whereby analytical methods6,7or
powerful computer search algorithms8,9
locate the best possible code under a given set of assumptions. The associated error value (Dopt) defines the expectation under ‘perfect’ selection for error minimization (‘100% optimal’). Conversely, the mean error value of a large sample of hypothetical codes (Dmean) defines the expectation in the absence of selection (‘0% optimal’). The location of Dcodebetween these extremes (the ‘percentage minimization distance’) is interpreted as a precise linear measure of the strength of selection.
The engineering approach is flawed because possible codes form a Gaussian distribution of error values4,5(Fig. 1a).
Evolution towards an error minimizing ideal would thus not proceed linearly: the fitter a code becomes, the less likely it is to be improved by further changes. Quite simply, a percentage minimization distance of 50% is not twice as adaptive as a distance of 25%, any more than a Z value lying two standard deviations from the mean is twice as significant as a Z value one standard deviation away.
Does the existence of better theoretical alternative codes imply weak selection for error minimization? First, amino acid polarity (used to measure code error values) will not represent selective similarity with complete accuracy. More generally, a highly conservative estimate of the number of possible codes in the absence of biosynthetic restrictions is 20! (52.43310^18), ~10 times the number of seconds that have elapsed since the formation of the earth, rendering it unlikely that nature has tried more than a small fraction. Interpreting Dcode’s failure to represent a global optimum as evidence for weak selection is thus extremely naïve. Although many better codes are possible, the important point is that for every single one there are up to a million worse alternatives4.
Interestingly, however, the incorporation of biosynthetic
restrictions (reducing possible codes to some 270 million), coupled to a more
accurate measure of amino acid similarity (the PAM 74–100 matrix), suggests that nature’s choice might indeed be the best possible code10. Far
from ignoring the idea that selection acted in concert with biosynthetic code expansion, the concluding paragraphs of our review explicitly propose that the combination of stereochemical interactions and strong selection could have channeled biosynthetic expansion to produce the current repertoire of 20 coded amino acids1.
References
1 Knight, R.D. et al. (1999) Selection, history and chemistry: the three faces of the genetic code. Trends Biochem. Sci. 24, 241–247
2 Haig, D. and Hurst L.D. (1991) A quantitative measure of error minimisation within the genetic code. J. Mol. Evol. 33, 412–417
3 Ardell, D.H. (1998) On error minimisation in a sequential origin of the genetic code. J. Mol. Evol. 47, 1–13
4 Freeland, S.J. and Hurst, L.D. (1998) The genetic code is one in a million. J. Mol. Evol. 47, 238–248
5 Freeland, S.J. and Hurst, L.D. (1998) Load minimisation of the genetic code: history does not explain the pattern. Proc. R. Soc. London Ser. B 265, 2111–2119
6 Wong, J.T-F. (1980) Role of minimisation of chemical distances between amino acids in the evolution of the genetic code. Proc. Natl. Acad. Sci. U. S. A. 77, 1083–1086
7 Di Giulio, M. (1989) The extension reached by the minimisation of polarity distances during the evolution of the genetic code. J. Mol. Evol. 29, 288–293
8 Di Giulio, M. et al. (1994) On the optimisation of the physiochemical distances between amino acids in the evolution of the genetic code. J. Theor. Biol. 168, 43–51
9 Di Giulio, M. and Medugno, M. (1999) Physiochemical optimization in the genetic code origin as the number of codified amino acids increases. J. Mol. Evol. 49, 1–10
10 Freeland, S.J. et al. Early fixation of an optimal genetic code. Mol. Biol. Evol. (in press)
STEPHEN J. FREELAND, ROBIN D.
KNIGHT AND LAURA F. LANDWEBER
Dept of Ecology and Evolutionary Biology, Princeton University, Princeton NJ, USA. Email: [email protected]
TIBS 25 –
FEBRUARY 2000
45
0968 – 0004/00/$ – See front matter © 2000, Elsevier Science Ltd. All rights reserved. PII: S0968-0004(99)01531-5LETTERS
(a)
(b)
Best code Canonical code Mean of random codes
F
requency within a r
andom sample
opt code mean
1 better code
999999 worse codes
Percentage
distance minimization
100% 50% 0%
Increasing code error value
TiBS 999 999
Increasing code error value
Figure 1
A comparison of methods. (a)The statistical (sampling) approach provides a direct esti-mate of the probability that a code as good as that used by Nature would evolve by chance; (b)the engineering approach measures code optimality on a linear scale. The figure illus-trates the very different values that resulted from using the two methods for the same set of random codes.
Letters to
TiBS
TiBS
welcomes letters on any topic of interest. Please note, however, that previously unpublished data and
criticisms of work published elsewhere cannot be accepted by this journal.
Letters should be sent to:
Emma Wilson, Editor
Trends in Biochemical Sciences
The PPR motif – a
TPR-related motif
prevalent in plant
organellar proteins
Genome sequencing projects in eukaryotes are revealing thousands of new genes of unknown function, many of which fall into gene families. We
discovered one such family while systematically screening predicted
Arabidopsisproteins for those likely to be targeted to mitochondria or chloroplasts. This large gene family (almost 200 genes in the 70% of the Arabidopsisgenome sequenced so far) is characterized by the presence of tandem arrays of a degenerate 35-amino-acid repeat (Fig. 1). The same family has been identified independently on the basis of other criteria by Aubourg
et al.1Two-thirds of these Arabidopsis
proteins are predicted to be targeted to either mitochondria or chloroplasts (N. Peeters and I.D. Small, unpublished). None of them have been characterized in any way to our knowledge, but a few related sequences in other organisms have been studied. The maize gene crp1
(Ref. 2) is a member of the same family, and similar repeats are found in PET309
from Saccharomyces cerevisiae3and cya-5
from Neurospora crassa4. All three genes
encode proteins involved in some way in processing or translation, or both, of particular organellar mRNAs (Ref. 2). None of these three proteins show obvious sequence similarity to each other or to the
Arabidopsisproteins outside the zone of repeats. The repeat structure in these proteins appears to have been initially overlooked, although Fisk et al.2state in a
note added in proof that these proteins contain TPR (tetratricopeptide) motifs. In fact, although the 35-amino-acid repeats do resemble TPR motifs, they have significant and characteristic differences. To distinguish them from TPR motifs, we propose to call them PPR
(pentatricopeptide) motifs. Using BLAST (Ref. 5) searches on the non-redundant GenBank peptide database, we built up a list of more than 100 sequences likely to contain PPR motifs and then used the MEME program6on this set of sequences
to define a profile corresponding to the PPR motif (Fig. 1). This profile was then used by MOTIFSEARCH [part of the Genetics Computer Group (GCG)
Wisconsin Package, Version 10.0, Madison, WI, USA] to screen the SwissProt/TrEMBL, PIR and GenPept databases. A total of 213 sequences were found with a combined probability score of less than 1024and
containing at least one pair of motifs in tandem (the latter criterion being a very stringent requirement). The number of
motifs per peptide ranges from 2 to 26, with a mean of 9.1. CRP1, Pet309p and CYA-5 were found in this search, along with a few other human or yeast proteins of unknown function (Table 1) and a salt-inducible protein from tobacco8. The vast
majority of the other sequences are from
Arabidopsis. There are numerous expressed sequence tags (ESTs) from related genes from several plant species, including rice, so this gene family is likely to be widespread in higher plants (EST-encoded peptides were not included in
the set searched with MOTIFSEARCH). No prokaryotic proteins were found to possess tandem copies of this motif and none of the known TPR-containing proteins were revealed via this search.
A similar search was carried out starting with known TPR-motif proteins (taken from Ref. 9), using MEME to determine a profile corresponding to the TPR motif (Fig. 1). A search using this profile with MOTIFSEARCH and the same criteria as for the PPR search (i.e.
PROTEIN SEQUENCE MOTIFS
TIBS 25 –
FEBRUARY 2000
46
0968 – 0004/00/$ – See front matter © 2000, Elsevier Science Ltd. All rights reserved. PII: S0968-0004(99)01520-0w
156 I CfN LL Id AYg Q K F Q Y K E A E SL YV QlL E S R Y V P T E 190 191 D TYA LL IK AYC M A G L I E R A E VvL V EMQ N H H V S P K T 225 230 V Y N A YiE G L M K r K G N T E E A I Dv fQ RMK R D R C K P T T 264 265 E TYN Lm IN LYg K A S K S Y M s W KL YC EMR S H Q C K P N I 299 300 C TYT AL vN AfA r E G L C E K A E Ei fE QlQ E D G L E P d V 334 335 Y VYN AL me sYs r A G Y P Y G A A Ei fS LMQ H M G C E P d R 369 370 A sYN Im vd AYg r A G L H S d A E Av fE EMK R L G I A P T M 404 405 K s H M LL lS AYs K A R D V T K C E AiV K EMS E N G V E P d T 439 440 F V L N sm lN LYg r L G Q F T K M E KiL A EME N G p C T A d I 474 475 S TYN IL IN IYg K A G F L E R I E EL fV ElK E K N F R P d V 509 510 V T T s RIG AYs r K K L Y V K C L Ev fE EMI D S G C A P d G 544 545 G T A K VL lS A C s S E E Q V E q V T SvL R TMH K G V T V S S L 579
. TYN AL IN AYA K . G . . E E A . .L Y. .M. . . G . . P N . PPR
consensus
. . AY. . . G . .Y. . . . .YE . A . . .Y. K AL.LN P N N
278 P D AYE I G M R L M E S G A K L S E A G L AfE A AvQ Q d P K H 311 312 V D AwL K L G E V Q T Q N E K E S D g I A A L E K CLELd P T N 345 422 A D V Q T G L G V LfY S M E Efd K T I D CfK A AiEve P d K 455 456 A L NwN R L G A A L A N Y N K P E E A V E AYS r ALQLN P N F 489 490 V R A R Y N L G V SfI N M G RYK E A V E H L L T giSLH E V e 523 TPR
consensus
Helix A Helix B
50 aa
50 aa
* * *
*
*
*
* *
*
*
*
*
Figure 1
TIBS 25 –
FEBRUARY 2000
47
combined probability ,1024, at least onepair of tandem repeats) revealed 288 proteins from a wide range of organisms containing 2–15 motifs (mean 5.2). There is almost no overlap in the sets of sequences returned by MOTIFSEARCH using the PPR and TPR profiles and so these two motifs are truly distinct. Nevertheless, the two motifs can be aligned relatively easily (Fig. 1). TPR motifs consist of a pair of anti-parallel a helices, and tandem arrays of TPR motifs are expected to form a superhelix enclosing a groove, which is likely to be a protein-binding site9. Helix B in the TPR
motif is on the outside of the superhelix, whereas helix A is towards the interior, and thus most of the amino acid side chains projecting into the ligand-binding site are from helix A. The PPR motif is also predicted to consist of two ahelices (using PredictProtein; http://dodo.cpmc. columbia.edu/predictprotein) on an alignment of 1000 putative PPR motifs), and as the conserved tyrosines of the TPR involved in inter-helix packing are also present in the PPR motif, the general arrangement of the helix pairs is
probably similar. We predict therefore that tandem PPR motifs also form a superhelix enclosing a groove or tunnel. The differences between PPR and TPR motifs are essentially at the end of helix B, in the connecting loop between tandem motifs and throughout most of helix A ( i.e. the parts of the motif that line the central cavity). In TPR motifs, the residues projecting into the central groove vary considerably between proteins reflecting the variety of ligands bound, whereas in PPR motifs the side chains lining the central groove are almost exclusively hydrophilic (Fig. 1), and models of several PPR-containing proteins show that the bottom of this groove is positively charged. This is reminiscent of both the positively charged groove in Armadillo-repeat proteins10that bind acidic proteins or
phosphoproteins, or both, and the shorter
basic TPR or TPR-related motifs in some phosphatases and phosphoprotein-binding 14-3-3 proteins8. All of these
proteins form a class containing superhelical structures composed of tandem repeats of an a-helical structural unit11. PPR-motif proteins can clearly be
considered as new members of this class. What could be the function of these PPR-containing proteins and why are there so many of them in plants? By analogy with other helical-repeat proteins, one would predict that PPR proteins have protein-binding properties and, indeed, CRP1 is present in chloroplasts in a protein complex2. Mutants of crp1have a
clear phenotype12, implying that other
members of the PPR family cannot complement the function, suggesting that they are sequence-specific-binding proteins. From the discussion above, it seems likely that the ligands will be hydrophilic and acidic. Moreover, PPR-containing proteins have on average twice as many repeats as TPR proteins,
suggesting multiple or rather extended ligands. It is hard to imagine enough specific organellar protein ligands to explain the huge number of different PPR proteins in Arabidopsis, but given the link between this general type of motif and binding to phosphoproteins, a remote possibility is that some or all the PPR motifs are RNA-binding rather than protein-binding motifs. The width of the central groove is sufficient to hold a single RNA strand, and the positively charged surface at the bottom of the groove could bind the phosphate backbone. The phenotype of crp1, pet309and cya5mutant strains shows that all three must interact directly or indirectly with RNA. Whatever the nature of the direct ligands of these proteins, the peculiarities of RNA processing in plant organelles – notably RNA editing, which appears to require a potentially large number of undiscovered nuclear-encoded sequence-specific factors13– might suggest a reason for the
explosive radiation of this sequence
family in plants. More experimental and structural data on these proteins will be needed to identify their true ligands in vivoand their physiological role.
References
1 Aubourg, S.et al. In Arabidopsis thaliana, 1% of the genome is coding for a novel protein family unique to plants. Plant Mol. Biol. (in press)
2 Fisk, D.G. et al. (1999) Molecular cloning of the maize gene crp1 reveals similarity between regulators of mitochondrial and chloroplast gene expression. EMBO J. 18, 2621–2630
3 Manthey, G.M. and McEwen, J.E. (1995) The product of the nuclear gene PET309 is required for translation of mature mRNA and stability or production of intron-containing RNAs derived from the mitochondrial COXI locus of Saccharomyces cerevisiae. EMBO J. 14, 4031–4043
4 Coffin, J.W. et al. (1997) The Neurospora crassa cya-5 nuclear gene encodes a protein with a region of homology to the Saccharomyces cerevisiae PET309 protein and is required in a post-transcriptional step for the expression of the mitochondrially encoded COXI protein. Curr. Genet. 32, 273–280
5 Altschul, S.F. et al. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410
6 Bailey, T.L. and Elkan, C. (1994) Fitting a Mixture Model by Expectation Maximization to Discover Motifs in Biopolymers. In Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology, pp. 28–36, AAAI Press
7 Claros, M.G. and Vincens, P. (1996) Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 241, 770–786
8 Chang, P-F. (1996) Alterations in cell membrane structure and expression of a membrane-associated protein after adaptation to osmotic stress. Physiol. Plant. 98, 505–516
9 Das, A.K. et al. (1998) The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein-protein interactions. EMBO J. 17, 1192–1199
10 Huber, A.H. et al. (1997) Three-dimensional structure of the Armadillo repeat region of b-catenin. Cell 90, 871–882
11 Groves, M.R. and Barford, D. (1999) Topological characteristics of helical repeat proteins. Curr. Opin. Struct. Biol. 9, 383–389
12 Barkan, A. et al. (1994) A nuclear mutation in maize blocks the processing and translation of several chloroplast mRNAs and provides evidence for the differential translation of alternative mRNA forms. EMBO J. 13, 3170–3181
13 Bock, R. and Koop, H.U. (1997) Extraplastidic site-specific factors mediate RNA editing in chloroplasts. EMBO J. 16, 3282–3288
IAN D. SMALL AND NEMO PEETERS
Station de Génétique, INRA, Route de St-Cyr,
PROTEIN SEQUENCE MOTIFS
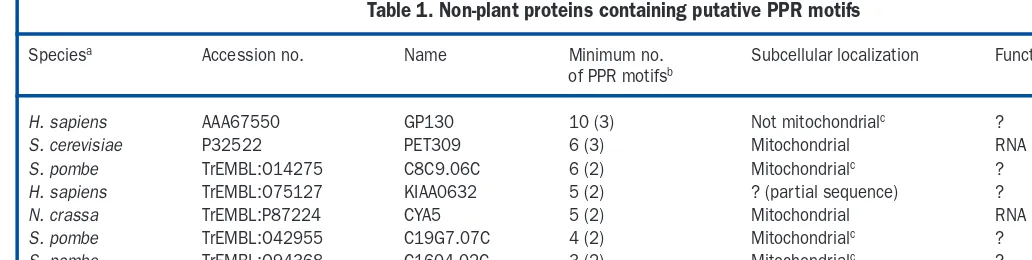
Table 1. Non-plant proteins containing putative PPR motifs
Speciesa Accession no. Name Minimum no. Subcellular localization Function
of PPR motifsb
H. sapiens AAA67550 GP130 10 (3) Not mitochondrialc ?
S. cerevisiae P32522 PET309 6 (3) Mitochondrial RNA processing/translation
S. pombe TrEMBL:O14275 C8C9.06C 6 (2) Mitochondrialc ?
H. sapiens TrEMBL:O75127 KIAA0632 5 (2) ? (partial sequence) ?
N. crassa TrEMBL:P87224 CYA5 5 (2) Mitochondrial RNA processing/translation
S. pombe TrEMBL:O42955 C19G7.07C 4 (2) Mitochondrialc ?
S. pombe TrEMBL:O94368 C1604.02C 3 (2) Mitochondrialc ?
S. cerevisiae P48237 YGL150c 3 (1) Mitochondrialc ?
S. cerevisiae S52526 YPLOO5w 3 (1) Mitochondrialc ?
aAbbreviations: H. sapiens, Homo sapiens; N. crassa, Neurospora crassa; S. cerevisiae, Saccharomyces cerevisiae; S. pombe, Schizosaccharomyces pombe.
bPPR motifs were detected using the MEME profile. The proteins could contain other, less conserved copies of the PPR motif. The longest stretch of strictly