualification trials) before the process is put into commercial use. Most validation efforts require some degree of prospective experimentation to generate validation support data. This particu-lar type of process validation is normally carried out in connection with the introduction of new drug products and their manufacturing processes. The for-malized process validation program should never be undertaken unless and until the following operations and procedures have been completed satisfactorily:
1. The facilities and equipment in which the process validation is to be conducted meet CGMP requirements (completion of installation qualification)
2. The operators and supervising personnel who will be “running” the validation batch(es) have an understanding of the process and its re-quirements
3. The design, selection, and optimization of the formula have been completed
4. The qualification trials using (10×size) pilot-laboratory batches have been completed, in which the critical processing steps and process variables have been identified, and the provisional operational control limits for each critical test parameter have been provided
5. Detailed technical information on the product and the manufacturing process have been provided, including documented evidence of prod-uct stability
6. Finally, at least one qualification trial of a pilot-production (100×size) batch has been made and shows, upon scale-up, that there were no significant deviations from the expected performance of the process
The steps and sequence of events required to carry out a process validation assignment are outlined inTable 8. The objective of prospective validation is to prove or demonstrate that the process will work in accordance with a validation master plan or protocol prepared for pilot-product (100×size) trials.
Table 8 Master Plan or Outline of a Process Validation Program
Objective Proving or demonstrating that the process works Type of validation Prospective, concurrent, retrospective, revalidation Type of process Chemical, pharmaceutical, automation, cleaning Definition of process Flow diagram, equipment/components, in-process,
fin-ished product
Definition of process output Potency, yield, physical parameters
Definition of test methods Method, instrumentation, calibration, traceability, preci-sion, accuracy
Analysis of process Critical modules and variables defined by process capa-bility design and testing program
Control limits of critical vari- Defined by process capability design and testing
pro-ables gram
Preparation of validation pro- Facilities, equipment, process, number of validation tri-tocol als, sampling frequency, size, type, tests to perform,
methods used, criteria for success Organizing for validation Responsibility and authority
Planning validation trials Timetable and PERT charting, material availability, and disposal
Validation trials Supervision, administration, documentation Validation finding Data summary, analysis, and conclusions
Final report and recommenda- Process validated, further trials, more process design,
tions and testing
may be the already successfully concluded first pilot batch at 100×size, which is usually prepared under the direction of the organizational function directly responsible for pilot scale-up activities. Later, replicate batch manufacture may be performed by the pharmaceutical production function.
The strategy selected for process validation should be simple and straight-forward. The following factors are presented for the reader’s consideration:
1. The use of different lots of components should be included, i.e., APIs and major excipients.
2. Batches should be run in succession and on different days and shifts (the latter condition, if appropriate).
3. Batches should be manufactured in equipment and facilities desig-nated for eventual commercial production.
5. Failure to meet the requirements of the validation protocol with re-spect to process inputs and output control should be subjected to re-qualificationfollowing a thorough analysis of process data and formal review by the CMC Coordination Committee.
B. Retrospective Validation
The retrospective validation option is chosen for established products whose manufacturing processes are considered stable and when on the basis of eco-nomic considerations alone and resource limitations, prospective validation pro-grams cannot be justified. Prior to undertaking retrospective validation, wherein the numerical in-process and/or end-product test data of historic production batches are subjected to statistical analysis, the equipment, facilities and subsys-tems used in connection with the manufacturing process must be qualified in conformance with CGMP requirements. The basis for retrospective validation is stated in 21CFR 211.110(b): “Valid in-process specifications for such charac-teristics shall be consistent with drug product final specifications and shall be derived from previous acceptable process average and process variability esti-mates where possible and determined by the application of suitable statistical procedures where appropriate.”
The concept of using accumulated final product as well as in-process nu-merical test data and batch records to provide documented evidence of product/ process validation was originally advanced by Meyers [26] and Simms [27] of Eli Lilly and Company in 1980. The concept is also recognized in the FDA’s Guidelines on General Principles of Process Validation[1].
Using either data-based computer systems [28,29] or manual methods, retrospective validation may be conducted in the following manner:
1. Gather the numerical data from the completed batch record and in-clude assay values, end-product test results, and in-process data. 2. Organize these data in a chronological sequence according to batch
manufacturing data, using a spreadsheet format.
3. Include data from at least the last 20–30 manufactured batches for analysis. If the number of batches is less than 20, then include all manufactured batches and commit to obtain the required number for analysis.
4. Trim the data by eliminating test results from noncritical processing steps and delete all gratuitous numerical information.
5. Subject the resultant data to statistical analysis and evaluation. 6. Draw conclusions as to the state of control of the manufacturing
One or more of the following output values (measured responses), which have been shown to be critical in terms of the specific manufacturing process being evaluated, are usually selected for statistical analysis.
1. Solid Dosage Forms
1. Individual assay results from content uniformity testing 2. Individual tablet hardness values
3. Individual tablet thickness values 4. Tablet or capsule weight variation
5. Individual tablet or capsule dissolution time (usually at t50%) or disinte-gration time
6. Individual tablet or capsule moisture content
2. Semisolid and Liquid Dosage Forms
1. pH value (aqueous system) 2. Viscosity
3. Density
4. Color or clarity values
5. Average particle size or distribution 6. Unit weight variation and/or potency values
The statistical methods that may be employed to analyze numerical output data from the manufacturing process are listed as follows:
1 Basic statistics (mean, standard deviation, and tolerance limits) [21] 2. Analysis of variance (ANOVA and related techniques) [21] 3. Regression analysis [22]
4. Cumulative sum analysis (CUSUM) [23] 5. Cumulative difference analysis [23]
6. Control charting (averages and range) [24,25]
Control charting, with the exception of basic statistical analysis, is proba-bly the most useful statistical technique to analyze retrospective and concurrent process data. Control charting forms the basis of modern statistical process con-trol.
C. Concurrent Validation
Test parameter Data source
Average unit potency End-product testing
Content uniformity End-product testing
Dissolution time End-product testing
Weight variation End-product testing
Powder-blend uniformity In-process testing
Moisture content In-process testing
Particle or granule size distribution In-process testing
Weight variation In-process testing
Tablet hardness In-process testing
pH value In-process testing
Color or clarity In-process testing
Viscosity or density In-process testing
Not all of the in-process tests enumerated above are required to demon-strate that the process is in a state of control. Selections of test parameters should be made on the basis of thecriticalprocessing variables to be evaluated.
D. Revalidation
Conditions requiring revalidation study and documentation are listed as follows:
1. Change in acriticalcomponent (usually refers to raw materials) 2. Change or replacement in acriticalpiece of modular (capital)
equip-ment
3. Change in a facility and/or plant (usually location or site)
4. Significant (usually order of magnitude) increase or decrease in batch size
5. Sequential batches that fail to meet product and process specifications
In some situations performance requalification studies may be required prior to undertaking specific revalidation assignments.
The FDA process validation guidelines [1] refer to a quality assurance system in place that requires revalidation whenever there are changes in packag-ing (assumed to be the primary container-closure system), formulation, equip-ment or processes (meaning not clear) which could impact on product effective-ness or product characteristics and whenever there are changes in product characteristics.
The reader should realize that there is no one way to establish proof or evidence of process validation (i.e., a product and process in control). If the manufacturer is certain that its products and processes are under statistical con-trol and in compliance with CGMP regulations, it should be a relatively simple matter to establish documented evidence of process validation through the use of prospective, concurrent, or retrospective pilot and/or product quality informa-tion and data. The choice of procedures and methods to be used to establish validation documentation is left with the manufacturer.
This introduction was written to aid scientists and technicians in the phar-maceutical and allied industries in the selection of procedures and approaches that may be employed to achieve a successful outcome with respect to product performance and process validation. The authors of the following chapters ex-plore the same topics from their own perspectives and experience. It is hoped that the reader will gain much from the diversity and richness of these varied approaches.
REFERENCES
1. Guidelines on General Principles of Process Validation, Division of Manufacturing and Product Quality, CDER, FDA, Rockville, Maryland (May 1987).
2. Current Good Manufacturing Practices in Manufacture, Processing, Packing and Holding of Human and Veterinary Drugs, Federal Register 43(190), 45085 and 45086, September 1978.
3. Good Manufacturing Practices for Pharmaceuticals, Willig, S. H. and Stoker, J. R., Marcel Dekker, New York (1997).
4. Commentary, Pre-approval Inspections/Investigations, FDA,J.Parent.Sci.& Tech. 45:56–63 (1991).
5. Mead, W. J., Process validation in cosmetic manufacture,Drug Cosmet.Ind., (Sep-tember 1981).
6. Chapman, K. G., A history of validation in the United States, Part I,Pharm.Tech., (November 1991).
7. Nash, R. A., The essentials of pharmaceutical validation inPharmaceutical Dosage Forms:Tablets, Vol. 3, 2nd ed., Lieberman, H. A., Lachman, L. and Schwartz, J. B., eds., Marcel Dekker, New York (1990).
8. Nash, R. A., Product formulation,CHEMTECH, (April 1976).
9. Pharmaceutical Process Validation, Berry, I. R. and Nash, R. A., eds., Marcel Dekker, New York (1993).
10. Nash, R. A., Making the Paper Match the Work, Pharmaceutical Formulation & Quality(Oct/Nov 2000).
11. Guidance for Industry, Scale-Up & Postapproval Changes, CDER, FDA (Nov 1995).
14. Nash, R. A., Streamlining Process Validation, Amer. Pharm. Outsourcing May 2001.
15. Ishikawa, K., What is Total Quality Control? The Japanese Way, Prentice-Hall, Englewood Cliffs, NJ (1985).
16. Nash, R. A., Practicality of Achieving Six Sigma or Zero-Defects in Pharmaceutical Systems,Pharmaceutical Formulation & Quality, Oct./Nov. 2001.
17. CGMP: Amendment of Certain Requirements, FDA Federal Register, May 3, 1996.
18. Box, G. E. and Hunter, J. S.,Statistics for Experimenters, John Wiley, N.Y. (1978). 19. Hendrix, C. D., What every technologist should know about experimental design,
CHEMTECH(March 1979).
20. Chapman, K. G., The PAR approach to process validation, Pharm. Tech., Dec. 1984.
21. Bolton, S.,Pharmaceutical Statistics:Practical and Clinical Applications, 3rd ed., Marcel Dekker, New York (1997).
22. Schwartz, J. B., Optimization techniques in product formulation.J. Soc. Cosmet. Chem. 32:287–301 (1981).
23. Butler, J. J., Statistical quality control,Chem.Eng. (Aug. 1983). 24. Deming, S. N., Quality by Design,CHEMTECH, (Sept. 1988).
25. Contino, AV., Improved plant performance with statistical process control,Chem. Eng. (July 1987).
26. Meyer, R. J., Validation of Products and Processes, PMA Seminar on Validation of Solid Dosage Form Processes, Atlanta, GA, May 1980.
27. Simms, L., Validation of Existing Products by Statistical Evaluation, Atlanta, GA, May 1980.
28. Agalloco, J. P., Practical considerations in retrospective validation,Pharm.Tech. (June 1983).
1
Regulatory Basis for
Process Validation
John M. Dietrick
U.S. Food and Drug Administration, Rockville, Maryland, U.S.A.
Bernard T. Loftus
U.S. Food and Drug Administration, Washington, D.C., U.S.A.
I. INTRODUCTION
Bernard T. Loftus was director of drug manufacturing in the Food and Drug Administration (FDA) in the 1970s, when the concept of process validation was first applied to the pharmaceutical industry and became an important part of current good manufacturing practices (CGMPs). His comments on the develop-ment and impledevelop-mentation of these regulations and policies as presented in the first and second editions of this volume are summarized below [1].
II. WHAT IS PROCESS VALIDATION?
The term process validation is not defined in the Food, Drug, and Cosmetic Act (FD&C) Act or in FDA’s CGMP regulations. Many definitions have been of-fered that in general express the same idea—that a process will do what it purports to do, or that the process works and the proof is documented. A June 1978 FDA compliance program on drug process inspections [2] contained the following definition:
A validated manufacturing process is one which has been proved to do what it purports or is represented to do. The proof of validation is obtained through the collection and evaluation of data, preferably, beginning from the process development phase and continuing through the production phase. Validation necessarily includes process qualification (the qualifica-tion of materials, equipment, systems, buildings, personnel), but it also in-cludes the control on the entire process for repeated batches or runs.
The first drafts of the May 1987 Guideline on General Principles of Process Validation [3] contained a similar definition, which has frequently been used in FDA speeches since 1978, and is still used today: “A documented program which provides a high degree of assurance that a specific process will consistently pro-duce a product meeting its pre-determined specifications and quality attributes.”
III. THE REGULATORY BASIS FOR PROCESS VALIDATION
Once the concept of being able to predict process performance to meet user requirements evolved, FDA regulatory officials established that there was a le-gal basis for requiring process validation. The ultimate lele-gal authority is Section 501(a)(2)(B) of the FD&C Act [4], which states that a drug is deemed to be adulterated if the methods used in, or the facilities or controls used for, its manufacture, processing, packing, or holding do not conform to or were not operated or administrated in conformity with CGMP. Assurance must be given that the drug would meet the requirements of the act as to safety and would have the identity and strength and meet the quality and purity characteristics that it purported or was represented to possess. That section of the act sets the premise for process validation requirements for both finished pharmaceuticals and active pharmaceutical ingredients, because active pharmaceutical ingredi-ents are also deemed to be drugs under the act.
The CGMP regulations for finished pharmaceuticals, 21 CFR 210 and 211, were promulgated to enforce the requirements of the act. Although these regulations do not include a definition for process validation, the requirement is implicit in the language of 21 CFR 211.100 [5], which states: “There shall be written procedures for production and process control designed to assure that the drug products have the identity, strength, quality, and purity they purport or are represented to possess.”
IV. THE REGULATORY HISTORY OF
PROCESS VALIDATION
in 1962 with Section 501(a)(2)(B) as an amendment. Prior to then, CGMP and process validation were not required by law. The FDA had the burden of prov-ing that a drug was adulterated by collectprov-ing and analyzprov-ing samples. This was a significant regulatory burden and restricted the value of factory inspections of pharmaceutical manufacturers. It took injuries and deaths, mostly involving cross-contamination problems, to convince Congress and the FDA that a revi-sion of the law was needed. The result was the Kefauver–Harris drug amend-ments, which provided the additional powerful regulatory tool that FDA re-quired to deem a drug product adulterated if the manufacturing process was not acceptable. The first CGMP regulations, based largely on the Pharmaceutical Manufacturers Association’s manufacturing control guidelines, were then pub-lished and became effective in 1963. This change allowed FDA to expect a preventative approach rather than a reactive approach to quality control. Section 505(d)(3) is also important in the implementation of process validation require-ments because it gives the agency the authority to withhold approval of a new drug application if the “methods used in, and the facilities and controls used for, the manufacture, processing, and packing of such drug are inadequate to preserve its identity, strength, quality, and purity.”
Another requirement of the same amendments was the requirement that FDA must inspect every drug manufacturing establishment at least once every 2 years [6]. At first, FDA did this with great diligence, but after the worst CGMP manufacturing situations had been dealt with and violations of the law became less obvious, FDA eased up its pharmaceutical plant inspection activi-ties and turned its resources to more important problems.
The Drug Product Quality Assurance Program of the 1960s and 1970s involved first conducting a massive sampling and testing program of finished batches of particularly important drugs in terms of clinical significance and dollar volume, then taking legal action against violative batches and inspecting the manufacturers until they were proven to be in compliance. This approach was not entirely satisfactory because samples are not necessarily representative of all batches. Finished product testing for sterility, for example, does not assure that the lot is sterile. Several incidents refocused FDA’s attention to process inspections. The investigation of complaints of clinical failures of several prod-ucts (including digoxin, digitoxin, prednisolone, and prednisone) by FDA found significant content uniformity problems that were the result of poorly controlled manufacturing processes. Also, two large-volume parenteral manufacturers ex-perienced complaints despite quality control programs and negative sterility test-ing. Although the cause of the microbiological contamination was never proven, FDA inspections did find deficiencies in the manufacturing process and it be-came evident that there was no real proof that the products were sterile.
offi-cials were not thinking in terms of process validation. One of the first entries into process validation was a 1974 paper presented by Ted Byers, entitled “De-sign for Quality” [7]. The term validation was not used, but the paper described an increased attention to adequacy of processes for the production of pharma-ceuticals. Another paper—by Bernard Loftus before the Parenteral Drug Associ-ation in 1978 entitled “ValidAssoci-ation and Stability” [8]—discussed the legal basis for the requirement that processes be validated.
The May 1987Guideline on General Principles of Process Validation[3] was written for the pharmaceutical, device, and veterinary medicine industries. It has been effective in standardizing the approach by the different parts of the agency and in communicating that approach to manufacturers in each industry.
V. UPDATE
affirmed the requirement for process validation in the current good manufactur-ing regulations, and ordered the defendants to perform process validation studies on certain drug products, as well as equipment cleaning validation studies. This case and the court’s ruling were widely circulated in the pharmaceutical industry and became the subject of numerous FDA and industry seminars.
The court also criticized the CGMP regulations for their lack of specific-ity, along with their ambiguity and vagueness. Responding to this criticism, FDA drafted revisions to several parts of these regulations. The proposed revi-sions were published in theFederal Register on May 3, 1996 [10]. One of the main proposed changes was intended to emphasize and clarify the process vali-dation requirements. The proposal included a definition of process valivali-dation (the same definition used in the 1987 guideline), a specific requirement to vali-date manufacturing processes, and minimum requirements for performing and documenting a validation study. These were all implied but not specific in the 1978 regulation. In proposing these changes, FDA stated that it was codifying current expectations and current industry practice and did not intend to add new validation requirements. Comments from all interested parties were requested under the agency’s rule-making policies, and approximately 1500 comments were received. Most of the responses to the changes regarding process validation supported the agency’s proposals, but there were many comments regarding the definitions and terminology proposed about which processes and steps in a pro-cess should or should not require validation, the number of batches required for process validation, maintenance of validation records, and the assignment of responsibility for final approval of a validation study and change control deci-sions. Because of other high-priority obligations, the agency has not yet com-pleted the evaluation of these responses and has not been able to publish the final rule. In addition to the official comments, the proposed changes prompted numerous industry and FDA seminars on the subject.
Process validation is not just an FDA or a U.S. requirement. Similar re-quirements are included in the World Health Organization (WHO), the Pharma-ceutical Inspection Co-operation Scheme (PIC/S), and the European Union (EU) requirements, along with those of Australia, Canada, Japan, and other interna-tional authorities.
Most pharmaceutical manufacturers now put substantial resources into process validation for both regulatory and economic reasons, but despite contin-ued educational efforts by both the agency and the pharmaceutical industry, FDA inspections (both domestically and internationally) continue to find some firms manufacturing drug products using unvalidated or inadequately validated processes. Evidently there is still room for improvement, and continued discus-sion, education, and occasional regulatory action appears warranted.
harmonized international standards and requirements. Many manufacturers are also working on strategies to reduce the cost of process validation and incorpo-rate validation consideration during product design and development. New tech-nologies under development for 100% analysis of drug products and other inno-vations in the pharmaceutical industry may also have a significant effect on process validation concepts and how they can be implemented and regulated.
REFERENCES
1. Loftus, B. T., Nash, R. A., ed.Pharmaceutical Process Validation. vol. 57. New York: Marcel Dekker (1993).
2. U.S. Food and Drug Administration.Compliance Program no.7356.002.
3. U.S. Food and Drug Administration.Guideline on General Principles of Process Validation. Rockville, MD: FDA, 1987.
4. Federal Food Drug and Cosmetic Act, Title 21 U.S. Code, Section 501 (a)(2)(B). 5. Code of Federal Regulations, Title 21, Parts 210 & 211.Fed Reg 43, 1978. 6. U.S. Code, Federal Food Drug and Cosmetic Act, Title 21, Section 510 (h). 7. Byers, T. E. Design for quality, Manufacturing Controls Seminar, Proprietary
Asso-ciation, Cherry Hill, NJ, Oct. 11, 1974.
8. Loftus, B. T. Validation and stability, meeting of Parenteral Drug Association, 1978.
9. U.S. v.Barr Laboratories,Inc., et al., Civil Action No. 92-1744, U.S. District Court for the District of New Jersey, 1973.
2
Prospective Process Validation
Allen Y. Chao
Watson Labs, Carona, California, U.S.A.
F. St. John Forbes
Wyeth Labs, Pearl River, New York, U.S.A.
Reginald F. Johnson and Paul Von Doehren
Searle & Co., Inc., Skokie, Illinois, U.S.A.
I. INTRODUCTION
Validation is an essential procedure that demonstrates that a manufacturing cess operating under defined standard conditions is capable of consistently pro-ducing a product that meets the established product specifications. In its proposed guidelines, the U.S. Food and Drug Administration (FDA) has offered the following definition for process validation [1].
Process validation is establishing documented evidence that provides a high degree of assurance that a specific process (such as the manufacture of pharmaceutical dosage forms) will consistently produce a product meeting its predetermined specifications and quality characteristics.
Many individuals tend to think of validation as a stand-alone item or an afterthought at the end of the entire product/process development sequence. Some believe that the process can be considered validated if the first two or three batches of product satisfy specifications.
brief discussion of specific ways in which experimental programs can be defined to ensure that critical process development and validation objectives are met.
II. ORGANIZATION
Prospective validation requires a planned program and organization to carry it to successful completion. The organization must have clearly defined areas of responsibility and authority for each of the groups involved in the program so that the objective of validating the process can be met. The structure must be tailored to meet the requirements in the specific organization, and these will vary from company to company. The important point is that a defined structure exists, is accepted, and is in operation. An effective project management struc-ture will have to be established in order to plan, execute, and control the pro-gram. Without clearly defined responsibilities and authority, the outcome of process validation efforts may not be adequate and may not comply with CGMP requirements.
III. MASTER DOCUMENTATION
An effective prospective validation program must be supported by documenta-tion extending from product initiadocumenta-tion to full-scale producdocumenta-tion. The complete documentation package can be referred to as the master documentation file.
It will accumulate as a product concept progresses to the point of being placed in full-scale production, providing as complete a product history as possi-ble. The final package will be the work of many individual groups within the organization. It will consist of reports, procedures, protocols, specifications, ana-lytical methods, and any other critical documents pertaining to the formulation, process, and analytical method development. The package may contain the ac-tual reports, or it may utilize cross-references to formal documentation, both internal and external to the organization.
The ideal documentation package will contain a complete history of the final product that is being manufactured. In retrospect, it would be possible to trace the justification or rationale behind all aspects of the final product, process, and testing.
mentation file should contain all information that was generated during the en-tire product development sequence to a validation process.
IV. PRODUCT DEVELOPMENT
Product development usually begins when an active chemical entity has been shown to possess the necessary attributes for a commercial product. The product development activities for the active chemical entity, formulation, and process form the foundation upon which the subsequent validation data are built.
Generally, product development activities can be subdivided into formula-tion and process development, along with scale-up development.
A. Formulation Development
Formulation development provides the basic information on the active chemical, the formula, and the impact of raw materials or excipients on the product. Typi-cal supportive data generated during these activities may include the following:
1. Preformulation profile or characterization of the components of the formula, which includes all the basic physical or chemical information about the active pharmaceutical ingredients (API, or the chemical en-tity) and excipients
2. Formulation profile, which consists of physical and chemical charac-teristics required for the products, drug-excipient compatibility stud-ies, and the effect of formulation on in vitro dissolution
3. Effect of formulation variables on the bioavailability of the product 4. Specific test methods
5. Key product attributes and/or specifications 6. Optimum formulation
7. Development of cleaning procedures and test methods
Formulation development should not be considered complete until all those fac-tors that could significantly alter the formulation have been studied. Subsequent minor changes to the formulation, however, may be acceptable, provided they are thoroughly tested and are shown to have no adverse effect on product.
B. Process Development
pro-posed manufacturing plant. The process development program should meet the following objectives:
1. Develop a suitable process to produce a product that meets all a. Product specifications
b. Economic constraints
c. Current good manufacturing practices (CGMPs)
2. Identify the key process parameters that affect the product attributes 3. Identify in-process specifications and test methods
4. Identify generic and/or specific equipment that may be required
It is important to remember that cleaning procedures should at least be in the final stages of development, as equipment and facilities in the pilot or proposed manufacturing plant are involved, and the development of the cleaning verifica-tion test methods must be complete.
Process development can be divided into several stages.
Design
Challenging of critical process parameters Verification of the developed process
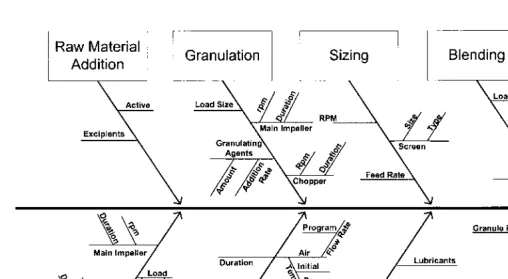
Typical activities in these areas are illustrated inFigure 2.
1. Design
This is the initial planning stage of process development. The design of the process should start during or at the end of the formulation development to define the process to a large extent. One aspect of the process development to remember is end user (manufacturing site) capabilities. In other words, the practicality and the reality of the manufacturing operation should be kept in perspective. Process must be developed in such a manner that it can easily be transferred to the manufacturing site with minimal issues. During this stage, technical operations in both the manufacturing and quality control departments should be consulted.
Key documents for the technical definition of the process are the flow diagram, the cause-and-effect diagram, and the influence matrix. The details of the cause-and-effect diagram and the influence matrix will be discussed under experimental approach in a later section.