PENGENALAN DASAR
TEKNIK
BIO-MOLEKULER
UU No 28 tahun 2014 tentang Hak Cipta Fungsi dan sifat hak cipta Pasal 4
Hak Cipta sebagaimana dimaksud dalam Pasal 3 huruf a merupakan hak eksklusif yang terdiri atas hak moral dan hak ekonomi.
Pembatasan Pelindungan Pasal 26
Ketentuan sebagaimana dimaksud dalam Pasal 23, Pasal 24, dan Pasal 25 tidak berlaku terhadap:
i. Penggunaan kutipan singkat Ciptaan dan/atau produk Hak Terkait untuk pelaporan peristiwa aktual yang ditujukan hanya untuk keperluan penyediaan informasi aktual; ii. Penggandaan Ciptaan dan/atau produk Hak Terkait hanya untuk kepentingan penelitian
ilmu pengetahuan;
iii. Penggandaan Ciptaan dan/atau produk Hak Terkait hanya untuk keperluan pengajaran, kecuali pertunjukan dan Fonogram yang telah dilakukan Pengumuman sebagai bahan ajar; dan
iv. Penggunaan untuk kepentingan pendidikan dan pengembangan ilmu pengetahuan yang memungkinkan suatu Ciptaan dan/atau produk Hak Terkait dapat digunakan tanpa izin Pelaku Pertunjukan, Produser Fonogram, atau Lembaga Penyiaran.
Sanksi Pelanggaran Pasal 113
1. Setiap Orang yang dengan tanpa hak melakukan pelanggaran hak ekonomi sebagaimana dimaksud dalam Pasal 9 ayat (1) huruf i untuk Penggunaan Secara Komersial dipidana dengan pidana penjara paling lama 1 (satu) tahun dan/atau pidana denda paling banyak Rp100.000.000 (seratus juta rupiah).
2. Setiap Orang yang dengan tanpa hak dan/atau tanpa izin Pencipta atau pemegang Hak Cipta melakukan pelanggaran hak ekonomi Pencipta sebagaimana dimaksud dalam Pasal 9 ayat (1) huruf c, huruf d, huruf f, dan/atau huruf h untuk Penggunaan Secara Komersial dipidana dengan pidana penjara paling lama 3 (tiga) tahun dan/atau pidana denda paling banyak Rp500.000.000,00 (lima ratus juta rupiah).
PENGENALAN DASAR
TEKNIK
BIO-MOLEKULER
Chris Adhiyanto
Laifa Hendarmin
Rini Puspitaningrum
PENGENALAN DASAR TEKNIK BIO-MOLEKULER Chris Adhiyanto
Laifa Hendarmin Rini Puspitaningrum
Editor:
dr. Hari Hendarto, SpPD-KEMD, Ph.D.
Desain cover Zahra Muthmainnah Solihin, S.Pd. Sumber freepik.com Tata letak :
Amira Dzatin Nabila
Proofreader :
Avinda Yuda Wati
Ukuran :
vi, 82 hlm, Uk: 15.5x23 cm
ISBN : Cetakan Pertama:
Bulan 2020
Hak Cipta 2020, Pada Penulis Isi diluar tanggung jawab percetakan
Copyright © 2020 by Deepublish Publisher
All Right Reserved Hak cipta dilindungi undang-undang Dilarang keras menerjemahkan, memfotokopi, atau
memperbanyak sebagian atau seluruh isi buku ini tanpa izin tertulis dari Penerbit.
PENERBIT DEEPUBLISH (Grup Penerbitan CV BUDI UTAMA)
Anggota IKAPI (076/DIY/2012)
Jl.Rajawali, G. Elang 6, No 3, Drono, Sardonoharjo, Ngaglik, Sleman Jl.Kaliurang Km.9,3 – Yogyakarta 55581
Telp/Faks: (0274) 4533427 Website: www.deepublish.co.id
www.penerbitdeepublish.com E-mail: [email protected]
Kata Pengantar
Buku Pengenalan Dasar Teknik Bio-Molekuler ini merupakan pengembangan dan perbaikan dari buku sebelumnya yang berjudul
Genetika Molekuler dan Aplikasinya. Dalam buku ini, kami
menambahkan beberapa metode teknik pembelajaran dalam melakukan penelitian bio-molekuler khususnya yang berkaitan dengan DNA, RNA dan Protein.
Buku ini kami tulis berdasarkan hasil pengalaman kerja dalam membimbing mahasiswa S-1, S-2 dan S-3 dari Program Studi Ilmu Kedokteran, Farmasi, Gizi dan Biologi, yang telah dilakukan di Laboratorium Riset Fakultas Kedokteran UIN Syarif Hidayatullah Jakarta sejak 2013 hingga sekarang. Selain itu, isi buku ini juga merupakan hasil pengalaman yang kami peroleh saat melakukan penelitian kolaborasi dengan universitas di luar Indonesia.
Kami harapkan, buku ini dapat membantu dan menjadi dasar pengetahuan melakukan penelitian bio-molekuler mahasiswa S-1, S-2 maupun S-3 dalam melakukan penelitian bio-molekuler.
Kami sebagai penulis merasa masih banyak kekurangan yang ada dalam buku ini, karenanya kami akan berupaya menambah informasi dan pengetahuan yang kami miliki agar ke depannya dapat kami tambahkan teknik maupun informasi tambahan lainnya dalam buku yang sama.
Semoga buku ini dapat membantu bagi para peneliti pemula dalam bidang bio-molekuler.
Jakarta, Februari 2020 Tim Penulis
Daftar Isi
Kata Pengantar ... v
Daftar Isi ... vi
BAB I. ISOLASI GENOM ... 1
1.1. Pendahuluan ... 1
1.1.1. Persiapan Genom Manusia ... 2
1.1.2. Menghancurkan Sel ... 2
1.1.3. Membuang Protein ... 3
1.1.4. Membuang RNA ... 4
1.1.5. Pemekatan DNA ... 4
BAB II. ELEKTROFORESIS DAN IDENTIFIKASI GENOM DNA ... 13
BAB III. ISOLASI RNA ... 16
BAB IV. PCR (POLYMERASE CHAIN REACTION) ... 23
BAB V. REAL-TIME PCR/KUALITATIF PCR (RT-PCR/ QPCR) ... 34
BAB VI. MERANCANG PRIMER ... 60
BAB VII. ISOLASI PROTEIN ... 67
7.1. Pemurnian Protein ... 69
7.2. SDS-Page ... 70
Daftar Pustaka ... 78
BAB I.
ISOLASI GENOM
1.1. Pendahuluan
Mendapatkan DNA berkualitas tinggi dan utuh seringkali merupakan langkah pertama dan paling penting dalam banyak aplikasi biologi molekuler, seperti kloning DNA, sekuensing, PCR, dan elektroforesis. Mengisolasi keseluruhan DNA utuh dari berbagai macam sampel jaringan memiliki kesulitan yang berbeda karena tergantung dari sifat fisik dan biokimia jaringan yang dituju. Sebagai contoh, banyak metode ekstraksi DNA akan bekerja dengan baik untuk jaringan seperti hati, namun pada jaringan lain seperti jantung, lemak, otak, dan limpa, isolasi DNA hingga saat ini masih merupakan hal yang sulit untuk dilakukan. Isolasi yang paling umum dan mudah, khususnya isolasi genom pada hewan adalah dengan menggunakan sampel darah atau cairan tubuh dari organisme tersebut.
Beberapa metode telah banyak dikembangkan dengan tingkat kesukaran, kemurnian hasil dan konsentrasi hasil yang berbeda-beda. Namun demikian, teknik konvensional masih merupakan teknik yang terbaik. Permasalahannya adalah teknik konvensional ini menghasilkan produk buangan atau limbah yang berbahaya. Teknik konvensional biasa menggunakan fenol-kloroform sebagai proses menghilangkan materi lain selain nukleotida, misal protein. Sebagaimana diketahui fenol merupakan bahan yang sangat korosif sehingga pembuangan limbah yang tidak tepat, akan mencemari lingkungan sekitarnya.
Pada teknik dasar bio-molekuler, kami akan memaparkan beberapa teknik yang dapat digunakan untuk isolasi genom dari bahan cairan tubuh. Pemilihan sumber cairan tubuh, bukan jaringan hanya pada kemudahan dalam pengerjaan isolasi. Perbedaan isolasi dari jaringan adalah pada langkah bagaimana teknik menghancurkan
jaringan tersebut. Dengan menguasai teknik dasar isolasi genom dari bahan cairan tubuh, akan memudahkan kita dalam mempelajari teknik isolasi genom dari sumber jaringan.
1.1.1. Persiapan Genom Manusia
Tujuan dari percobaan ini adalah mengisolasi dan mempurifikasikan dalam jumlah banyak genom manusia. Sumber yang paling mudah dan tidak korosif adalah saliva ataupun usapan lapisan mukosa mulut pipi bagian dalam.
Telah kita ketahui bahwa konsentrasi jumlah DNA dalam sel hanya sedikit, ditemukan sebagian besar di inti sel (90%). Karena jumlah yang sangat sedikit, maka diperlukan keterampilan dalam memisahkan DNA dengan komponen sel lainnya. Ada banyak metode memurnikan atau purifikasi DNA dengan komponen lain, terutama protein. Namun semua proses memiliki prinsip dasar yang sama, yaitu: 1. Menghancurkan sel. 2. Membuang protein. 3. Membuang RNA. 4. Memekatkan DNA. 1.1.2. Menghancurkan Sel
Menghancurkan sel merupakan salah satu langkah penting dalam pemurnian DNA. Sel dapat dihancurkan atau dipecah menggunakan teknik sonikasi, grinding/giling, cacah atau dengan tekanan tinggi agar sel hancur. Namun, langkah ini sering mengakibatkan DNA terpotong-potong jika metode yang digunakan kurang baik, sehingga kemurnian DNA akan turun.
Langkah yang terbaik adalah menggunakan bahan kimia seperti pemberian detergen/sabun atau menggunakan teknik enzimatik. Sabun akan melarutkan lipid membran sel. Sabun juga akan memiliki peran sebagai inhibitor DNAse dan dapat mendenaturasi protein, sehingga membantu dalam membuang protein dari larutan uji. Detergen yang biasanya digunakan sebagai pelarut lisis sel hewan
adalah detergen yang bersifat anionic seperti SDS (sodium deodesil sulfat) atau Sarkosil (sodium deodesil sarkosinat).
1.1.3. Membuang Protein
Langkah selanjutnya dalam proses isolasi dan pemurnian genom adalah membuang semua materi atau komponen protein, dikenal sebagai deproteinasi. Prinsip dasar pada proses ini adalah membuang kemampuan kelarutan protein, sehingga protein akan menjadi tidak larut (mengendap) sedangkan asam nukleat tetap larut.
Banyak pelarut organik yang dapat digunakan untuk mengendapkan protein. Metode fenol dan kloroform, umumnya digunakan dalam mengendapkan protein. Metode fenol mempunyai prinsip sebagai berikut: Fenol adalah kristal pada suhu kamar, namun dengan adanya 20 persen air, ia membentuk suspensi berair yang mengandung misel fenol yang dikelilingi oleh molekul air. Molekul protein banyak mengandung residu asam amino hidrofobik yang terpusat di pusat molekul. Ketika larutan yang mengandung protein terlarut dicampurkan dengan fenol yang bersifat lebih hidrofobik akan menyebabkan fenol akan berdifusi ke dalam inti protein, sehingga protein membengkak dan protein menjadi terbuka (unfold) dan ter-denaturasi. Protein yang terdenaturasi dengan kelompok hidrofobik yang terpapar dan dikelilingi oleh misel fenol, jauh lebih mudah larut dalam fase fenol daripada fasa berair. Akibatnya, protein dipartisi ke fasa fenol yang meninggalkan asam nukleat dalam fasa berair. Asam nukleat tidak memiliki gugus hidrofobik sama sekali dan tidak dapat larut dalam fase fenol. Selain itu, fenol juga berperan dalam membuang fraksi lipid dalam larutan sampel DNA tersebut.
Metode kloroform digunakan karena kloroform tidak larut dalam air sehingga mencegah hilangnya DNA ke fase organik. Deproteinisasi oleh kloroform didasarkan atas ter-denaturasi rantai polipeptida pada interfase air-kloroform. Konsentrasi protein yang tinggi pada interfase akan menyebabkan protein mengendap. Namun deproteinisasi ini sangat tergantung pada pembentukan daerah interfase yang besar, sehingga bantuan mekanik yang besar yaitu
pengocokan/penggoyangan yang kuat. Selain itu, metode ini hanya cocok untuk mendapatkan DNA dengan total ukuran 20 ribu-50 ribu pasang basa (umumnya DNA virus). Metode fenol dan kloroform memiliki keburukan dalam hal limbah. Sifat toksin yang tinggi, memerlukan penangan limbah yang khusus.
Metode lain adalah menggunakan enzim. Protein dapat dibuang dalam larutan sampel genom menggunakan enzim protease. Enzim ini akan memecah atau mencerna semua protein. Ada dua macam enzim yang sering digunakan yaitu proteinase K dan pronase. Kedua enzim ini sangat stabil dan diperoleh dari jamur. Enzim ini akan bekerja aktif pada larutan yang rendah seperti detergen anionik, tinggi konsentrasi garam EDTA, pH 6-10 dan suhu antara 500-600C.
Perbedaan antar kedua enzim ini adalah pronase bersifat
self-digesting, sehingga harus selalu ditambahkan pada saat proses
berlangsung. Metode lain adalah dengan penggunaan asam. Prinsip dasarnya adalah penambahan larutan asam seperti asam cuka akan menyebabkan koagulasi protein, sehingga protein akan mudah diendapkan. Proses ini terjadi akibat asam akan mengacaukan jembatan garam dengan adanya muatan ionik.
1.1.4. Membuang RNA
Untuk membuang RNA saat isolasi DNA, maka digunakan metode enzimatik. Namun, tidak semua RNA akan hilang, sejumlah kecil RNA akan tetap ditemukan pada proses pemurnian DNA. Ada dua enzim yang digunakan, yaitu RNase A dan RNase T1. Mekanisme keduanya didasarkan pada posisi pemotongan basa nuklotida urasil, sitosin dan guanine.
1.1.5. Pemekatan DNA
Pengendapan dan pemekatan DNA telah dimulai pada tahap deproteinisasi menggunakan campuran fenol-kloroform-alkohol. Prinsip pengendapan pada alkohol didasarkan pada kemampuan penurunan kelarutan asam nukleat dalam air. Molekul air yang polar yang mengelilingi molekul DNA akan mempengaruhi kelarutan DNA.
Etanol akan mengubah potensial ionik dari DNA sehingga akan membuang molekul air yang berinteraksi dengan DNA, akibatnya DNA akan mengendap. Penambahan etanol 95% dalam larutan sampel DNA akan menarik molekul air-DNA sehingga DNA akan mengendap. Penggunaan etanol 95% atau 100% murni, akan mempengaruhi dalam hal pembiayaan. Untuk itu, digunakan etanol dengan konsentrasi lebih rendah. Namun, proses ini memerlukan penambahan larutan senyawa garam dalam larutan DNA agar menetralkan muatan fosfat-DNA sehingga mengeliminasi interaksi air-DNA. Senyawa larutan garam yang sering digunakan adalah natrium atau amonium. Namun harap diingat, pengeliminasi materi garam dari larutan DNA yang tidak sempurna akan mempengaruhi proses PCR karena senyawa garam ini akan mengganggu proses enzimatik pada proses PCR.
Penggunaan amonium asetat yang memiliki kelarutan tinggi dalam etanol, sangat dianjurkan dalam memperoleh DNA yang murni. Setelah larutan DNA diendapkan, garam amonium akan mudah dilarutkan dengan penambahan etanol 70%. Sedangkan untuk menghilangkan fraksi etanol pada DNA, dapat digunakan pemanasan. Dalam temperatur 600C, etanol akan mudah dan cepat menguap.
Prinsip protokol isolasi genom ini dapat diterapkan baik menggunakan darah, sel maupun jaringan. Perbedaan pada jaringan, seperti jaringan tumbuhan, organ dan sebagainya hanya bagaimana cara kita untuk memecah jaringan tersebut, seperti untuk isolasi genom DNA dari jaringan hati, maka kita harus menggerus jaringan tersebut menggunakan teknik penggerusan dengan lumping. Ekstrak gerusan tersebut akan digunakan pada metode isolasi seperti di atas.
Saat ini, telah banyak dikembangkan teknik isolasi genom DNA menggunakan KIT dari berbagai manufaktori. Masing-masing KIT memiliki kelebihan dan kekurangan, sebagai contoh KIT isolasi DNA genom menggunakan miniprep column memiliki kelebihan dalam memangkas waktu kerja, tidak menggunakan bahan fenol yang berbahaya dan mudah dalam pengerjaannya, namun kekurangannya adalah dalam harga yang lebih mahal. Umumnya KIT isolasi genom
yang diproduksi di Eropa maupun Amerika, dijual lebih mahal dibandingkan produk dari di luar negara tersebut.
Hal yang perlu diperhatikan dalam isolasi genom
Penanganan material sampel
Untuk isolasi DNA genom dari sel dan jaringan, sebaiknya gunakan sampel baru fresh sample. Jika kita tidak dapat langsung mengisolasi, sampel tersebut wajib dibekukan dalam cairan nitrogen atau lemari beku -70C. Hal ini bertujuan mencegah degradasi DNA akibat aktivitas enzim pendegradasi. Jika sampel yang kita koleksi adalah darah atau cairan tubuh, penyimpanan pada suhu ruang hanya bertahan selama 1-2 hari. Penyimpanan pada lemari beku suhu 4C, hanya bertahan hingga 7 hari. Penyimpanan untuk 1 bulan, sampel dapat dibekukan pada suhu -20C. Penyimpanan terbaik adalah pada suhu -80C, sampel dapat bertahan lebih dari 5 tahun. Umumnya, penyimpanan lama, akan menurunkan kemurnian dari DNA genom. Perlu diingat, untuk sampel darah, antikoagulan yang diperbolehkan adalah selain heparin. Pemakaian heparin sebagai antikoagulan, akan mengganggu proses tahap PCR. Bahan sampel terutama dalam bentuk cairan, harap disimpan dalam bentuk alikuot atau ditaruh di lebih dari 1 tabung. Hal ini bertujuan mencegah kerusakan bahan sampel akibat proses cair-beku-cair (“thawing”) saat melakukan isolasi genom DNA.
Genom DNA hasil isolasi, sebaiknya disimpan pada suhu 2-8C. Penyimpanan pada suhu beku akan merusak genom DNA atau genom DNA akan terpotong-potong. Untuk genom DNA plasmid, dapat disim-pan pada suhu -20C. Hindari pengeringan yang berlebih (pemanasan yang lama) saat menguapkan etanol ditahap isolasi genom DNA. Sebaiknya keringkan atau uapkan etanol dengan membuka tutup tube dan taruh pada suhu ruangan (namun memerlukan waktu o.n atau “over night”). Untuk melarutkan genom DNA, gunakan buffer Tris 10 mM pH 7-8. Hindari penggunaan air atau ddH2O. Agar memudahkan genom DNA cepat larut, dapat diinkubasi suhu 65C selama 10 menit. Pemanasan ini juga akan merusak DNase, sehingga genom DNA tidak rusak. Hindari vorteks saat melarutkan genom DNA.
Cara menghitung konsentrasi dan kemurnian genom DNA
Genom DNA hasil isolasi dapat diukur konsentrasi dan kemurniannya dengan cara mengetahui mengukur nilai OD dari genom tersebut menggunakan teknik spektrofotometri. Genom DNA diencerkan dan dibaca pada panjang gelombang 230 nm, 260 nm dan 280 nm. Saat ini, telah ada alat spektrofotometer yang dapat digunakan untuk mengukur kemurnian dan konsentrasi genom DNA dalam jumlah yang sangat sedikit, antara 0.5 – 2.0 uL. Alat ini akan menghitung konsentrasi DNA secara otomatis dan tanpa perlu pengenceran. Untuk mengetahui kemurnian DNA, diukur berdasarkan perbandingan absorbansi pada panjang gelombang 260/280 nm dan 260/230 nm. Kemurnian yang baik adalah jika nilai perbandingan 260/280 nm : 1.8 – 2.1 dan 260/230 nm 2-2.2. Jika nilai kurang atau lebih dari kisaran tersebut, menunjukkan masih ada senyawa organik atau garam dalam sampel DNA hasil isolasi. Untuk mengatasi kemurnian yang tidak baik, dapat dilakukan pengulangan tahap isolasi, atau dilakukan pengenceran dengan tujuan untuk memperkecil senyawa bukan DNA dalam sampel. Namun, pengenceran akan menurunkan konsentrasi dari sampel DNA hasil isolasi. Sampel DNA dengan konsentrasi rendah (0.01 ng/uL) masih dapat digunakan untuk tahapan RT PCR (qPCR) selama kemurniannya dalam kisaran yang baik.
Isolasi genom dari liur
Isolasi genom dari liur atau saliva, merupakan salah satu teknik yang sederhana, tidak melukai dan mudah pengerjaannya. Bahan dan alat yang dibutuhkan adalah liur, tabung reaksi 30 mL, etanol, buffer saline (PBS), larutan lisis, PCI dan amonium asetat serta alat sentrifuse dan tabungnya. Kadang teknik dikombinasikan dengan pemberian asam cuka encer.
Prosedur kerja:
1. Sepuluh milliliter larutan NaCl fisiologis (buffer saline) digunakan untuk berkumur selama 60 detik.
3. Tabung kemudian disentrifuse pada kecepatan maksimum (5000 rpm) selama 20 menit. Lebih baik jika menggunakan sentrifuse dingin.
4. Hati-hati dalam membuang supernatant, jangan sampai pellet sel terlepas dari dinding tabung.
5. Tambahkan 1 mL mL larutan lisis buffer yang terdiri dari campuran 0.5% SDS, 0.1 M EDTA, 10 mM Tris-HCl.
6. Tambahkan 50 uL Proteinase K
7. Kocok perlahan, jangan berbuih (ingat kita menggunakan sabun), sampai pellet terlepas dari dinding tabung. Kemudian inkubasi 560C selama 3 jam atau biarkan dalam suhu kamar
selama 1 malam.
8. Setelah 3 jam atau dibiarkan satu malam, tambahkan larutan 1 mL PCI (fenol-8OH-TE) dengan perbandingan volume yang sama. Kocok kuat-kuat selama 30 detik, lalu sentrifuse kecepatan maksimum selama 10 menit. (gambar 1)
9. Ambil lapisan atas dan pindahkan ke tabung baru yang berisi kloroform dengan volume yang sama. Hati-hati dalam memindahkan DNA yang terlarut dalam fase cair (larutan bening), jangan sampai tercampur dengan daerah interfase yang berwarna putih, dan lapisan bawah yang merupakan fase fenol (larutan kuning). Tabung baru yang telah berisi hasil sentrifuse no 8 dan kloroform, kemudian dikocok dan disentrifuse kecepatan maksimum selama 10 menit. Pindahkan larutan atas ke tabung baru. Fungsi kloroform dalam percobaan ini untuk membersihkan fenol dari sampel. 10. Tambahkan setengah volume (kira-kira 1 mL) 7.5 M amonium
asetat, tabung dibolak-balik 10 kali.
11. Tambahkan etanol 95% dingin sebanyak 2 x jumlah volume larutan sebelumnya, misalkan jumlah volume total DNA dan amonium asetat 3 ml, maka tambahkan etanol 6 mL. Bolak balik dengan pelan, maka akan muncul helaian DNA seperti kapas. Ambil helaian DNA secara hati-hati dengan
menggunakan batang kaca dan pindahkan ke tabung yang sudah mengandung 3 etanol 70% dingin.
12. Sentrifuse pada kecepatan maksimum selama 5 menit. DNA akan mengendap di bagian bawah tabung seperti lapisan atau butiran putih.
13. Buang hati-hati etanol tersebut, buang sebanyak mungkin, kemudian keringkan di oven dengan tutup tabung terbuka, pada suhu 700C.
14. Setelah kering, pellet DNA dapat dilarutkan dengan 100 uL buffer TE (Tris EDTA). Biarkan semalam dalam temperatur 40C
sehingga DNA larut.
15. Untuk memeriksa keberhasilan kita, dapat digunakan uji elektroforesis dan nanodrop untuk menghitung kemurnian-konsentrasi DNA.
Gambar 1. Isolasi DNA pada Pemisahan Fenol
(www.genetargetsolutions.com.au/product/5prime-phase-lock-gel/)
Isolasi Genom dari darah dengan metode fenol-kloroform
Bahan : - dH2O
- Larutan salin (NaCl 0.8%)
- Larutan lisis terdiri dari 14 g NH4Cl dan 0.144 g NH4HCO3
- Proteinase K - 10% SDS
- STE pH 7.4, yang terdiri dari 2.92 g NaCl; 3.03 g Trizma base; 0.19 g EDTA dibuat dalam 500 mL air steril
- TE yang terdiri dari 1.21 g Trizma base; 0.37 g Na-EDTA dibuat dalam 900 mL. Larutan TE ini adalah stock, jika akan dibuat pH 8, maka larutan TE tersebut ditetesi 1 N HCl hingga pH 8, kemudian ditambahkan air steril hingga 1 L. Hal yang sama dilakukan jika untuk membuat pH 7.6
- TE-Saturated Phenol pH 8 dengan penambahan 0.1% 8-hydroxyquinoline - Kloroform - 99.5% etanol - 3 M Na-Asetat - 70 % etanol Prosedur: Cell Lysis.
Darah (sekitar 2ml) disentrifus pada kecepatan 5,000 rpm selama 5 menit pada suhu ruang.
Tiga lapisan akan terbentuk (serum,
Buffy coat dan eritrosit/RBC).
Serum dibuang (jika dibutuhkan, simpan di tabung lain).
Tambahkan salin untuk mencuci pellet, sentrifus pada kecepatan 5,000 rpm selama 5 menit pada suhu 40C (atau suhu
ruang).
Buang lapisan atas (supernatan) dan pindahkan buffy coat ke tabung baru (tabung mikro1.5 mL).
Ulangi langkah 4 dan 5 sehingga akan diperoleh buffy coat (sel darah putih) dalam jumlah yang banyak.
Tambahkan lysing solution (volume maksimum) ke dalam tabung yang berisi buffy coat, kocok dan campur dengan baik
Serum RBC Buffy coat
selama 5 menit. Kemudian disentrifus dengan kecepatan 15,000 rpm selama 5 menit pada 40C.
Ulangi langkah ke 7 tiga kali, dan harus yakin tidak ada eritrosit yang tersisa (hanya buffy coat).
putih terlarut sempurna.
Tambahkan 20 L 10% SDS, campur dengan baik.
Tambahkan 4 L proteinase K, dan campur dengan baik (kocok pelan-pelan, jangan berbuih).
Biarkan semalaman pada 370C atau simpan pada inkubator
suhu 600C untuk 1-2 jam.
Phenol-Chloroform Reaction
Tambahkan 400 L TE-saturated phenol, kocok perlahan-lahan selama 5 menit.
Sentrifus pada 15,000 rpm selama 5 menit dalam suhu kamar.
Dapat terlihat 3 lapisan, bagian lapisan atas (DNA), tengah (protein yang terdegradasi), bawah (phenol).
Pindahkan lapisan atas (DNA) ke dalam tabung mikro baru 1.5 mL menggunakan pipet tetes.
chloroform dingin dan campur dengan
baik (bolak-balik) untuk 5 menit dan sentrifus pada 15000 rpm selama 5 menit. (chloroform dapat berfungsi untuk membuang atau menetralkan phenol, phenol harus disingkirkan karena akan medetnaturasi protein enzim yang akan digunakan pada step PCR atau analisis lain.)
mikro baru 1.5 mL menggunakan pipet bersih.
Ulangi dua langkah sebelumnya hingga seluruh phenol yang ada hilang.
Tamba ethanol 99.9
%.
Sentrifuse pada 15,000 rpm selama 5 menit. Buang supernatan
ethanol.
Tambahkan 1mL 70% ethanol, dan sentrifus pada 15,000 rpm selama 5 menit.
Buang ethanol seluruhnya.
DNA dapat dikeringkan menggunakan sentrifus vakum selama 5 menit hingga tidak ada ethanol yang tersisa.
Jika tidak ada sentrifus vakum, cukup letakkan pada blok inkubator suhu 600C dan biarkan hingga kering.
Larutkan DNA pellet menggunakan larutan 100
per 1ml darah).
Simpan pada 40C.
Selain metode konvensional, ada beberapa metode yang memudahkan peneliti dalam isolasi DNA, yaitu menggunakan spin kolom.
BAB II.
ELEKTROFORESIS
DAN IDENTIFIKASI GENOM DNA
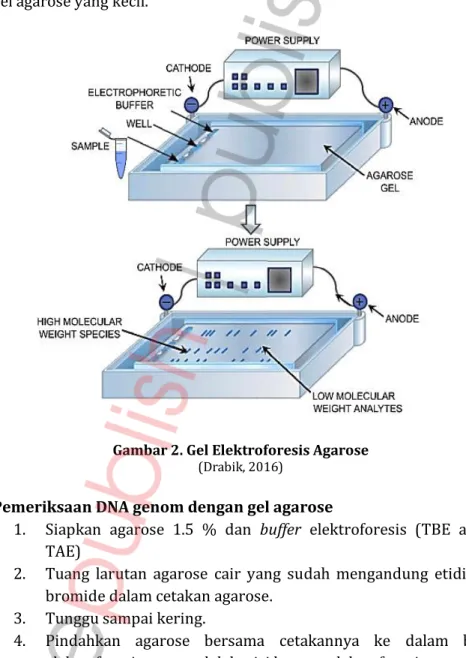
Gel elektroforesis adalah suatu teknik yang digunakan untuk memisahkan fragmen DNA dari makromolekul lain (RNA dan protein) berdasarkan ukuran dan muatannya. Molekul ini akan bergerak melalui gel menuju arah yang berbeda muatan dan kecepatan. Untuk molekul DNA, semua molekulnya memiliki muatan listrik yang sama yaitu negatif, sehingga pergerakannya akan dipengaruhi oleh ukurannya.
Gel elektroforesis untuk pemisahan DNA adalah agarose yang merupakan polisakarida. Ketika agarose dilarutkan dalam buffer garam kemudian dipanaskan dan setelah terlarut sempurna, didinginkan, maka akan membentuk suatu bahan solid, namun tidak keras dan permukaan yang licin. Pada prinsipnya agarose yang mengeras menyerupai makanan agar-agar atau jello. Pada pengamatan molekular, agarose gel merupakan matriks yang molekulnya satu sama lain disatukan oleh ikatan hidrogen dan membentuk semacam pori-pori atau lubang saringan kecil.
Untuk mengamati fragmen DNA, maka gel agarose tersebut ditempatkan dalam suatu wadah berisi larutan buffer garam serta dialirkan listrik melalui panel elektroda positif dan negatif. Saat pembuatan gel agarose, dalam cetakan akan ditaruh sisir DNA, sehingga saat gel agarose mengeras, akan ada sumur-sumur tempat menuangkan cairan DNA. Adanya gugus fosfat pada DNA, maka DNA akan bermuatan negatif. Oleh karena itu, posisi sumur untuk DNA diletakkan dalam wadah elektroforesis di area kutub elektroda negatif. Saat aliran listrik mengalir dalam gel agarose, DNA akan bergerak ke arah kutub elektroda positif. Pergerakan kecepatan DNA dalam gel agarose dipengaruhi oleh ukuran dari fragmen DNA
tersebut. Jika ukuran fragmen DNA adalah kecil, maka pergerakan menuju kutub elektroda positif akan cepat, karena molekul DNA akan mudah melewati pori-pori gel agarose. Jika ukuran fragmen DNA-nya besar, maka kecepatan akan melambat akibat hambatan dari pori-pori gel agarose yang kecil.
Gambar 2. Gel Elektroforesis Agarose
(Drabik, 2016)
Pemeriksaan DNA genom dengan gel agarose
1. Siapkan agarose 1.5 % dan buffer elektroforesis (TBE atau TAE)
2. Tuang larutan agarose cair yang sudah mengandung etidium bromide dalam cetakan agarose.
3. Tunggu sampai kering.
4. Pindahkan agarose bersama cetakannya ke dalam bak elektroforesis yang sudah berisi larutan elektroforesis.
5. Masukkan 1 uL genom DNA hasil isolasi kita.
6. Jalankan elektroforesis (ingat DNA berjalan dari kutub negatif ke positif).
7. Setelah 30 menit, hasil elektroforesis dapat dilihat di lampu UV. (gambar 2)
Gambar 3. Elektroforesis Genom DNA
BAB III.
ISOLASI RNA
RNA (asam ribonukleat) adalah polimer yang ada dalam sel hidup dan banyak virus, yang terdiri dari rantai panjang tunggal untaian fosfat dan unit ribosa dengan basis nitrogen adenin, guanin, sitosin, dan urasil, yang terikat pada gula ribosa. RNA digunakan dalam semua langkah sintesis protein di semua sel hidup dan membawa informasi genetik untuk banyak virus.
Isolasi RNA dengan kualitas tinggi adalah langkah penting yang diperlukan untuk melakukan berbagai eksperimen biologi molekuler. TRIzol Reagen adalah reagen siap pakai yang digunakan untuk isolasi RNA dari sel dan jaringan. TRIzol bekerja dengan menjaga integritas RNA selama homogenisasi jaringan, sementara pada saat yang sama mengganggu dan memecah sel dan komponen sel. Penambahan kloroform, setelah sentrifugasi, memisahkan larutan menjadi fase berair dan organik. RNA hanya tersisa dalam fase air.
Setelah mentransfer fase berair, RNA dapat diperoleh kembali dengan presipitasi dengan isopropil alkohol. Tetapi DNA dan protein dapat pulih dengan pemisahan berurutan setelah penghilangan fase berair. Pengendapan dengan etanol membutuhkan DNA dari interfase, dan presipitasi tambahan dengan isopropil alkohol membutuhkan protein dari fase organik. Total RNA yang diekstraksi oleh TRIzol Reagent bebas dari kontaminasi protein dan DNA. RNA ini dapat digunakan dalam analisis Northern blot dan lain-lain.
Pada umumnya, prosedur isolasi RNA tradisional diutamakan terhadap agen penghambat kerja RNase (biasanya denaturants kuat seperti garam guanidine, sodium dodecylsulfate (SDS), atau senyawa berbasis fenol yang dirancang untuk menurunkan risiko degradasi RNA dalam sampel). Namun pada kenyataannya, sebelum dan sesudah isolasi, integritas RNA berada pada risiko tertinggi.
Teknik isolasi RNA atau total RNA hampir sama dengan teknik isolasi DNA, namun harus diperhatikan sifat fisik dari RNA yang labil dibanding DNA sehingga mudah mengalami kerusakan jika kerja kita tidak baik. Hal yang wajib diperhatikan adalah proses pengambilan dan pengantaran sampel, metode yang akan digunakan untuk isolasi RNA, dan proses penyimpanan hasil isolasi RNA.
Berikut ini, akan dijelaskan proses isolasi RNA dari berbagai sumber. Namun sebelumnya akan dijelaskan tahapan yang perlu diperhatikan:
1. Koleksi sampel dan perlindungan terhadap sampel
Harus diingat, RNA sama seperti DNA, dapat diperoleh dari jaringan atau sel yang berinti. Teknik isolasi untuk mendapatkan RNA tergantung dari jenis sampel yang kita gunakan. Menemukan metode koleksi dan perlindungan sampel RNA yang tepat akan memaksimalkan hasil dan kualitas RNA yang diisolasi. Pada umumnya, untuk memperoleh hasil RNA yang baik, sampel yang diisolasi harus segera dibekukan baik dengan nitrogen cair atau es kering. Bagaimana jika tidak mempunyai bejana nitrogen cair ataupun es kering. Itu kita harus memikirkan cara mengontrol kondisi agar hasil yang diperoleh tetap terjaga walaupun mungkin kualitasnya sedikit menurun. Hal yang paling baik adalah, sampel yang diperoleh langsung digunakan untuk diisolasi RNA-nya. Namun, jika kita tidak dapat segera melakukannya, maka siapkan es balok atau es batu dalam termos. Sampel dapat disimpan sementara dalam termos es selama maksimal 6 jam hingga kita memindahkan sampel tersebut dalam lemari pendingin beku. Untuk sampel RNA yang bersumber dari jaringan atau organ, dapat disimpan dalam formalin dingin (jika larutan tersebut tersedia).
2. Preparasi RNA
Banyak teknologi yang sudah diproduksi massal untuk mengisolasi RNA. Setiap metode ada kelebihan atau kekurangannya. Umumnya pada kemudahan dalam kerja isolasi RNA. Sejumlah
teknologi persiapan RNA tersedia secara luas yang dapat diklasifikasikan ke dalam empat teknik umum: metode ekstraksi organik, format keranjang spin, metode partikel magnetik, dan metode lisis langsung. Sementara semua dapat digunakan untuk menyiapkan RNA berkualitas tinggi yang cocok untuk berbagai macam teknik analisis, ada beberapa faktor yang perlu dipertimbangkan dalam memilih teknologi pemurnian yang tepat.
Metode Ekstraksi Organik
Metode ekstraksi organik dianggap sebagai standar emas untuk persiapan RNA. Selama proses ini, sampel dihomogenisasi dalam larutan yang mengandung fenol dan kemudian disentrifugasi. Selama sentrifugasi, sampel dipisahkan menjadi tiga fase, yaitu fase organik ada di area lebih rendah, fase tengah di area tengah yang mengandung protein terdenaturasi dan gDNA, serta fase berair di area atas yang mengandung RNA. Fase berair di atas dipindahkan dan RNA diendapkan oleh pengendapan alkohol dan rehidrasi. Kelebihan menggunakan metode ekstraksi organik adalah denaturasi nukleasi yang cepat dan menstabilkan RNA.
Metode Filter-sentrifuse
Dasar metode ini adalah filter atau penyaringan menggunakan membran berbahan serat kaca, silika atau penukar ion. Sampel dilisiskan dalam buffer yang mengandung inhibitor RNase (biasanya garam guanidin), dan asam nukleat terikat ke membran dengan melewatkan lisat melalui membran menggunakan gaya sentrifugal. Larutan pencucian kemudian dilewatkan melalui membran dan dibuang. Pelarut elusi yang tepat diterapkan dan sampel dikumpulkan ke dalam tabung dengan sentrifugasi. Beberapa teknik dapat dilakukan dengan sentrifugasi atau vakum sentrifugasi.
3. Jumlah RNA
Untuk mencapai keberhasilkan dalam kerja dengan RNA, maka perlu dilakukan identifikasi hasil isolasi RNA, yaitu dengan
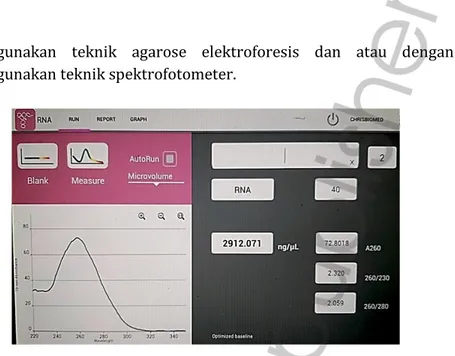
menggunakan teknik agarose elektroforesis dan atau dengan menggunakan teknik spektrofotometer.
Gambar 4. Pengukuran konsentrasi dan kemurnian RNA dengan UV Spektrofotometer nano drop.
(Koleksi pribadi)
Tampak konsentrasi 2912,071 ng/uL dengan kemurnian 2,05.
Prinsip dasar menggunakan agarose, serupa dengan identifikasi hasil isolasi DNA. Molekul RNA juga memiliki muatan negatif karena adanya gugus fosfat sebagai tulang punggungnya, dengan demikian proses pergerakan elektroforesisnya adalah dari kutub negatif ke kutub positif. Untuk mengetahui kemurnian dan konsentrasi RNA, digunakan spektrofotometer. Saat ini telah ada teknologi dalam mengukur kemurnian dan konsentrasi RNA menggunakan alat UV spektrofotometer nano drop. Jika kita tidak memiliki UV Spektrofotometer nano drop dapat menggunakan UV Spektro biasa. Absorbansi sampel RNA yang diencerkan diukur pada panjang gelombang 260 dan 280 nm. Konsentrasi RNA dihitung dengan menggunakan hukum Beer-Lambert, memprediksi perubahan linier dalam absorbansi dengan konsentrasi. Menggunakan persamaan ini, maka pembacaan pada panjang gelombang 260 nm dari 1.0 setara dengan ~ 40 ug/mL RNA untai tunggal. Rasio absorbansi pada 260 nm/280 nm digunakan untuk menilai kemurnian RNA. Nilai 1.8 – 2.1
menunjukkan RNA dengan kemurnian yang sangat baik. Jika kita mempunyai alat UV Spektrofotometer nano drop, kita hanya meneteskan 1 uL sampel larutan RNA hasil isolasi dan mesin akan langsung menghitung konsentrasi serta kemurniannya.
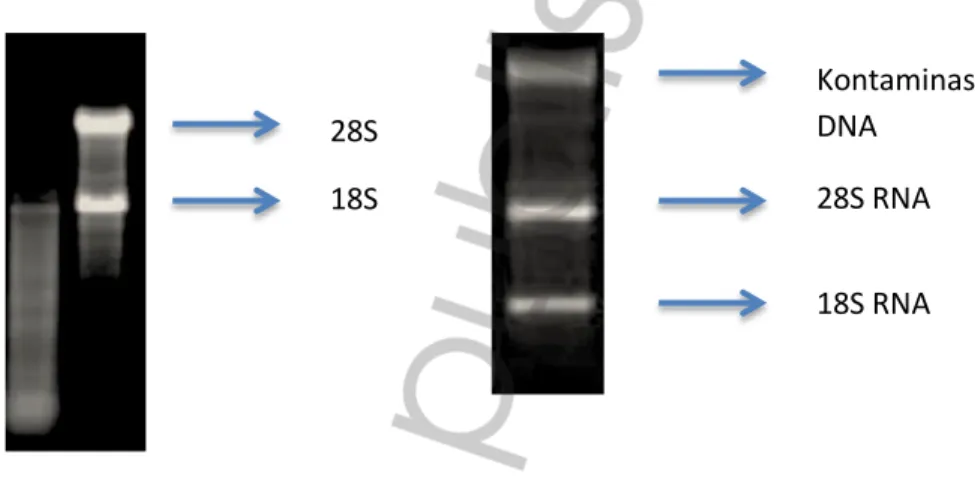
Gambar 5. Hasil elektroforesis RNA. Hasil isolasi yang baik, akan memberikan gambaran 2 pita, yang menunjukkan RNA ukuran 28 S dan
18S
(Dokumentasi pribadi)
Isolasi RNA menggunakan TRIzol merupakan teknik yang umum dilakukan saat ini. Selain menggunakan TRIzol dengan teknik konvesnional, ada juga teknik TRIzol yang dikombinasikan dengan tabung kolom seperti pada isolasi DNA. Namun, secara prinsip dasar kedua metode sama yaitu memisahkan RNA dari bahan biologis lainnya.
Isolasi RNA dari darah menggunakan TRIzol-kloroform
Bahan: - Darah
- 10x RBC lisis buffer steril (89.9 g NH4Cl, 10 g KHCO3, 2 mL 0.5
M EDTA dalam 1 L dengan pH 7.3), sebagai larutan stok. - TRIzol - PBS - Isopropanol 28S 18S Kontaminasi DNA 28S RNA 18S RNA
- Etanol
- RNAse-free water
Prosedur:
1. 1 mL darah ditambahkan dalam 9 mL 1x RBC lisis buffer. Bolak-balik pelan dan biarkan 10 menit pada suhu 40C.
2. Sentrifuse 1500 rpm selama 10 menit pada suhu ruang.
3. Buang supernatan dengan hati-hati, jangan sampai pellet terbawa.
4. Tambahkan 1 x RBC lisis pada pellet, lakukan pemipetan “up and down atau sedot sebul” hingga pellet larut dalam larutan RBC lisis tersebut dan pindahkan ke tabung sentrifuse 2 mL. 5. Sentrifuse 3000 rpm selama 5 menit pada suhu 40C
(sebaiknya).
6. Buang supernatan, kemudian tambahkan 1 mL PBS, lakukan pemipetan “sedot sebul”, kemudian ulangi lakukan sentrifuse seperti pada tahap sebelumnya.
7. Tambahkan 1200 uL TRIzol dalam tabung berisi pellet, lalu lakukan pemipetan “sedot sebul” sehingga pellet menjadi terlarut dalam TRIzol.
8. Tambahkan 200 uL kloroform, vortex 10 detik, kemudian sentrifuse pada kecepatan 13000 rpm selama 10 menit pada suhu 40C.
9. Dalam tabung akan ada 3 lapisan larutan, lapisan bening atas; lapisan tipis putih tengah dan lapisan merah muda bawah. RNA berada di lapisan bening atas, DNA berada di lapisan putih tengah (interface) dan protein berada di lapisan bawah, larutan yang berwarna merah muda.
10. Ambil lapisan bening bagian atas dengan hati-hati tanpa membawa lapisan putih dan merah muda, kemudian pindahkan ke tabung baru yang bersih-steril.
11. Tambahkan 500 uL larutan isopropanol dingin dan lakukan pemipeting “sedot-sebul” atau dikocok. Isopropanol akan mengendapkan RNA. Biarkan 10 menit.
12. Taruh sampel di -200C jika tidak digunakan.
13. Sentrifuse 13000 rpm selama 10 menit pada suhu 40C. Buang
supernatan.
14. Buang supernatan dengan hati-hati, jangan sampai pellet putih terbawa. Kemudian tambahkan 500 uL 75% etanol dingin. 15. Sentrifuse selama 10 menit pada kecepatan 13000 rpm dengan
temperatur 40C.
16. Buang supernatan, keringkan tabung jangan sampai ada etanol sisa.
17. Tambahkan 30 uL air yang bebas RNA dalam tabung, biarkan selama 2 jam agar larut.
18. RNA sudah diperoleh. Simpan pada suhu -200C.
Metode lain yang dapat digunakan adalah kombinasi TRIzol dan kit isolasi PCR. Kami menggunakan Direct-zol RNA Miniprep Plus dari Zymo Research. Metode kombinasi ini membantu mengurangi waktu kerja dan mudah dalam pelaksanaannya.
BAB IV.
PCR (POLYMERASE CHAIN REACTION)
Hibridiasasi atau aneling (penempelan) merupakan dasar dari kemampuan untai tunggal asam nukleat untuk melakukan pengikatnya spesifik dengan untai komplementernya (pasangannya). Ketika untai ganda DNA mengalami denaturasi atau dipanaskan, maka untai akan terpisah, jika pemanasan dihilangkan atau dilakukan penurunan temperatur, maka untai terpisah akan bergabung kembali. Prinsip dasar ini yang menjadi dasar pada proses PCR, di mana saat terjadi penurunan suhu pada proses PCR, akan terjadi proses penempelan untai oligonukleotida. Proses penempelan ini dipengaruhi oleh faktor yang mempengaruhi proses pembentukan ikatan hidrogen antara basa nukelotida pasangannya, yaitu temperatur, pH, larutan garam dan sebagainya.
Telah diketahui bahwa basa nukleotida G akan berpasangan dengan basa nukleotida C dengan bantuan tiga (3) jembatan hidrogen, sedangkan basa nukleotida A akan berpasangan dengan T melalui dua jembatan hidrogen. Untuk memutuskan jembatan yang menghubungkan basa-basa tersebut diperlukan energi (untuk laboratorium dalam hal ini adalah pemanasan yang tinggi).
PCR merupakan suatu metode in vitro dalam sintesis DNA. Prinsip dasar metode ini adalah perbanyakan fragmen DNA menggunakan enzim polymerase pada temperatur yang tinggi yang dilakukan secara berulang. Pada proses PCR dibutuhkan oligonukleotida pendek (primer DNA) yang berperan dalam mengawali proses ini. Primer akan menempel atau hybrid pada untai tunggal DNA saat temperatur diturunkan setelah terjadi pemisahan untai ganda DNA. Produk hasil PCR dapat diamati menggunakan teknik eletroforesis agarose.
PCR memiliki beragam aplikasi, tidak hanya dalam penelitian dasar tetapi juga dalam bidang diagnosa medis, forensik, dan pertanian. Seperti dijelaskan di halaman ini, beberapa contoh aplikasi PCR meliputi: 1. Ekspresi gen 2. Deteksi genotipe 3. Kloning 4. Mutagenesis 5. Analisis metilasi 6. Sekuensing
7. Kesehatan, forensik dan aplikasi lainnya
Hingga saat ini, metode PCR telah berkembang dari metode PCR yang umum hingga metode PCR yang dapat digunakan langsung untuk melihat apakah sampel tersebut memiliki mutasi atau tidak, tergantung aplikasi apa yang akan dipakai apakah untuk identifikasi molekuler, sekuensing atau rekayasa genetika. Beberapa PCR yang telah dikembangkan saat ini:
1. PCR standar, metode dasar PCR dan hasil produk PCR dapat digunakan untuk tahap selanjutnya seperti kloning, sekuensing, restriksi enzim.
2. ARMS PCR, atau Amplification Refractory Mutation System (ARMS) PCR adalah aplikasi PCR di mana menggunakan primer spesifik. Metode ini yang sangat berguna untuk identifikasi mutasi titik atau polimorfisme.
3. GAP-PCR, mutasi delesi pada gen cluster seperti -globin, dapat dideteksi oleh PCR standar menggunakan sepasang primer yang komplemen dengan untai DNA yang di dalamnya ada area delesi. Untuk delesi yang kecil, kurang dari 1 kb, maka akan dihasilkan dua buah produk fragmen, satu fragmen besar dan satu fragmen kecil (fragmen yang ada delesi kurang dari 1 kb). Untuk delesi yang lebih besar (2 kb), jarak antara kedua primer yang mengapit produk PCR (amplikon) terlalu besar, sehingga sukar didapatkan dua fragmen produk (hanya yang terbentuk fragmen yang delesi/fragmen kecil, sedang framen
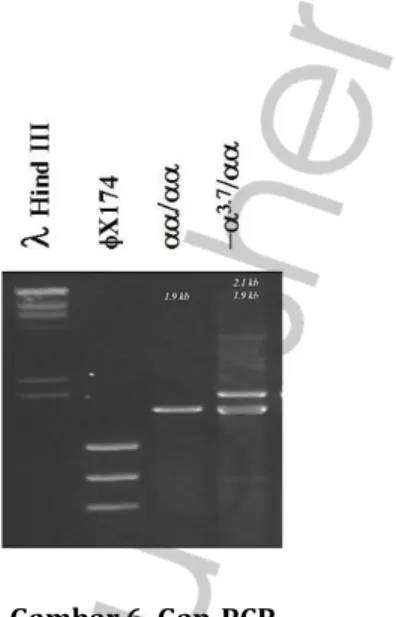
normal tidak terbentuk), untuk itu diperlukan teknik khusus untuk memperoleh kedua fragmen tersebut. Oleh karena itu, teknik GAP-PCR digunakan. Teknik ini banyak digunakan untuk deteksi alfa talasemia, di mana delesi yang terjadi sangat besar (gambar 3).
4. RFLP PCR, metode ini digunakan jika ada area pemotongan enzim restriksi. Pada alel homozigot/mutan yang memiliki situs pemotongan, maka produk PCR akan dapat dipotong oleh enzim restriksi tertentu (atau sebaliknya). Untuk alel hetero, maka produk PCR yang diamati adalah kombinasi antara pita yang terpotong dan yang tidak terpotong (gambar 4).
5. Kuantitatif-PCR, metode ini digunakan untuk menghitung kuantitas atau jumlah produk spesifik hasil PCR, biasanya dikenal dengan sebutan real-time PCR (“RT-PCR”). RT PCR di sini, berbeda dengan Reverse Transcription (RT) PCR. Pada
Reverse Transcription PCR (RT-PCR), pcr didisain untuk
amplifikasi DNA dari RNA. Hasil jumlah amplifikasi DNA dari RNA dapat diamati dengan menggunakan “RT-PCR”.
6. Multipleks tandem PCR, metode untuk mendeteksi banyak target pada satu sampel. Pada metode ini, satu sampel akan menggunakan banyak primer spesifik dan proses PCR dijalan serentak. Karena menggunakan banyak primer, maka akan dihasilkan banyak amplikon (produk PCR) dengan ukuran yang beragam (gambar 5).
Masih banyak lagi jenis PCR, namun kami hanya menjelaskan PCR yang sering digunakan untuk identifikasi penyakit. Semua metode ini dikembangkan untuk memudahkan peneliti atau pekerja laboratorium dalam mengidentifikasi sampel DNA seperti bidang forensik, mikrobiologi dan sebagainya.
Gambar 6. Gap-PCR
(http://www.ithanet.eu/ithapedia/index.php/Protocol:Gap-PCR)
Gambar 7. ARMS PCR
(koleksi pribadi CYP1A1*2A)
Aplikasi PCR atau teknologi amplifikasi telah banyak digunakan, biasanya di klinik/rumah sakit hingga membuat area spesifik dari DNA dan diperbanyak untuk digunakan dalam kloning teknologi. Di bawah ini merupakan ringkasan kelebihan dan kekurangan teknologi amplifikasi:
Kelebihan teknologi amplifikasi:
1. Hasil cepat diperoleh (namun tergantung metode ataupun kit PCR yang digunakan dan dikembangkan oleh pihak developer).
2. Lebih sensitif dan spesifik dibandingkan metode deteksi konvensional.
3. Reprodusibilitinya sangat baik (kembali tergantung jenis kit PCR dari berbagai developer).
4. Hasil amplifikasi dapat dihitung secara kuantitatif atau semi kuantitatif, biasanya untuk teknik “RT-PCR”.
5. Mudah dikembangkan teknologi terbaru untuk dalam pemeriksaan penanda kelainan genetik.
Kekurangan teknologi amplifikasi:
1. Memerlukan ruangan dengan alur proses kerja satu arah (untuk mencegah kontaminasi).
2. Mudah terkontaminasi dalam melakukan pekerjaannya.
3. Target sekuensing DNA sasaran harus diketahui terlebih dahulu, dan harus melakukan perancangan primer serta kondisi alat yang sesuai dengan tujuan target yang diinginkan. 4. Biaya alat dan bahan untuk metode “RT PCR” yang mahal.
Prinsip dasar prosedur kerja PCR semua adalah sama, yaitu harus ada primer, dNTP, Taq polimerase enzim, dH2O, buffer PCR dengan atau tanpa MgCl, dan DNA template (sampel DNA genom). Setiap teknik, pada dasarnya yang berbeda adalah:
1. Prosedur temperatur hibridisasi (penempelan) dan elongasi (pemanjangan) produk PCR serta siklus dan waktu.
2. Desain primer (metode ARMS akan berbeda dengan metode PCR standar).
3. Ada tidaknya area restriksi.
4. Perlu tidaknya menggunakan probe (primer yang berpendar).
Pemanfaatan PCR metode konvensional 1. PCR -globin
a. Bahan:
Primer:
Forward primer F5’-TAGCAATTTGTACTGATG GTATGG-3’ dan
10X PCR buffer
Tag Polimerase
dNTP
DNA template b. Cara kerja
Siapkan semua bahan dalam tabung PCR
Untuk program PCR yang digunakan: 1. Denaturasi awal 950C selama 3 menit.
2. Denaturasi PCR 980C selama 20 detik.
3. Hibridisasi PCR 600C selama 15 detik.
4. Elongasi PCR 720C selama 60 detik.
5. Ulangi no 2 – 4 untuk 35 siklus. 6. Elongasi akhir 720C selama 60 detik.
7. Pendinginan 240C.
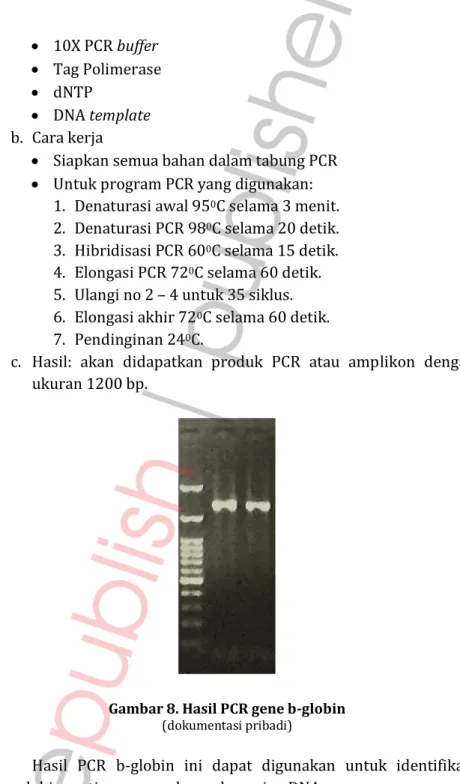
c. Hasil: akan didapatkan produk PCR atau amplikon dengan ukuran 1200 bp.
Gambar 8. Hasil PCR gene b-globin
(dokumentasi pribadi)
Hasil PCR b-globin ini dapat digunakan untuk identifikasi hemoglobinopati menggunakan sekuensing DNA.
2. PCR ARMS gen ALAD a. Bahan ALAD F 5’-GCCTCAGTCTTCCCTCCTATTTAGT-3’ ALAD R 5’-TCCCTTCTTAGCCCTTCCTTTGATT-3’ ALAD C 5’-TTCTGTCCTGGGGGCTTGAG-3’ ALAD N 5’-TCAGCATCTCTTCCAGCCGC-3’ ALAD M 5’-TCAGCATCTCTTCCAGCCGG-3’
Bahan lain sama dengan PCR standar b. Cara kerja
PCR tahap 1:
1. Campur semua bahan kecuali primer CNM. 2. Denaturasi awal 940C selama 3 menit.
3. Denaturasi PCR 940C selama 30 detik.
4. Hibridisasi PCR 600C selama 30 detik.
5. Elongasi PCR 720C selama 30 detik.
6. Ulangi no 2 – 4 untuk 40 siklus. 7. Elongasi akhir 720C selama 60 detik.
8. Pendinginan 240C.
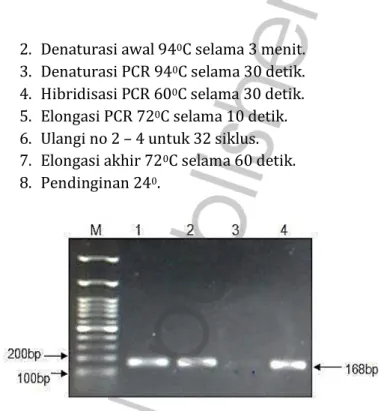
9. Produk PCR adalah 306 bp.
Gambar 9. Hasil PCR Tahap 1
(Dokumentasi pribadi) PCR tahap 2
1. Campur semua bahan ditambah primer CN untuk pengecekan sampel normal dan CM untuk sampel mutan.
2. Denaturasi awal 940C selama 3 menit.
3. Denaturasi PCR 940C selama 30 detik.
4. Hibridisasi PCR 600C selama 30 detik.
5. Elongasi PCR 720C selama 10 detik.
6. Ulangi no 2 – 4 untuk 32 siklus. 7. Elongasi akhir 720C selama 60 detik.
8. Pendinginan 240.
Gambar 10. Hasil PCR Tahap 2. Lajur 1 dan 3 (sampel PCR A dan B) menggunakan primer mutan, sedangkan lajur 2 dan 4 (sampel PCR A
dan B) menggunakan primer normal. Hasil menunjukkan sampel A genotipe GG (ALAD – 1) dan sampel B genotipe GC (ALAD – ½)
(Dokumentasi pribadi)
3. PCR RFLP CYP1A1*2A
a. Bahan
Primer A 5'-TAGGA GTCTT GTCTC ATGCC T-3'
Primer B 5'-CAGTG AAGAG GTGTA GCCGC T-3'
Enzim restriksi Msp I.
Bahan lain sama dengan bahan untuk PCR standar. b. Cara kerja
Campur semua bahan kecuali enzim restriksi MspI, menjadi satu.
Ambil 24 uL larutan campuran, masukkan ke dalam tabung PCR, tambahkan 1 uL sampel DNA. Kemudian dimasukkan ke dalam alat PCR.
Program PCR untuk CYP1A1*2A restriksi MspI, adalah 1. Denaturasi awal, 950C 3 menit.
2. Denaturasi PCR, 950C 3 menit.
3. Hibridisasi PCR, 600C 15 detik.
4. Elongasi PCR, 720C 2 detik.
5. Ulangi no 2 – 4 sebanyak 35 siklus. 6. Elongasi akhir 720C 5 detik.
Periksa hasil PCR menggunakan agarose elektroforesis. Jika didapatkan hasil produk PCR 340 bp, proses PCR telah sukses.
Ambil 5 uL produk PCR di atas, masukkan dalam tabung PCR yang telah ada larutan campuran MspI dan buffernya (masing-masing tergantung dari Kit perusahaan yang menjual).
Inkubasi pada mesin PCR untuk suhu 370C selama 4 – 24
jam.
Hasil pemotongan diamati menggunakan 1.5 % agarosa elektroforesis.
A: Band with 340 bp B: DNA Marker size 100 bp C,D, F: Band with 340, 200 and 140 bp
E: Band with 140 and 200 bp
4. Multiplex PCR deteksi 0-talassemia
Pada metode ini, kita dapat mendeteksi adanya mutasi menggunakan lebih dari 1 primer dalam waktu yang bersamaan. Primer-primer yang digunakan hanya mengenali atau berkomplementasi dengan target. Metode ini dikenal juga sebagai MARMS atau multiplex amplification refractory mutation system.
a. Bahan
- Kodon 17-M: 5’-CTC ACC ACC AAC TTC AGC CAC GTT CAG CTA-3’
- Kodon 41/42-M: 5’-GAG TGA CAG ATG CCC AAA GGA CTC AAC CT-3’
- Kodon 71/72-M: 5’-AGG TTG TCC AGG TGA GCC AGG CCA TCA ATT-3’
- IVS1nt1-M: 5’-TTA AAC CTG TCT TGT AAC CTT GAT ACC GAA-3’
- S-primer: 5’-ACC TCA CCC TGT GGA GGC AC-3’ - P1-primer: 5’-GCG ATC TGG GCT CTG TGT TCT-3’ - P2-primer: 5’-GTT CCC TGA GCC CCG ACA CG-3’ b. Prosedur Material uL 10x PCR buffer X 2.5 50 mM MgCl2 X 0.9 10 mM dNTPs X 0.75 5 uM S primer X 3.0 5 uM P1 X 0.5 5 uM P2 X 0.5 5 uM mutan primer @ 0.5 uL X 2
5U/uL HotStart Taq DNA X 0.13
dH2O X 7.72
DNA Sampel 7
c. Program
Nama Suhu (0C) Waktu Siklus
Denaturasi awal 95 00:15:00 1 x Denaturasi 94 00:01:00 Annealing 65 00:01:00 35 x Extensi 72 00:02:00 Extensi final 72 00:07:00 1 x Pendinginan 24 99:99 1 x
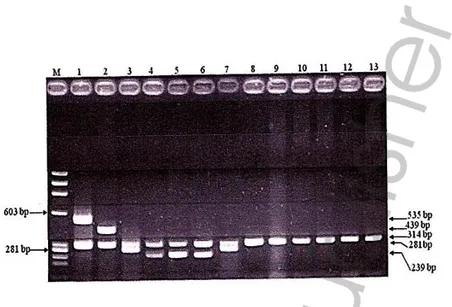
Gambar 12. Hasil PCR MARMS b-globin deteksi b-thalassemia. M: DNA Ladders/Markers; 1: kontrol b-thal CD71/72, 535 bp; 2: kontrol b-thal CD41/42, 439 bp; 3: kontrol IVS1nt1, 281 bp; 4: kontrol b-thal CD17, 239 bp; 8: kontrol internal kontrol fragmen, 314; 5-7 dan 9-13 sampel
pasien
BAB V.
REAL-TIME PCR/
KUALITATIF PCR (RT-PCR/QPCR)
Telah dijelaskan sebelumnya secara singkat bahwa PCR adalah teknik perbanyakan area DNA pada suatu gen, yang menjadi sasaran oleh peneliti. Secara teoritis, PCR akan meningkatkan jumlah DNA secara eksponensial atau dengan kata lain melipat gandakan jumlah molekul target pada setiap amplifikasi siklus. Pada teknik PCR, pada umumnya hanya untuk mengetahui bahwa DNA dari yang dituju telah berhasil diamplifikasi. Sedangkan real-time PCR (RT-PCR) atau qPCR bertujuan secara kuantitatif atau mengetahui jumlah DNA sasaran. Harap dibedakan pengertian antara RT-PCR di sini dengan RT-PCR yang reverse transcription PCR (RT-PCR) yang berfungsi dalam mengubah RNA menjadi cDNA kemudian dilakukan qPCR.
Jika diaplikasikan dalam bidang kesehatan, teknik PCR ditujukan untuk mengetahui ada tidaknya gen bakteri/virus dalam tubuh pasien, dengan kata lain, jika ditemukan gen bakteri pada darah pasien, maka pasien tersebut terinfeksi oleh bakteri tersebut. Sedangkan qPCR bertujuan apakah dengan pengobatan akan menurunkan atau tidak jumlah gen bakteri, dengan kata lain jika suatu obat berhasil mengobati, maka jumlah gen mikroorganisme (bakteri) akan turun.
Dalam RT-PCR, jumlah DNA diukur dalam setiap siklus melalui pewarna berfluoresen (berpendar) yang akan meningkatkan sinyal sejalan dengan bertambahnya jumlah produk amplikon. Senyawa berfluoresen ini akan bergabung (hibridisasi) dalam untai DNA selama proses PCR. Data sinyal akan dikumpulkan dalam fase eksponensial dan diproses melalui perangkat lunak (software) dari alat PCR serta diterjemahkan dalam plot amplifikasi yang mewakili akumulasi produk selama durasi proses PCR.
Perbedaan yang mendasar dalam deteksi hasil PCR dan qPCR dapat dilihat pada gambar di bawah ini.
Gambar 13. PCR konvensional dan RT-PCR (qPCR)
(Vilborg Matre, 2020)
Pada gambar di atas tampak bahwa pada siklus ke 45 dari PCR konvensional, pita agarose akan menunjukkan hasil yang tajam dibandingkan pada siklus sebelumnya. Kotak hijau merupakan area amplifikasi PCR konvensional jika dipindahkan pada grafik fluoresensi di RT-PCR. Ini berarti, gambaran pita siklus 45 pada elektroforesis PCR konvensional merupakan produk akhir dari amplifikasi (gambar pita tampak jelas dibandingkan siklus lain). Sedangkan pada teknik RT-PCR, proses amplifikasi langsung terdeteksi oleh sistem di awal peningkatan grafik (kotak merah). Jika pada PCR, hasil amplifikasi tampak jelas pada siklus 45, di mana pada siklus ini dihasilkan jumlah fragmen DNA sasaran (amplikon) sejumlah 6.9 x1019, maka pada
RT-PCR, hasil amplikon dapat terbaca oleh sensor alat berjumlah 9x106.
Hal ini menunjukkan RT-PCR lebih sensitif dan cepat dalam hasil
Akhir produk amplifikasi
amplifikasi DNA gen sasaran dibandingkan PCR konvensional. Atau dengan kata lain, RT-PCR akan memonitor secara simultan proses amplifikasi DNA target selama PCR berlangsung, sehingga akan lebih cepat dalam memberikan hasil dibandingkan PCR konvensional. Selain cepat, RT-PCR mempunyai kelebihan dalam konsentrasi jumlah DNA sasaran, artinya jika konsentrasi sampel DNA target sangat banyak (misalnya 100 ng/uL) maka siklus awal sensor membaca produk amplifikasi akan lebih cepat atau pendek dibandingkan konsentrasi sampel DNA target yang sangat sedikit (misalnya 1 ng/uL). Oleh karena itu, metode RT-PCR dibidang medis digunakan untuk mengetahui apakah dalam tubuh suatu obat berhasil digunakan untuk membunuh bakteri/mikroba, di mana jika awal saat penderita sakit, jumlah mikrobanya banyak yang berarti jumlah DNA sasaran sangat tinggi (siklus PCR akan lebih cepat, maka saat pengobatan sukses, jumlah mikroba (dalam hal ini DNA sasaran) turun, akan digambarkan dengan siklus amplifikasi yang makin lama.
Gambar 14. Jumlah Siklus, Konsentrasi Sampel
(Life Technology, 2010)
Pada gambar di atas, warna merah mewakili sampel dengan konsentrasi 108 ng/uL, biru mewakili 107 ng/uL hingga warna kuning
yang mencukupi jumlahnya, terbaca oleh alat pada siklus 14, begitu juga pada konsentrasi yang lain hingga NTC (atau air), maka pola kemunculan grafik fluorosensi makin lama (siklus makin tinggi). Pola grafik seperti ini dapat digunakan dalam bidang medis untuk mengetahui apakah suatu obat dapat menurunkan atau memusnahkan mikroba, di mana jika jumlah mikroba masih tinggi, siklus amplifikasi masih terdeteksi.
Dengan demikian, kita dapat melihat keuntungan RT-PCR dibandingkan PCR konvensional sebagai berikut:
Dengan RT-PCR, kita dapat memantau kemajuan reaksi PCR dari waktu ke waktu.
Dengan RT-PCR, kita dapat mengukur jumlah produk PCR (amplikon) dengan tepat pada setiap siklus sehingga memungkinkan kuantifikasi jumlah yang akurat dari bahan sampel.
Proses amplifikasi dan deteksi terjadi dalam satu tube, sehingga dapat menghilangkan manipulasi pasca-PCR (manipulasi foto hasil elektroforesis PCR untuk menghasilkan pita yang jelas).
RT-PCR selain dapat digunakan untuk mengetahui jumlah amplifikasi, dapat juga digunakan untuk mengetahui adanya mutasi dalam urutan DNA target suatu gen. Salah satu yang telah digunakan dan dikembangkan adalah teknik hibrid-probe untuk mengetahui jenis mutasi yang sering ditemukan pada penderita talasemia. Pada teknik ini, digunakan probe yang akan menempel di salah satu nukleotida DNA target dan hasilnya dianalisis berdasarkan kurva melting (kurva leleh).
Analisis kurva leleh adalah suatu metode di mana saat suhu DNA dinaikkan, untai DNA akan berdisosiasi yang akhirnya akan meningkatkan intensitas absorbansi cahaya fluorosensi menuju kondisi hiperkromisitas absorbansi (atau dengan kata lain penyerapan sinar flourosen meningkat ketika dua untai DNA terpisahkan). Kurva leleh atau kurva melting merupakan kondisi di mana suhu meningkat dan 50% DNA mengalami denaturasi sehingga
akan berdisosiasi (berpisah). Adanya kurva melting ini, dapat digunakan dalam mengetahui ada tidaknya mutasi atau perubahan tunggal pada basa nukleotida DNA. Misalnya pada dua sampel, 30 urutan DNA target sampel A memiliki basa no 25 adalah A, namun sedangkan sampel B adalah C, maka jika ditotal jumlah energi yang dibutuhkan untuk memisahkan dua untai DNA pada sampel A dan B dalam kondisi kurva melting, akan berbeda. Perbedaan ini menjadi penanda adanya perubahan basa nukleotida pada 2 sampel yang berbeda namun untuk target gen yang sama.
Gambar 15. Kurva melting pada 3 sampel dengan DNA gen target yang sama
Pada gambar di atas memperlihatkan 4 warna yang berbeda. Setiap warna mewakili mutasi dan no sampel. Jika warna merah muda (pink) adalah sampel no 1, maka kuning adalah sampel nomor 2, ungu nomor 3 dan biru nomor 4 (atau air). Keempat sampel direaksikan dengan bahan pereaksi yang sama, namun menghasilkan gambaran puncak grafik yang berbeda. Jika kita perumpamakan jumlah target DNA adalah 200 pasang basa (bp) dengan posisi mutasi A menjadi G pada basa ke 70, maka warna merah muda dan biru mewakili salah satu basa nukleotida tersebut (A atau G). Jika kita perumpamakan warna merah muda adalah sampel 1 yang memiliki basa A pada posisi
70, maka ungu adalah sampel 2 yang memiliki basa G pada posisi 70. Perubahan basa akan menyebabkan perubahan pada temperatur melting untuk masing-masing sampel. Sehingga akan mengubah kurva melting. Sampel 1 dapat dinyatakan homozigot untuk basa A (harap ingat kembali pengertian konsep pasangan alel). Begitu juga dengan sampel 2 yang dinyatakan homozigot untuk G. Sedangkan untuk garis grafik kuning atau sampel 3, tampak ada dua puncak grafik yang berada diposisi sampel 1 dan 2, maka sampel 3 dinyatakan sebagai heterozigot karena mempunyai kedua basa di pasangan alel-nya.
RT-PCR atau qPCR juga dapat digunakan untuk pemeriksaan SNP genotipe. SNP genotipe adalah pengukuran variasi genetik polimorfisme nukleotida tunggal (SNPs) antara anggota suatu spesies. Ini adalah bentuk genotipe, yang merupakan pengukuran variasi genetik yang lebih umum. SNP adalah salah satu jenis variasi genetik yang paling umum. SNP adalah mutasi pasangan basa tunggal pada lokus tertentu, biasanya terdiri dari dua alel (di mana frekuensi alel yang jarang adalah> 1%). SNP ditemukan terlibat dalam etiologi banyak penyakit manusia dan menjadi minat khusus dalam farmakogenetika. Karena SNP dilestarikan selama evolusi, mereka telah diusulkan sebagai penanda untuk digunakan dalam analisis sifat kuantitatif (QTL) dan dalam studi asosiasi di tempat mikrosatelit. Penggunaan SNP sedang diperluas dalam proyek HapMap, yang bertujuan untuk menyediakan set minimal SNP yang diperlukan untuk genotipe genom manusia. SNP juga dapat memberikan sidik jari genetik untuk digunakan dalam pengujian identitas. Meningkatnya minat terhadap SNPs telah tercermin oleh perkembangan besar-besaran beragam metode genotipe SNP. Prinsip teknik SNP genotipe merupakan kombinasi teknik RFLP dan hampir sama dengan hibidrisasi probe, hanya analisisnya tidak menggunakan kurva melting namun “end point genotyping”. Berbedaan dengan RFLP adalah menggunakan enzim restriksi pada yang dapat memotong area SNP dan RFLP menggunakan teknik PCR konvensional kombinasi dengan elektroforesis agarose (telah dijelaskan di pembahasan PCR sebelumnya). Persamaannya adalah dianalisis pada akhir proses PCR.
Gambar 16. Hasil SNP genotipe menggunakan LC 480-Roche dan analisis “end point genotyping”, warna biru mewakili homo G, hijau adalah homo
A dan merah adalah hetero GA. Abu-abu adalah negatif sedangkan merah muda adalah alel yang tidak diketahui.
(Koleksi pribadi)
Gambar 17. Hasil RFLP pada target gen yang sama dengan gambar sebelumnya
Dijelaskan sebelumnya bahwa identifikasi SNP dapat dilakukan dengan real time PCR. Pemanfaatan teknologi ini akan memudahkan peneliti dalam hal waktu dalam bekerja dan analisis. Berbedaan yang paling mendasar dalam SNP-RFLP dan SNP-RT PCR adalah biaya. Jika dalam RFLP, enzim yang dipakai dapat digunakan untuk identifikasi target gen lain selama enzim tersebut mengenali area pemotongan. Sedangkan untuk SNP-RT PCR, karena menggu-nakan probe khusus dan spesifik, maka probe tersebut hanya menge-nai fragmen target pada daerah sarannya dan probe SNP tidak dapat digunakan untuk fragmen target yang lain. Dengan demikian metode SNP RFLP menjadi lebih murah. Namun dalam hal waktu pengerjaan, SNP-RT PCR lebih cepat karena dalam 1 “plate tube PCR” yang berisi 96 sumur, kita dapat melakukan uji SNP untuk 96 sampel secara ber-samaan. Dan dalam waktu kurang lebih 2 jam, kita dapat memperoleh hasil serta analisis hasil secara bersamaan. Sedangkan untuk SNP RFLP, memerlukan waktu lebih lama untuk memeriksa 96 sampel (kurang lebih 1-2 hari). Berikut ini, beberapa contoh percobaan yang dapat digunakan untuk uji menggunakan RT PCR (qPCR).
1. Identifikasi makanan mengandung produk non halal (babi)
Metode ini kami kembangkan dari percobaan yang telah dilakukan oleh Tanabe (2007).
Bahan:
- Sampel DNA dari produk makanan (seperti bakso), lihat prosedur isolasi genom dari makanan siap saji.
- Primer sapi : Forward 5’- CCCGATTCTTCGCTTTCCAT -3’ Reverse 5’- CTACGTCTGAGGAAATTCCTGTTG -3’ - Primer babi : Forward 5’ - CTTGCAAATCCTAACAGGCCTG -3’
Reverse 5’ CGTTTGCATGTAGATAGCGAATAAC -3’ (Tanabe, 2007)
- Kit Sybrgreen (harap membaca manual dari masing-masing manufaktori produsen kit) untuk LC480 (LightCycler@480 SBYR Green).
Prosedur:
- Encerkan masing-masing primer hingga konsentrasi 10 uM - DNA sampel diukur konsentrasinya dengan spektrofotometer.
Jika konsentrasi di atas 60 ng/uL dan kemurnian 1.8 – 2, lakukan pengenceran 1:9. Jika konsentrasi kurang dari 60 ng/uL dan kemurnian 1.8 – 2, lakukan pengenceran 2:8. Pada dasarnya, pengenceran dilakukan berdasarkan nilai kemurniannya. Jika makin rendah kemurniannya, maka jumlah pelarut untuk pengenceran lebih banyak dari jumlah sampel DNA. Sebaiknya, lakukan pengenceran dengan menggunakan pelarut elusi DNA.
- Buat larutan “mix PCR” yang terdiri dari:
- Masukkan 22 uL “mix PCR” ke dalam masing-masing sumur di “well plate”, kemudian tambahkan 3 uL sampel DNA yang telah diencerkan.
- Tutup “plate well” dengan plastik penutupnya. - Masukkan ke dalam alat real time PCR
- Lakukan pengaktifan alat real time PCR sesuai masing-masing prosedur alat PCR-nya.
- Untuk LC480, telah melakukan optimasi sebelumnya dengan tahapan:
o Lakukan optimasi suhu annealing menggunakan teknik PCR konvensional.
o Amati hasil PCR konvensional menggunakan teknik agarose elektroforesis dan lihat pada lampu UV.
o Hasil optimasi suhu yang tepat, akan digunakan untuk real
time PCR identifikasi kandungan B2 (babi) pada bakso.
o Sebelum melakukan identifikasi kandungan B2 pada bakso, lakukan pembuatan kontrol positif bakso yang menggandung B2. Hal ini bertujuan untuk menguji sensitivitas metode dalam melacak adanya B2 dalam bakso. o Program real time PCR yang digunakan sebagai berikut
Target (0C) Waktu Siklus Denaturasi awal 95 00:10:00 1 x PCR 95 60 72 00:00:10 00:00:20 00:00:30 30 x Kurva Melting 95 60 97 00:00:05 00:01:00 1 x Pendinginan 40 00:00:10 1 x Hasil percobaan:
Mencari suhu optimun untuk “annealing”
Gambar 18. 1= 590C; 2= 59.50C; 3= 600C; 4= 60.50C; 5= 610C; 6= 61.50C;
7= 62.50C; 8= DNA Ladder 100 bp, besar amplikon sekitar 100 – 150 bp
Gambar 19. Garis hijau adalah NTC atau kontrol negatif, garis sigmoid merah adalah sampel DNA bakso sapi menggunakan primer DNA sapi
(Dokumentasi pribadi)
Pada gambar di atas, kami ingin mengidentifikasi apakah primer DNA sapi yang kami kutip dari Tanabe akan menempel pada target dan menghasilkan amplikon produk PCR dengan sampel bakso sapi. Munculnya kurva garis sigmoid menunjukkan terjadi amplifikasi bakso sapi dengan primer sapi.
Gambar 20. Garis grafik sigmoid warna merah adalah primer babi dengan sampel beberapa bakso babi, dengan konsentrasi terendah 6.67
ng/uL. Garis warna hijau adalah NTC atau dH2O
(Dokumentasi pribadi).
Pada gambar di atas memperlihatkan bahwa primer babi dapat mendeteksi bakso yang mengandung DNA babi dengan konsentrasi DNA 6.67 ng/uL. Ini menunjukkan jika sampel mengandung produk non halal (babi), maka akan dapat terdeteksi menggunakan metode ini. Selanjutnya kita dapat gunakan primer tersebut untuk menguji produk bakso, apakah mengandung babi atau tidak.
Gambar 21. Primer babi digunakan untuk identifikasi produk bakso yang ada babinya. Garis kurva sigmoid adalah kontrol bakso babi,
sedangkan garis hijau adalah bakso yang diuji
(Dokumentasi pribadi).
Dengan melihat gambar di atas, maka kita dapat melakukan percobaan untuk identifikasi makanan yang mengandung babi. Selain melihat kurva kenaikan amplifikasi, ada cara lain untuk melihat apakah makanan dalam hal ini bakso mengandung babi, dengan melihat kurva leleh atau kurva melting.
Gambar 22. Kurva leleh atau kurva melting. Grafik garis biru adalah menggunakan primer sapi untuk bakso sapi, sedangkan garis merah adalah menggunakan primer babi untuk bakso babi. Grafik garis hijau adalah primer sapi dengan bakso babi, sedangkan garis abu-abu adalah
primer babi dengan bakso sapi
(Dokumentasi pribadi).
2. Identifikasi SNP RS9642880 menggunakan prosedur dan pereaksi Tagman
Real time PCR juga dapat digunakan untuk identifikasi adanya
satu mutasi pada suatu fragmen target gen, yang biasanya disebut dengan SNP (senip). Banyak mesin real time PCR yang dapat digunakan untuk mendeteksi SNP dengan menggunakan sistem pereaksi terbuka (open system) atau bahan pereaksi uji tidak berasal dari satu pabrik dari mesin, sebagai contoh LC 480 dari Roche dapat digunakan untuk identifikasi SNP menggunakan bahan pereaksi uji bukan dari 1 perusahaan yang sama. Di sini kami akan memberikan langkah prosedur dalam identifikasi SNP RS9642880 menggunakan pereaksi Taqman dan mesin LC 480 dari Roche.