Penerapan Algoritme Viterbi pada Hidden Markov Model (HMM)
untuk Prediksi Struktur Sekunder Protein
Dian Puspita Sari
1), Toto Haryanto
1)*1)
Laboratorium Komputasi Terapan, Departemen Ilmu Komputer FMIPA IPB
Jl. Meranti Wing 20 Level 5 Kampus IPB Dramaga, 16680
email : [email protected], [email protected]
ABSTRACT
This research aims to predict protein secondary structure using Hidden Markov Model (HMM). Viterbi Algorithm is conducted in HMM. A total of 780 data, will be conducted with 600 training data and 180 testing data. The data obtained from Protein Data Bank (PDB) with assigned by DSSP for protein secondary structure label. The protein secondary structure data consist of 394052 residues with 152782(39%) alpha-helix (H), 82355(21%) betha-sheets (B), and 158915(40%) coil (C). Imbalance data are observed for getting its influence in classification. Oversampling technique was used for handling imbalance data. However, global accuracy (Q3) score after oversampling show that result of predictions not significantly increase.
Key words
HMM, oversampling, protein secondary structure prediction, Viterbi Algorithm
1. Pendahuluan
Protein merupakan salah satu biomakromolekul yang mempunyai peran penting dalam makhluk hidup. Secara hierarki protein terbagi menjadi tiga tingkat yaitu, struktur primer, struktur sekunder, dan struktur tersier. Struktur primer adalah urutan asam amino yang membentuk rantai polipeptida. Struktur sekunder adalah sejumlah rangkaian asam amino yang membentuk struktur tiga dimensi alpha-helix (H), betha-sheet (B), maupun coil (C) yang merupakan hasil dari sekuens asam amino yang berikatan dengan ikatan peptida [1]. Struktur tersier adalah gabungan dari struktur sekunder setelah terjadi pelipatan (folding). Fungsi dari protein dapat diketahui jika sudah membentuk struktur tersier dalam bentuk 3D. Namun struktur tersier dapat ditentukan apabila struktur sebelumnya sudah diketahui.
Secara konvensional, Struktur protein dapat diketahui dengan kristalografi sinar-X dan Nuclear Magnetic Resonance (NMR) spectroscopy. Namun kedua teknik
tersebut memakan waktu dan relatif mahal. Dengan demikian, pendekatan berbasis komputasi banyak digunakan untuk melakukan prediksi struktur protein. Prediksi struktur sekunder protein dilakukan untuk menemukan struktur 3D protein berdasarkan struktur primer protein. Ada dua metode prediksi struktur sekunder protein, yaitu metode pemodelan komparatif dan pemodelan de novo atau ab initio. Pemodelan protein komparatif memprediksi struktur protein berdasarkan struktur protein lain yang telah diketahui, sedangkan metode ab initio struktur protein ditentukan dari sekuens primernya tanpa membandingkan dengan struktur protein lain [2].
Berbagai metode digunakan untuk memprediksi struktur sekunder protein yang berbasis komputasi seperti menggunakan Hidden Markov Model (HMM), Hidden Semi Markov Model (HSMM), BP Neural Network dan Quasi-Newton algorithm, algoritme SOM dan SOGR, dan Neural Network. Hidden Markov Model (HMM) merupakan suatu kelas dari model probabilistik yang secara umum dapat diaplikasikan untuk permasalahan deret sekuens yang bersifat linear [3].
Prediksi struktur sekunder protein dengan model HMM dilakukan oleh Martin et al yang menggunakan 2024 sekuens didapatkan tingkat akurasi 34.5% untuk data uji dan 58.3% untuk data latih [2]. Akurasi yang didapat masih kecil karena data yang digunakan masih tidak seimbang. Hidden Markov Model (HMM) merupakan model yang digunakan dalam penelitian ini dengan menggunakan algoritme Viterbi untuk melakukan prediksi struktur sekunder. Data yang digunakan dalam penelitian ini merupakan data yang tidak seimbang, sehingga dilakukan strategi sampling dengan untuk mengatasinya. Penelitian lain dilakukan oleh He dan Edwardo yang mengusulkan metode sampling untuk menangani data yang tidak seimbang. Metode sampling untuk menangani imbalanced data antara lain adalah oversampling dan
2. Hidden Markov Model (HMM)
HMM adalah salah satu pendekatan yang digunakan untuk memodelkan kumpulan sekuens tersebut. HMM telah banyak dikembangkan pada banyak permasalahan seperti speech recognition. Aplikasi pada HMM pada akhirnya akan direduksi untuk menyelesaikan tiga jenis permasalahan, yaitu [5]:
1. Jika diberikan suatu model λ = (A,B,π) , bagaimana menghitung peluang dari sekuens observasi O =
O1,O2,...OT yang dinotasikan dengan P(O | λ).
2. Jika diberikan suatu model λ = (A,B,π) , bagaimana memilih state sekuens I = I1,I2,...IT sehingga
P(O,I | λ) sebagai peluang bersama dari sekuens
observasi O = O1,O2,...OT dan state sekuens
tersebut memiliki nilai maksimum.
3. Mendapatkan parameter model HMM yang optimal sehingga peluang suatu observasi memiliki nilai maksimum, dengan
λ adalah model HMM
A adalah Matriks peluang transisi, B adalah Matriks peluang emisi dan
π adalah Matriks peluang awal / Matriks priority O = O1,O2,...OT adalah variabel observasi
P(O | λ) adalah peluang variabel observasi jika
diberikan model
Hidden Markov Model (HMM) menggambarkan distribusi peluang dari sejumlah sekuens yang tidak terbatas. Pada prediksi struktur sekunder protein, himpunan alpha-helix (H), betha-sheet(B) dan coil (C) merupakan hidden state. Adapun sekuens dari simbol seperti : A, M, L, N dan sekuens asam amino lainnya merupakan variabel yang secara langsung dapat diobservasi atau dikenal dengan observable state.
3. Algoritme Viterbi
Algoritme Viterbi digunakan untuk mencari state yang optimal dari suatu sekuens observasi yang menyebabkan peluang observasi tersebut maksimal. Dalam hal ini termasuk pada permasalahan ke-2 pada HMM. Beberapa permasalahan yang dapat diselesaikan dengan algoritme ini antara lain konversi suara ke teks, deteksi intrusi dalam
bidang jaringan dan permasalahan di bidang
bioinformatika. Algoritme Viterbi diilustrasikan sebagai berikut [5]. Pada tahap inisialiasi, dilakukan perhitungan peluang observasi dalam hal ini asam amino sebagai struktur sekunder pada setiap state dengan notasi δ1 (i).
dalam hal ini i adalah adalah state struktur sekunder H, B dan C. Adapun nilai ψi (i) adalah argumen maksimum yang pada
saat inisialisasi masih bernilai 0.
Pada saat tahap inisialiasi, akan dilakukan perhitungan nilai δt (j) dengan j adalah state berikutnya setelah
sebelumnya state i. Nilai δt (j) akan dihitung berdasarkan nilai
δt-1 (i) dan akan diperoleh nilai maksimumya berdasarkan
persamaan max1≤ i ≤ N[δt-1 (i)aij].bj(ot). sementara nilai ψt(j)
akan argumen yang menyebabkan nilai δt (j) maksimum.
Algoritme Viterbi
4. Prediksi Struktur Sekunder
Protein merupakan elemen yang sangat esensial bagi makhluk hidup. Secara hieraki, struktur protein memiliki beberapa bentuk, yaitu struktur primer protein, struktur sekunder protein, struktur tersier dan struktur quartener. Protein merupakan elemen dasar yang terbentuk dari asam amino dasar. Terdapat 20 asam amino dengan struktur kimia yang berbeda. Asam amino terbentuk dari tiga huruf (triplet) dari kombinasi Asam Deoksiribosa (DNA) yang disebut dengan codon [6].
Struktur protein akan menentukan fungsi dari protein itu sendiri. Hal ini yang menjadikan penelitian mengenai prediksi struktur protein masih menjadi fokus riset di
bidang Bioinformatika khususnya pada bidang
proteomika. Prediksi struktur sekunder protein memiliki peranan untuk menentukan struktur berikutnya.
Pada dasarnya, terdapat tiga bentuk struktur sekunder protein yaitu alpha-helix(H), betha-sheet (B) dan coil (C) yang direduksi dari delapan struktur pada file yang dihasilkan dari program DSSP [7]. Prediksi struktur sekunder protein sebenarnya adalah memberikan label dari
inisialisasi
δ1 (i) = ᴨibi(o1) 1≤ i ≤ N
ψi (i) = 0
rekursif
δt (j) = max 1≤ i ≤ N[δt-1 (i)aij]bj(ot) 2 ≤ t ≤ T , 1 ≤ j ≤ N
ψt(j) = arg max1≤ i ≤ N [δt-1 (i)aij] 2 ≤ t ≤ T , 1 ≤ j ≤ N
terminasi
P* = max 1≤ i ≤ N [δT(i)]
δT* = arg max 1≤ i ≤ N [δT(i)]
lacak balik
qt* = ψt+1 (q*t+1 ), t = T - 1, T - 2,…, 1 dengan:
δ
t(i) = rangkaian terbaik dengan kemungkinan terbesar pada setiap
t dan state i. q = state o = observasi
ψ = path terbaik pada saat sampai state ke i P = peluang
b = matriks emisi a = matriks transisi
t = waktu perhitungan pengamatan t pertama dan berakhir pada status i.
(a) (b) (c)
setiap asam amino pembentuk protein. Ilustrasi prediksi struktur sekunder protein dapat dilihat pada Gambar 1. Pada gambar tersebut asam amino Met, Ala, Pro, Ile dan seterusnya sebagai struktur primer protein, sementara coil (C), betha-sheet (B) dan alpha-helix (H) sebagai struktur sekunder.
Gambar 1. Ilustrasi struktur sekunder protein
Secara visual, potongan struktur protein sekunder dapat dilihat pada Gambar 2 dengan menggunakan software Rasmol 2.7.4.2 [8].
Gambar 2. struktur sekunder protein : (a). alpha-helix (H), (b). betha-sheet (b) dan (c). coil (c)
5. Pengukuran Performansi Prediksi
Penilaian hasil prediksi struktur sekunder protein dilakukan dengan persamaan Q3 Score [9], Recall dan Precison. Persamaan Q3 diformulasikan pada (1).
3 = + + × 100%… … … … … …(1)
Dengan NH , NB dan NC adalah jumlah struktur
sekunder yang terprediksi baik alpha-helix, betha-sheet dan coil secara benar sesuai dengan kelas awalnya. Sementara Ntot adalah jumlah seluruh yang diujikan.
Adapun untuk menilai performa untuk kelas yang tidak seimbang menggunakan pengukuran Recall dan Precision [10]. Nilai tersebut diperoleh dengan terlebih dahulu membuat tabel confusion matrix untuk permasalahan dua kelas (Tabel 1)
Tabel 1 confusion matrix untuk klasifikasi dua kelas terprediksi positif terprediksi negatif Positif TP FP Negatif FN TN = ( + )… … … … … . . … .(2) = ( + )… … … … … . . .(3)
6. Hasil Percobaan
Prediksi struktur protein sekunder dilakukan melalui beberapa tahap. Dimulai dari pengambilan data, praproses data, ekstraksi ciri, pembentukan model HMM, proses prediksi dengan Algoritme Viterbi dan evaluasi.
6.1 Pengambilan data, praproses dan ektraksi ciri
Data yang digunakan pada penelitian ini diperoleh dari bank data protein atau protein data bank (PDB) pada alamat : http://www.rcsb.org/pdb/home/home.do . Protein memiliki beberapa kriteria dalam klasifikasi. Pada penelitian ini diambil kriteria protein berdasarkan Enzyme Classification. Pada kategori ini, terdapat enam kelas jenis protein, yaitu: Hydrolases, Transferases, Oxidoreductases, Lyases, Isomerases dan Ligases.
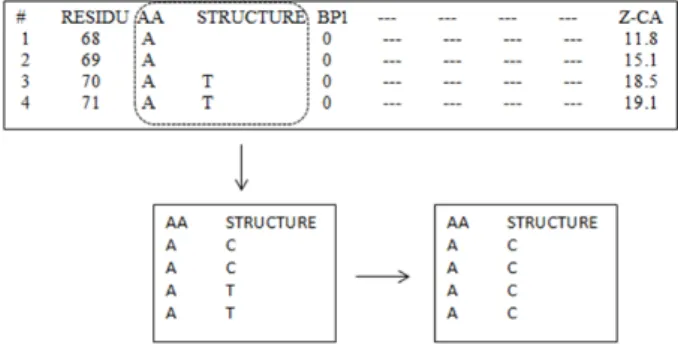
Dari enam kelas tersebut diambil masing-masing seratus data sehingga diperoleh sebanyak enam ratus data sebagai data latih. Adapun label kelas untuk mendapatkan data struktur sekunder, diperoleh dari file pada alamat ftp://ftp.cmbi.ru.nl/pub/molbio/data/dssp/. Pada alamat ini, diperoleh file dengan ekstensi .dssp yang berisi informasi sekuens asam amino dan struktur sekunder dari setiap asam amino. Format data file berkestensi .dssp dapat dilihat pada Gambar 3.
Gambar 3. Contoh format data protein pada file berekstensi. Pada file tersebut, tidak semua informasi diambil. Hanya informasi asam amino (AA) dan informasi struktur sekundernya (STRUCTURE) yang diambil informasinya seperti terlihat pada Gambar 3 di atas. Hasil dari praproses tersebut adalah pasangan antara asam amino dan struktur sekundernya.
Hasil akusisisi data diperoleh enam ratus pasang data dengan jumlah residu (asam amino) sebanyak 394052. Dari data tersebut, terdapat 152782 (40%) struktur alpha-struktur
sekunder struktur primer
helix (H), sebanyak 82355 (21%) struktur betha-sheet (B), dan 158915(39%) struktur coil (C).
Proporsi data struktur tersebut masih tidak seimbang sehingga pada penelitian ini dilakukan teknik
oversampling. Dengan oversampling ini struktur
betha-sheet (B) akan memiliki proporsi yang sesuai dengan kelas lainnya. Visualisasi distribusi data latih data awal dapat dilihat pada Gambar 4.
Gambar 4 Distribusi data awal 6.2 Pembentukan Model HMM
Model HMM dinotasikan dengan lambda ( ). Notasi model HMM mengikuti persamaan = ( , , ). Nilai
menunjukkan matrik transisi. Matrik ini
merepresentasikan hidden state dari struktur sekunder alpha-helix(H), betha-sheet(B) dan coil(C). Pembuatan Hidden Markov Model dilakukan setelah didapatkan kombinasi peluang antara struktur sekunder protein akan direpresentasikan sebagai matriks transisi yang bertindak sebagai hidden state dalam HMM. Nilai merupakan nilai peluang distribusi setiap struktur sekunder yang diperoleh dari data latih. Nilai untuk setiap struktur alpha-helix, betha-sheet dan coil masing-masing adalah 0.39, 0.21 dan 0.40.
Data protein hasil praproses yang diperoleh pada Gambar 3 berupa array berdimensi dua yang merupakan pasangan antara asam amino dan struktur sekundernya. Ilustrasinya dapat dilihat pada Gambar 5.
A A A … … … M L N
C C C … … … H H H
Gambar 5 Ilustrasi pembentukan model matrik transisi dari data protein hasil praproses
Matrik transisi yang akan dibentuk diperoleh dengan cara menghitung peluang perubahan struktur sekunder mulai saat t=0 sampai panjang sekuen N-1. Dari struktur sekunder H, B dan C tersebut akan terbentuk sembilan nilai peluang HH, HB, HC dan seterusnya sampai dengan CC. HH berarti peluang transisi struktur H
jika sebelumnya struktur H. Begitu pula seterusnya untuk seluruh nilai peluang transisi.
Adapun nilai peluang matrik transisi yang diperoleh berdasarkan data latih dapat dilihat pada Tabel 1.
Tabel 1 Matrik Transisi Struktur Sekunder alpha-helix(H), betha-sheet (B) dan coil (C) sebelum oversampling
H B C
H 0.895 0.008 0.096
B 0.009 0.764 0.225
C 0.094 0.114 0.790
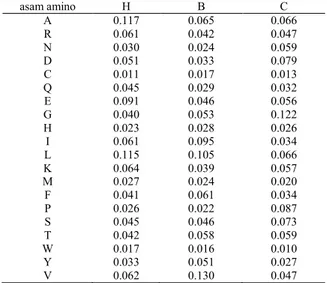
Peluang antara asam amino dan struktur protein sekundernya dinotasikan sebagai B yaitu matrik emisi. Dari Gambar 5 dapat dihitung juga nilai peluang setiap asam amino yang terdapat pada state struktur sekuder tertentu. Nilai peluang matrik emisi yang dihasilkan dapat dilihat pada Tabel 2.
Tabel 2 Matrik Emisi model yang dibentuk sebelum oversampling
asam amino H B C A 0.117 0.065 0.066 R 0.061 0.042 0.047 N 0.030 0.024 0.059 D 0.051 0.033 0.079 C 0.011 0.017 0.013 Q 0.045 0.029 0.032 E 0.091 0.046 0.056 G 0.040 0.053 0.122 H 0.023 0.028 0.026 I 0.061 0.095 0.034 L 0.115 0.105 0.066 K 0.064 0.039 0.057 M 0.027 0.024 0.020 F 0.041 0.061 0.034 P 0.026 0.022 0.087 S 0.045 0.046 0.073 T 0.042 0.058 0.059 W 0.017 0.016 0.010 Y 0.033 0.051 0.027 V 0.062 0.130 0.047
6.3 Evaluasi nilai recall, precision, akurasi dan Q3 score Berdasarkan model tersebut, diperoleh hasil evaluasi beberapa parameter recall, precision, akurasi dan Q3
score. Nilai-nilai recall, precision dan akurasi dapat
dilihat pada Tabel 3.
Tabel 3 Evaluasi hasil recall dan precision identifikasi struktur sekunder alpha-helix (H) betha-sheet
Tabel 3 memperlihatkan evaluasi dari data uji dengan distribusi data seperti pada Gambar 6.
recall Precision akurasi
H B C H B C H B C
40.5 0.4 63.1 40.8 26.7 44.7 54.4 81.9 49.9
Gambar 6 Distribusi data uji untuk melakukan evaluasi Berdasarkan hasil tersebut, dilakukan proses resampling pada data pelatihan untuk memberpaiki model HMM yang dibuat. Teknik ini diharapkan dapat meningkatkan nilai-nilai evaluasi tersebut. Adapun nilai Q3 score yang diperoleh dari hasil evaluasi sebesar 43.19 persen.
6.4 Strategi Sampling
Untuk memperbaiki model HMM, teknik oversampling terhadap data latih dilakukan pada data betha-sheet (B) yang memilliki proporsi lebih kecil. Duplikasi data ditentukan dari panjang segmen betha-sheet (B). Dari setiap file data latih disetiap data dipilih segmen betha-sheet (B) yang terpanjang. Kemudian diduplikasi sebanyak n kali dengan ketentuan terdapat minimal 100 residu betha-sheet (B) hasil duplikasi di setiap data, agar jumlah dari bethasheet (B) dapat mendekati jumlah dari alpha -helix (H) dan coil (C). Hasil dari duplikasi tersebut diletakkan di barisan paling bawah pasangan asam amino dengan strukturnya. Skema oversampling dapat dilihat pada Gambar 6.
Gambar 6. Ilustrasi proses oversampling struktur betha-sheet (B) Setelah dilakukan oversampling secara acak pada residu betha-sheet (B), jumlah dari betha-sheet (B) yang semula 82355 menjadi 151445 residu. Gambar 10 menunjukkan perbandingan dari alpha-helix (H), betha-sheet (B), dan coil (C) setelah dilakukan oversampling. Visualisasi data hasil oversampling dapat dilihat pada Gambar 7.
Gambar 7. Distribusi data latih setelah oversampling Dari gambar terlihat bahwa setelah dilakukan
oversampling, distribusi kelas struktur sekunder relatif
seragam. Beriktunya proses pembentukan model dapat dilanjutkan. Data hasil oversampling kemudian dilatih kembali untuk mendapatkan model HMM yang baru. Model HMM yang baru ini akan menghasilkan matrik emisi dan matrik transisi setelah proses oversampling. Matrik transisi setelah proses oversampling dapat dilihat pada Tabel 4.
Tabel 4 Matrik Transisi Struktur Sekunder alpha-helix(H), betha-sheet (B) dan coil (C) setelah oversampling
H B C
H 0.895 0.008 0.096
B 0.005 0.866 0.128
C 0.094 0.114 0.789
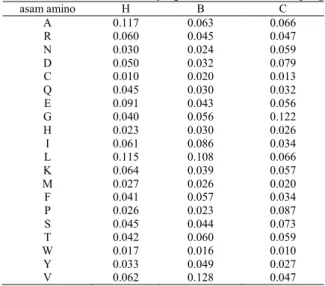
Adapun matrik emisi setelah proses oversampling pada model HMM dapat dilihat Tabel 5.
Tabel 5 Matrik Emisi model yang dibentuk setelah oversampling
asam amino H B C A 0.117 0.063 0.066 R 0.060 0.045 0.047 N 0.030 0.024 0.059 D 0.050 0.032 0.079 C 0.010 0.020 0.013 Q 0.045 0.030 0.032 E 0.091 0.043 0.056 G 0.040 0.056 0.122 H 0.023 0.030 0.026 I 0.061 0.086 0.034 L 0.115 0.108 0.066 K 0.064 0.039 0.057 M 0.027 0.026 0.020 F 0.041 0.057 0.034 P 0.026 0.023 0.087 S 0.045 0.044 0.073 T 0.042 0.060 0.059 W 0.017 0.016 0.010 Y 0.033 0.049 0.027 V 0.062 0.128 0.047
Setelah melakukan oversampling, dilakukan evaluasi kembali terhadap nilai recall, precision, akurasi dan Q3
score. Tabel 6 memperlihatkan hasil evaluasi tersebut
Tabel 6 Evaluasi hasil recall dan precision identifikasi struktur sekunder alpha-helix (H) betha-sheet setelah oversampling
Secara umum tidak terdapat perubahan yang signifikan terhadap nilai-nilai tersebut. Hanya saja pada beberapa nilai memang mengalami kenaikan. Nilai recall pada prediksi struktur betha-sheet (B) mengalami sedikit kenaikan dari 0,4 menjadi 3,9. Sementara nilai precision betha-sheet mengalai sedikit kenaikan dari 26,7 menjadi 30,4. Namun, terdapat trade off dari adanya kenaikan ini yaitu penurunan recall dan precision pada struktur alpha-helix (H) dan coil (C).
Dengan demikian, perlakuan dengan oversampling ini tidak memberikan kenaikan yang signifikan. Hal ini dibukatikan dengan nilai Q3 score yang diperoleh setelah dilakukan oversampling. Berdarkan hasil percobaan yang dilakukan, nilai Q3 score pada data uji dengan model HMM baru hasil oversampling sebesar 43,21 persen hanya memiliki selisih sebesar 0.02 jika dibandingkan dengan Q3
score sebelum dilakukan oversampling.
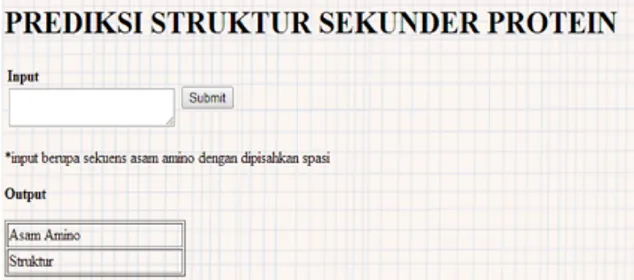
6.5 Implementasi Program
Prediksi struktur sekunder protein dengan menerapkan Algoritme Viterbi diaplikasikan dalam bentuk website karena umumnya prediksi struktur sekunder adalah aplikasi yang berfungsi sebagai layanan online di bidang bioinformatika Gambar 8 adalah contoh antarmuka untuk sistem prediksi tersebut.
Gambar 8 Antarmuka implementasi algoritme Viterbi untuk prediksi struktur sekunder protein
4. Kesimpulan dan Peluang Penelitian
Berdasarkan hasil penelitian dapat disimpulkan bahwa algoritme viterbi dapat diterapkan untuk permasalahan prediksi struktur sekunder protein. Teknik resampling tidak memberikan pengaruh yang signifikan untuk memperpaiki nilai Q3 score. Dengan demikian, diperlukan teknik ekstraksi informasi lain yang berbasis kimia dan fisika seperti Physicochemical sebagai penciri untuk
melakukan ekstraksi informasi untuk prediksi strukutur sekunder protein ini.
REFERENSI
[1] Atar E, Ersoy O, Ozyilmaz L. 2005. Prediction of protein secondary structure by SOM and SOGR algorithm. IEEE. doi: 10.1109/CIMA.2005.1662358.
[2] Martin J, Gibrat JF, Rodolphe J. 2005. Hidden markov model for protein secondary structure. Oxford University
Press. 14(9): 755-763.
[3] Eddy SR. 1998. Profile hidden markov model.
Bioinformatics Review.14:755-763
[4] He H, Edwardo AG. 2009. Learning from imbalanced data.
IEEE Transactions on Knowledge and Data Engineering.
21(9):1263-1284.
[5] Rabiner LR. 1989. A Tutorial on hidden markov model and selected applications in speech recognitions. Proceedings of
the IEEE. 77 (2), 257-286
[6] Polanski A, Kimmel M.2007. Bioinformatics. Germany (DE): Springer Sciene.
[7] Wang J, Ping Li J. 2008. Protein secondary structure prediction based on BP neural network and quasi-newton algorithm. IEE. doi : 10.1109/CACIA.20084769988 [8] Haryanto T, Buono A, Nugroho AS. 2011. Pengembangan
Hidden Semi Markov Model dengan Distribusi Durasi Empiris untuk Prediksi Struktur Sekunder Protein. Thesis. Institut Pertanian Bogor (IPB).
[9] Lakizadeh A, Marashi S-A. 2009. Addition Of Contact Number Information Can Improve Protein Secondary Structure Prediction By Neural Networks. EXCLI Journal 2009;8:66-73 ISSN 1611-2156.
[10] Han H, Wang WY, Mao BH. Borderline – SMOTE. 2005. A New Over-Sampling Method in Imbalanced Data Sets Learning. Advanced in Intelligent Computing: Lecture Notes
in Computer Science. Volume 3644. pp 878-887
Dian Puspita Sari, menyelesaikan program sarjana dengan memperoleh gelar Sarjana Komputer (S.Kom) di Departemen Ilmu Komputer Institut Pertanian Bogor (IPB) pada tahun 2014. *Toto Haryanto [penulis korespondensi], memperoleh gelar Sarjana Komputer (S.Kom) dan Magister Sains (M.Si) dari Institut Pertanian Bogor, Indonesia pada tahun 2006 dan 2011. Minat riset penulis pada bidang Bioinformatika dan Sistem Pakar. Saat ini sebagai Pengajar dan Peneliti di Departemen Ilmu Komputer Institut Pertanian Bogor (IPB). Peneliti juga sebagai anggota bagian grup riset Bioinformatika di Departemen Ilmu Komputer FMIPA IPB sekaligus administrator web kelompok riset ini .
Recall precision Akurasi
H B C H B C H B C
39.